

Журнал неврологии и психиатрии / 2007 / NEV_2007_02_13
.pdfОБЗОРЫ
Митохондриальные цитопатии
О.М. ПОЗДНЯКОВ, Л.Л. БАБАКОВА, Б.М. ГЕХТ
Mitochondrial cytopathies
O.M. POZDNYAKOV, L.L. BABAKOVA, B.M. GEKHT
НИИ общей патологии и патофизиологии РАМН, Москва
Âнастоящее время митохондриология выделилась в самостоятельное научное направление. Более того, открытие в последние годы ведущей роли митохондрий в чувствительности к лекарствам, их ключевой ролью в старении, апоптозе и нейродегенеративных расстройствах привело к созданию митохондриальной медицины. Важным ее разделом являются болезни, связанные с нарушением функции митохондрий, — митохондриальные цитопатии (МЦ).
Сейчас большинство исследователей признают, что митохондрии в клетках животных и хлоропласты в растительных являются отдаленными потомками архибактерий, которые на заре жизни внедрились в первобытные эукариотические клетки и постепенно превратились в эндосимбионтов [9]. Развитие геохимии, молекулярного филогенеза
èмолекулярной биологии когда-нибудь позволит расшифровать последовательность этих событий. Пока же мы имеем дело с их результатами и концепция симбиоза принимается как наиболее вероятная гипотеза, в пользу которой, помимо второстепенных, свидетельствуют два фундаментальных факта: во-первых, митохондрии — единственные органеллы, имеющие собственный геном [21], и во-вто- рых, генетический код митохондриальной (митДНК) и ядерной (яДНК) ДНК различен [27]. Последнее обстоятельство является веским аргументом против существовавшего ранее предположения о происхождении митохондрий в результате компартментализации части ядерного генома. В свете современных знаний удивительно, что еще в 1890 г. Р. Альтман назвал эти структуры (тогда еще не устоялось название «митохондрии») в книге «Elementarorganismen» «биопластами» и предположил, что они являются автономными элементарными живыми частицами, образующими, подобно бактериям, колонии в клетке-хозяине.
Главная функция митохондрий — продукция энергии для клеток в виде АТФ в результате окислительного фосфорилирования различных субстратов. Респираторная цепочка
состоит из пяти энзимных комплексов [19]. В функции митохондрий входят также β-окисление жирных кислот, цикл трикарбоновых кислот и ряд других метаболических функций. Они играют важную роль во внутриклеточной сигнализации, апоптозе, промежуточном метаболизме, а также в метаболизме аминокислот, липидов, холестерина, стероидов и нуклеотидов [7].
Âпроцессе симбиоза митохондрии утратили значительную часть самостоятельности и передали бóльшую часть своего генома ядрам клеток. В результате их жизнь и функционирование только в малой степени обеспечиваются собственной ДНК. Бóльшая часть митохондриальных белков кодируется в ядрах клеток и доставляется в митохондрии из цитоплазмы [35]. В постмитотических клетках, таких как мышечные волокна, нейроны и кардиомиоциты, митохон-
© Коллектив авторов, 2007
Zh Nevrol Psikhiatr Im SS Korsakova 2007;107:2:64—69
дрии имеют ограниченный срок жизни (несколько недель) [18]. В нормальных условиях их новообразование требует координации между митохондриальной ДНК, кодирующей 13 из 80 белковых субъединиц респираторной цепи, 2 белковых субъединицы мРНК и 22 митохондриальных тРНК (всего 37 генов), и ядерным геномом, кодирующим более 99% митохондриальных белков.
Таким образом, митохондрии, являющиеся потомками свободно живущих эубактерий, сохранили только минимальные остатки своего генома в эволюционном процессе эндосимбиоза. Бóльшая часть генома была или передана ядру эукариотического хозяина, или утрачена, так как животные клетки предоставляют митохондриям «и стол, и дом», используя в свою очередь энергию, запасаемую в продукте жизнедеятельности митохондрий в виде АТФ.
Фрагменты из кодирующей и некодирующей областей митДНК находятся как ископаемые остатки в ядерном геноме различных эукариотов [3]. Буквально в последние годы был обнаружен полиморфизм включения фрагментов митДНК в человеческой популяции и у шимпанзе и определена скорость фиксации этих последовательностей у че- ловека. Более того, была проведена корреляция этих данных с наблюдениями включения таких фрагментов, связанных с заболеваниями человека. Показано, что у человека фрагменты митДНК предпочтительно интегрируются в известные или предполагаемые гены и, таким образом, могут быть мутагенными и являться причиной новых заболеваний [24].
В нормальных условиях все митохондрии в клетке имеют одинаковую копию ДНК (гомоплазмия). Однако в митохондриальном геноме могут происходить мутации. К настоящему времени известно более 200 заболеваний, вызванных мутацией митДНК. Они подразделяются на две группы: то- чечные мутации белков, тРНК, рРНК в кодирующих областях, которые часто наследуются по линии матери, или же структурные перестановки — дупликации и делеции, которые обычно являются спорадическими [25].
Как уже говорилось, митДНК кодирует только 13 полипептидных субъединиц дыхательной цепи из 80. Вдобавок энзимы и другие факторы, необходимые для транскрипции, репликации и трансляции, поступают в митохондрии из цитоплазмы клетки. Поэтому неудивительно, что ядерные мутации также могут приводить к нарушению функции митохондрий и в первую очередь нарушению окислительного фосфорилирования в них. Поскольку знания о ядерном геноме на протяжении последних лет значительно расширились, идентифицируются все больше дефектов митохондриями, кодируемых ядром. Различают мутации структурных белков и тРНКаз, нарушающие функционирование респираторной цепи, и мутации, которые нарушают интергеномное взаимодействие между ядром и митохондриями и тем самым вызывают вторичные изменения митДНК [10, 30]. Такие изменения вызывают главным образом делеции митДНК [20, 23] или множественные делеции [12, 14, 31]. Они наследуются главным образом по Менделю.
64 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 2, 2007 |

МИТОХОНДРИАЛЬНЫЕ ЦИТОПАТИИ
Ввиду неравномерного распределения митохондрий в клетках и параллельного существования мутированной и немутированной митДНК в этих органеллах (гетероплазмия) расстройства могут проявляться огромным разнообразием симптомов даже при одинаковых мутациях.
К настоящему времени описано много вариантов нарушения процесса окислительного фосфорилирования в митохондриях человека. Дефект может быть связан с одним или несколькими энзимными комплексами. Как уже говорилось, в одной клетке могут сосущестовать митохондрии нормальные и с нарушенной функцией. За счет первых клетка может функционировать какое-то время. Если же продукция энергии в ней падает ниже определенного порога, происходит компенсаторная пролиферация всех митохондрий, включая дефектные. Естественно, при этом в худшем положении оказываются клетки, которые потребляют много энергии: мышечные волокна, кардиомиоциты, нейроны. Поэтому не случайно именно в биоптатах скелетных мышц были впервые обнаружены дефектные митохондрии.
Из-за утечки в дыхательной цепочке митохондрий постоянно продуцируют свободные радикалы на уровне 1— 2% поглощенного кислорода. Количество продукции радикалов зависит от мембранного потенциала митохондрий, на изменения которого влияет состояние АТФ-зависимых калиевых каналов митохондрий. Открытие этих каналов вле- чет за собой возрастание образования свободных радикалов. Свободные радикалы играют огромную роль в старении митохондрий и, следовательно, в старении эукариотических клеток [32]. Агрессивная среда вокруг митохондрий при зна- чительном увеличении их количества и нарушении функции может быть одним из факторов развития деструктивных изменений в клетках. Изменения мембранного потенциала митохондрий, а также образование свободных радикалов в свою очередь оказывают повреждающее влияние на другие белки митохондриальных мембран.
Митохондриальная ДНК содержит очень небольшую некодирующую область и хорошо доступна для радикалов, генерируемых респираторной цепочкой в ходе аэробного образования АТФ, а способность митохондрий к восстановлению мала. Поэтому уровень повреждения митДНК, возрастающий с возрастом, влияет на степень гетероплазмии. Принято считать, что 10% митохондрий с измененной ДНК не оказывает влияния на фенотип. Вместе с тем высокая скорость их обновления и короткая жизнь создают своеобразный способ восстановления путем замещения для коррекции повреждения свободными радикалами [2].
Первое упоминание о болезни, связанной с дефектом митохондрий, относится к 1962 г.: R. Luft и соавт. [15] описали случай заболевания, при котором имело место нарушение сопряжения дыхания и фосфорилирования в митохондриях скелетных мышц у пациента с нетиреоидным гиперметаболизмом. В последующие годы были описаны клинические, биохимические и морфологические аспекты митохондриальных энцефаломиопатий. Для развития этого направления большую пользу оказало использование модифицированной окраски по Гомори, с помощью которой удавалось выявлять в скелетных мышцах волокна с измененными митохондриями — так называемые ragged-red волокна (RRF). Особую роль сыграло введение в клиническую практику электронной микроскопии.
После открытия в 1988 г. мутаций митДНК [11, 36] концепция митохондриальных болезней вызвала буквально взрыв исследований в этой области, и 90-е годы по справедливости стали декадой митохондриального генома.
МЦ — гетерогенная группа системных расстройств, обусловленных мутациями митохондриального или ядерного генома, которые поражают преимущественно мышечную, нервную и нервно-мышечную системы. Митохондриальные мутации проявляются широким рядом клинических симптомов [16, 17, 28]. Эти мутации способны вовлекать тРНК, рРНК или структурные гены и могут выражаться биохими-
чески как дефекты всей электронно-транспортной цепи или как дефекты отдельных энзимов [37]. МЦ поражают множественные органные системы, но, как указывалось, предпочтительно поражаются органы с высокой метаболической активностью — мозг и скелетные мышцы. Таким образом, скелетные мышцы являются тканью выбора для выявления митохондриальных болезней [13].
На протяжении 90-х годов идентификация множества митохондриальных генных дефектов, обусловливающих клинически совершенно разные расстройства, ставила в тупик клиницистов в отношении диагностики гетерогенных и сложных синдромов. Действительно, здесь было о чем задуматься. Достаточно перечислить характерные признаки МЦ.
Скелетные мышцы: низкая толерантность к физической нагрузке, гипотония, проксимальная миопатия, включающая фациальные и фарингеальные мышцы, офтальмопарез, птоз.
Сердце: нарушения сердечного ритма, гипертрофиче- ская миокардиопатия.
ЦНС: атрофия зрительного нерва, пигментная ретинопатия, миоклонус, деменция, инсультоподобные эпизоды, расстройства психики.
Периферическая нервная система: аксональная нейропатия, нарушения двигательной функции гастроинтестинального тракта.
Эндокринная система: диабет, гипопаратиреоидизм, нарушение экзокринной функции панкреас, низкий рост.
Поскольку МЦ проявляются у человека целым рядом различных симптомов, клиницисты попробовали объединить некоторые группы наиболее часто встречающихся комбинаций симптомов в синдромы. Все они по странному сте- чению обстоятельств являются сокращениями английских названий симптомов. Поэтому приходится приводить основные синдромы на этом языке.
MELAS — Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis and Stroke-like episodes (митохондриальная миопатия, энцефалопатия, лактатный ацидоз и инсультоподобные эпизоды).
CPEO/PEO — External Ophthalmoplegia, Ophthalmoplegia plus syndrome (офтальмоплегия, связанная с поражением глазодвигательных мышц, офтальмоплегия плюс синдром).
KSS — Kearns-Sayre Syndrome — retinopathy, proximal muscle weakness, cardiac arrythmia and ataxia (ретинопатия, слабость проксимальных мышц, аритмия и атаксия).
MERRF — Myoclonic Epilepsy associated with Ragged Red Fibres (миоклоническая эпилепсия с обнаружением RRF).
LHON — Leber Hereditary Optic Neuropathy (врожденная нейропатия глазного нерва).
Leigh syndrome — infantile subacute necrotizing encephalopathy (инфантильная подострая некротизирующая энцефалопатия).
NAPR — Neuropathy, Ataxia and Pigmentary Retinopathy (нейропатия, атаксия и пигментная ретинопатия).
Здесь приведены основные синдромы, при этом следует иметь в виду, что симптомы при разных синдромах могут перекрещиваться. Проявления и тяжесть синдромов зависят от того, какая пропорция митохондрий повреждена в клетке.
Наиболее обычными при МЦ являются неврологиче- ские симптомы, поскольку, как указывалось выше, ткани, в наибольшей степени зависящие от окислительного фосфорилирования, составляют основу патогенеза этих страданий. Наиболее частой мутацией является 3243А>G точеч- ная мутация в MTTL 1 митДНК, представленная клиниче- ски митохондриальной энцефалопатией, лактатным ацидозом и инсультоподобными эпизодами (MELAS). У таких пациентов наблюдаются также миопатия и периферическая нейропатия.
МЦ могут быть спорадическими или наследственными, при этом они наследуются, как и гемофилия, по линии матери [26], только в отличие от гемофилии поражают лиц
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 2, 2007 |
65 |
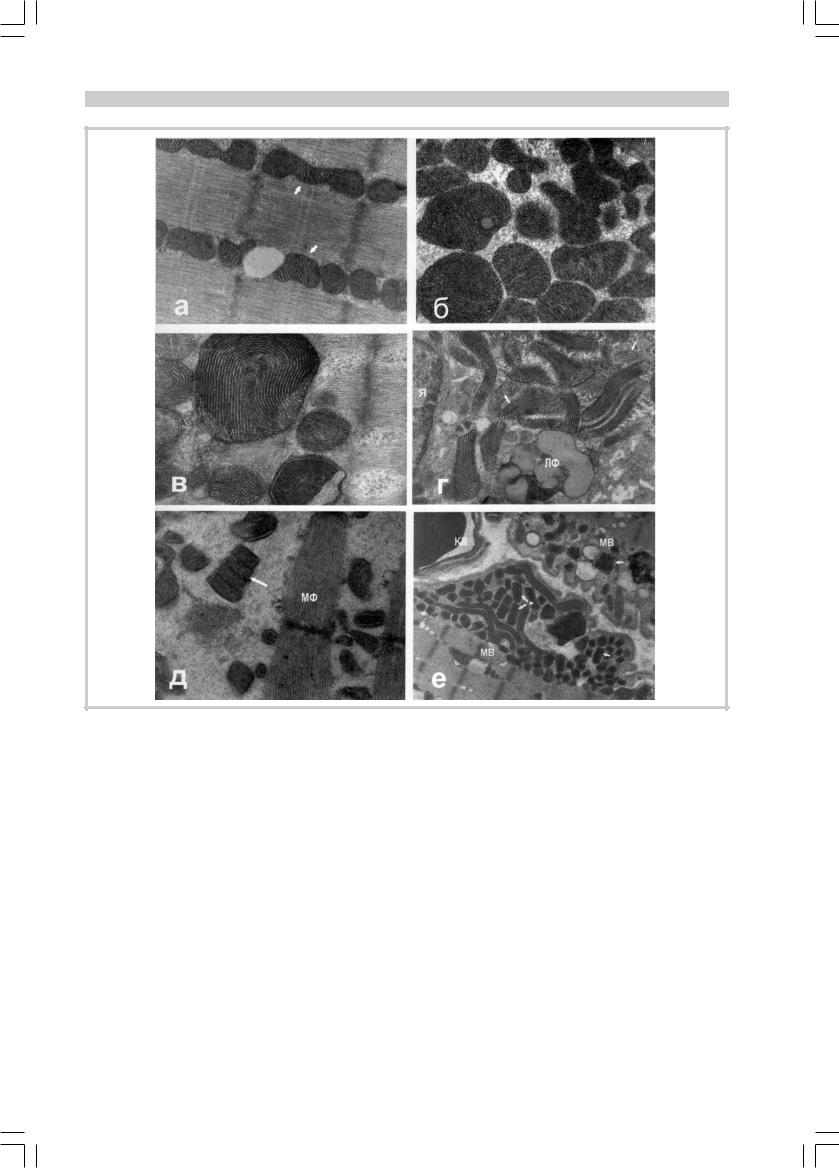
ОБЗОРЫ
Рис. 1. Ультраструктура митохондрий в скелетных мышцах больных митохондриальной миопатией (биопсийный материал).
а — митохондрии (стрелки) между миофибриллами в нормальной мышце человека. ½29 000; б — скопление митохондрий разной величины и формы с измененной структурой. ½26 000; в — митохондрия с дугообразными кристами, имитирующими картину отпечатка пальца. ½22 500; г — лентовидные митохондрии с продольно расположенными кристами; на поперечных срезах митохондрии видно трубчатое строение крист (стрелки). ½32 000; д — разнообразные по величине, форме и структуре митохондрии между атрофирующимися миофибриллами; репликация митохондрии (стрелка). ½13 000; е — субсарколеммальные скопления митохондрии в двух соседних мышечных волокнах; репликация митохондрий (стрелки). ½22 500. Обозначения на рис. 1—4: МВ — мышечное волокно; Я — ядро мышечного волокна; МФ — миофибрилла; ЛФ — липофусцин.
обоего пола. Применение некоторых фармакологических средств, например зидовудина (AZT), также может индуцировать проксимальную миопатию и появление RRF [5]. С возрастом в митДНК накапливаются мутации, в результате чего у пожилых людей также могут встречаться RRF [38]. МитДНК накапливает мутации более чем в десять раз быстрее по сравнению с ядерным геномом. Это связано с тем, что митДНК лишена защитных гистонов и, как уже упоминалось, ее окружение чрезвычайно богато реактивными видами кислорода, являющимися побочным продуктом метаболических процессов, протекающих в митохондриях. Кроме того, восстановительные механизмы митДНК малоэффективны по сравнению с ядерной [4].
МЦ встречаются не только у человека, но и у животных. В литературе приведен случай МЦ у 9-летней немецкой овчарки. Диагноз был установлен на основании данных
морфологического (RRF), биохимического и генетического исследований [22].
Окончательный диагноз МЦ ставится исходя из результатов биохимических и молекулярных исследований, что доступно в специально оборудованных центрах. К сча- стью, существует ряд рутинных клинических методов исследования, которые можно использовать при подозрении на МЦ.
Лактатный ацидоз является практически постоянным спутником митохондриальных болезней. Однако только этот признак является недостаточным для постановки диагноза, так как он может выявляться и при других патологических состояниях. В этом отношении может быть полезным измерение уровня лактата в венозной крови после умеренной физической нагрузки, например на велоэргометре.
66 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 2, 2007 |
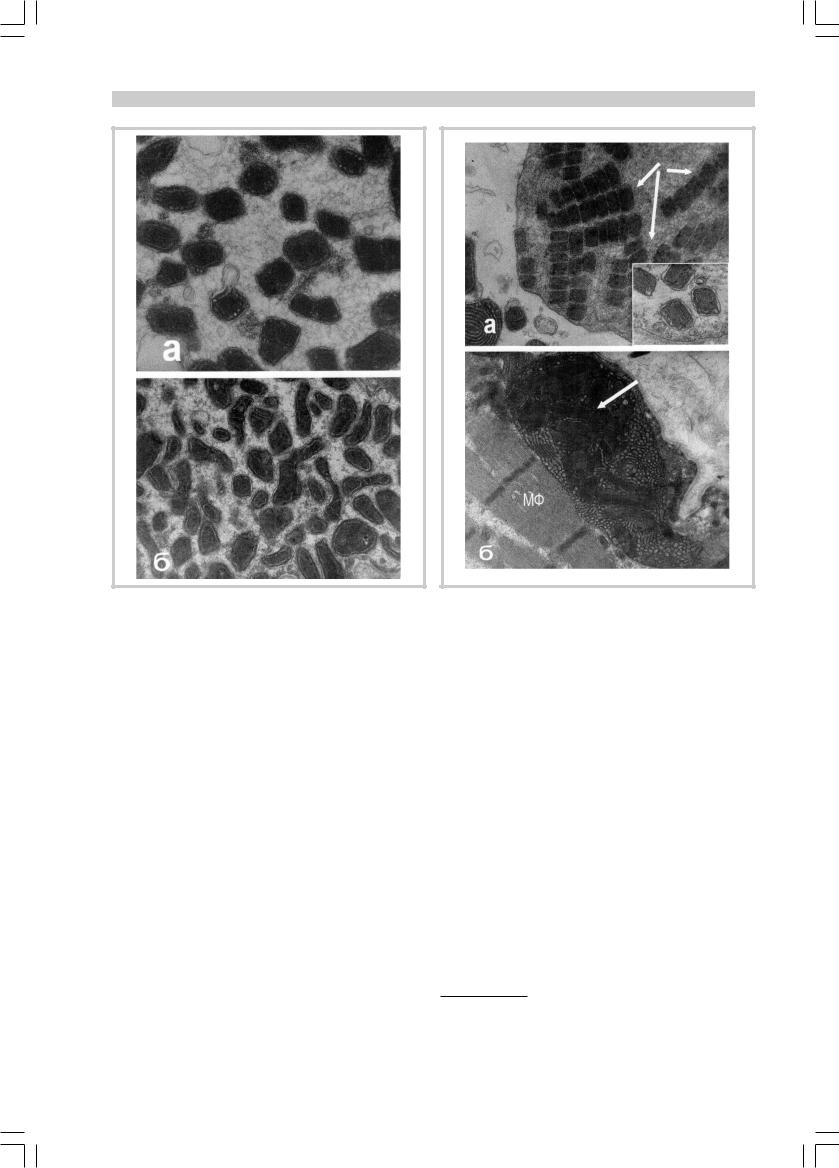
МИТОХОНДРИАЛЬНЫЕ ЦИТОПАТИИ
Рис. 2. Скопления аномальных мелких митохондрий с относительно однородной структурой и паракристаллическими включениями.
à — ½48 000; á — ½34 000.
Данные ЭМГ-исследования сами по себе также не могут быть маркером МЦ. Вместе с тем нормальная или близкая к нормальной ЭМГ у пациентов с выраженной мышеч- ной слабостью может быть подозрительной в отношении митохондриальной патологии. Данные ЭЭГ не являются достаточно специфическими.
Исследование биоптатов скелетных мышц является наиболее информативным методом при постановке диагноза МЦ. Помимо обнаружения RRF при трехцветной окраске по Гомори, полезными являются другие гистохимические и иммунологические исследования: окраска на цитохром с-оксидазу и сукцинатдегидрогеназу, иммунногистохими- ческие исследования с применением антител к отдельным субъединицам дыхательного комплекса. Прекрасные результаты дает электронно-микроскопическое исследование скелетных мышц. Поэтому данный метод надо использовать, если имеется такая возможность. Кроме того, мышечная ткань удобна для биохимического исследования респираторной цепочки, а также как материал для генетического исследования [6].
Нарушение функции митохондрий сопровождается выраженными изменениями их структуры. Как и следовало ожидать, эти изменения наиболее демонстративны в скелетных мышцах. В нормальных условиях митохондрии в скелетных мышцах, располагающиеся между миофибриллами, имеют удлиненную форму, электронно-плотный матрикс и относительно редкие пластинчатые кристы (рис. 1, а). Они могут также образовывать небольшие субсарколеммальные скопления. При электронно-микроскопическом исследовании
Рис. 3. Митохондрии необычной структуры, являющиеся, повидимому, показателем их «эволюции» при митохондриальной миопатии.
а — «колония» реплицирующихся митохондрий (стрелки); на врезке хорошо видно их паракристаллическое содержимое. ½36 000, врезка — ½65 000; б — гигантская митохондрия с паракристаллическими включениями (стрелка) и трубчатыми кристами на периферии мышечного волокна. ½24 500.
мышц больных МЦ1 состояние их митохондриального аппарата меняется кардинально. Это касается количества митохондрий, разнообразия их величины, формы и внутренней структуры. Иногда изменения структуры заходят настолько далеко, что такие образования можно с трудом идентифицировать как митохондрии. Патологически измененные митохондрии представлены на рис. 1—4, некоторые пояснения даны в подписях.
Наиболее характерные изменения митохондрий обусловлены удлинением крист. Иногда это приводит к удлинению самих митохондрий (лентовидные), в других случаях — к спиральному закручиванию крист. Изменения претерпевают и сами кристы, становясь из пластинчатых трубчатыми. Все это можно рассматривать как попытку скомпенсировать недостаточную эффективность функции дыхательной цепочки в митохондриях. Другой распространенной особенностью дефектных структур является наличие в них паракристаллических включений. Наконец, можно проследить эволюцию митохондрий от более простых к более сложным.
1 Материал был получен в ходе диагностических биопсий в отделе нервно-мышечной патологии человека НИИ общей патологии и патофизиологии РАМН [1].
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 2, 2007 |
67 |

ОБЗОРЫ
Рис. 4. Конечная стадия «эволюции» митохондрии при миохондриальной миопатии.
а, б — скопление полиморфных, вакуолизированных и распадающихся митохондрий на периферии мышечного волокна; гигантская митохондрия в центре содержит подобие ядерного материала (стрелка). а — ½14 500; б — ½12 400.
Морфологические, в частности электронно-микроско- пические исследования изменений митохондрий в клетках позволяют ответить на некоторые вопросы относительно патогенеза МЦ. Так, преобладание в скелетных мышцах патологически измененных митохондрий, относительное количество которых возрастает по мере прогрессирования заболевания, может быть результатом нарушения их нормального оборота, т.е. увеличения продолжительности жизни наряду с продолжающейся репликацией. О том же свидетельствует усложнение их структуры по мере увеличения размеров. При этом картины, которые можно трактовать как репликацию дефектных митохондрий, встречаются очень часто в отличие от нормальных мышц, что приводит к образованию скоплений одинаково измененных митохондрий.
Возникает также вопрос, что является причиной такого разнообразия структуры патологически измененных митохондрий. Результат ли это разных мутаций в митохондриальной или ядерной ДНК, или речь идет о разных стадиях изменений, или это комбинация того и другого? Наконец, можно лишний раз вспомнить о единстве структуры и функции.
Как уже упоминалось, при МЦ могут поражаться несколько систем (мышечная, нервная, сердечно-сосудистая и др.). Если речь идет о наследовании заболевания, то это
легко объяснить. Однако в случаях приобретенной МЦ неясным остается вопрос, вовлекаются ли разные системы одновременно или последовательно, и если последнее справедливо, то каков механизм (или механизмы) передачи мутаций от системы к системе.
МЦ являются относительно новым классом болезней человека. При этом их расчетная частота встречаемости составляет 1:8000. Поэтому при обнаружении хотя бы одного из описанных выше симптомов следует в первую очередь исследовать биоптаты скелетных мышц на предмет выявления RRF.
Что касается терапии этой формы патологии человека, то речь может идти пока только о симптоматической. Лече- ние включает также антиоксиданты (витамин Е, α-липое- вая кислота), доноры и акцепторы электронов (коэнзим Q10, рибофлавин), альтернативные источники энергии (креатин моногидрат), стратегию снижения уровня лактата (дихлорацетат) и физические упражнения [33].
Разработка методов генной терапии и вообще патогенетических методов лечения еще находится в стадии экспериментов [8]. Одним из наиболее перспективных направлений генной терапии является попытка изменить уровень гетероплазмии путем или селективной ингибиции репликации митохондрий, или разрушения мутантной ДНК [29, 34]. Такой подход базируется на факте, что требуется большое число копий мутантной митДНК, чтобы эффект мутации стал фенотипически явным. Аргументируется, что при эффективном уменьшении популяции мутантной ДНК увеличивается количество нормальной и это приводит в результате к нормализации фенотипа.
Выявление мутаций митДНК в качестве причины МЦ позволило начать их моделирование на экспериментальных животных. Модель митохондриальной миопатии была воспроизведена путем повреждения митохондриального фактора транскрипции А (Tfam) в скелетных мышцах мыши. У нокаутированных животных развивалась миопатия с мышеч- ными RRF и прогрессивным нарушением функции дыхательной цепочки в скелетных мышцах [39].
К настоящему времени по разным аспектам МЦ опубликовано огромное число работ, и вопросы их этиологии и патогенеза уже не представляются такими загадочными, как это было всего десятилетие назад. В данной статье представлены основные материалы по МЦ. По сути она является введением в эту сложную, актуальную и увлекательную проблему. И цель ее — ознакомление широких кругов специа- листов-неврологов с этой формой патологии человека, которая может встретиться у неврологических больных. Проблема МЦ возникла первоначально как чисто клиническая, однако ее разработка вышла далеко за эти рамки и обогатила митохондриологию в отношении биологии, функций митохондрий и механизмов их нарушения. В результате был определен новый класс болезней, уникальных по этиологии и патогенезу, связанных с особенностями происхождения этих клеточных органелл, их биологии и функции. В настоящее время неизвестны формы патологии, в основе которых лежит неконтролируемуя внутриклеточная репликация органелл клетки, заканчивающаяся ее гибелью. Положение усугубляется еще и тем, что при этом в первую очередь гибнут постмитотические, т.е. практически невосполнимые, клетки — нервные и мышечные. Все это, а также тот факт, что МЦ являются отнюдь не редкими заболеваниями и могут передаваться по наследству, делают клинический аспект проблемы чрезвычайно актуальным, тем более что с ухудшением экологии и умножением в среде обитания мутагенных факторов можно ожидать учащения этой формы патологии. Наследственный же характер части МЦ означает раннее начало их проявлений. Поэтому разработка методов их патогенетической терапии чрезвычайно важна.
68 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 2, 2007 |

МИТОХОНДРИАЛЬНЫЕ ЦИТОПАТИИ
ЛИТЕРАТУРА
1.Гехт Б.М., Бабакова Л.Л., Касаткина Л.Ф. и др. Клинико-элек- трофизиологический анализ митохондриальной миопатии. Журн невропатол и психиат 1987; 87: 7: 967—1075.
2.Beckman K.B., Ames B.N. The free radical theory of aging matures. Physiol Rev 1998; 78: 547—581.
3.Blanchard J.L., Schmidt G.W. Mitochondrial DNA migration events in yeast and humans: Integration by a common end-joining mechanism and alternative perspectives on nucleotide substitution patterns. Mol Biol Evol 1996; 13: 893.
4.Bohr V.A., Stevnsner T., de Souza-Pinto N.C. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene 2002; 286: 127—134.
5.Chariot P., Drogou I., de Lacroix-Szmania I. et al. Zidovudine-in- duced mitochondrial disorder with massive liver steatosis,myopathy, lactic acidosis, and mitochondrial DNA depletion. J Hepatol 1999;
30:1: 156—160.
6.Chaturvedi S., Bala K., Thakur R., Suri V. Mitochondrial encephalomyopathies: advances in understanding. Med Sci Monit 2005; 11: 7: 238—246.
7.Chinnery P.F., Schon E.A. Mitochondria. J Neurol Neurosurg Psychiat 2003; 74: 1188—1199.
8.D’Souza G.G., Weissig V. Approaches to mitochondrial gene therapy. Curr Gene Ther 2004; 4: 3: 317—328.
9.Dyall S.D., Brown M.T., Johnson P.J. Ancient invasions: from endosymbionts to organelles. Science 2004; 304: 5668: 253—257.
10.Hirano M.,Vu T.H. Defects of intergenomic communication: where do we stand? Brain Pathol 2000; 10: 3: 451—461.
11.Holt I., Harding A.E., Morgan-Hughes J.A. Deletion of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988; 331: 717—719.
12.Kaukonen J., Zeviani M., Comi G.P. et al. A third locus predisposing to multiple deletions of mtDNA in autosomal dominant progressive external ophthalmoplegia. Am J Hum Genet 1999; 65: 256—261.
13.Larsson N.G., Oldfors A. Mitochondrial myopathies. Acta Physiol Scand 2001; 171: 385—393.
14.Li F.Y., Tariq M., Croxen R. et al. Mapping of autosomal dominant progressive external ophthalmoplegia to a 7-cM critical region on 10q24. Neurology 1999; 53: 1265—1271.
15.Luft R., Ikkos D., Palmieri G. et al. A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a correlated clinical, biochemical, and morphological study. J Clin Invest 1962; 41: 1776—1804.
16.Mauro di S., Bonilla E., Zeviani M. et al. Mitochondrial myopathies. Ann Neurol 1985; 17: 521—538.
17.Mauro di S., Schon E.A. Mitochondrial respiratory-chain diseases. N Engl J Med 2003; 348: 2656—2668.
18.Menzies R.A., Gold P.H. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem 1971; 246: 2425— 2429.
19.Mitchell P. Keilins respiratory chain concept and its chemiosmotic consequences. Sience 1979; 206:1148—1159.
20.Moraes C.T., Shanske S., Tritschler H.J. et al. MtDNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases. Am J Hum Genet 1991; 48: 492—501.
21.Nass S., Nass M.M.K. Intramitochondrial fibres with DNA characteristics. J Cell Biol 1963; 19: 593—629.
22.Paciello O., Maiolino P., Fatone G., Papparella S. Mitochondrial myopathy in a german shepherd dog. Vet Pathol 2003; 40: 5: 507—511.
23.Poulton J., Morten K., Freeman-Emmerson C. et al. Deficiency of the human mitochondrial transcription factor h-mtTFA in infantile mitochondrial myopathy is associated with mtDNA depletion. Hum Molec Genet 1994; 3: 1763—1769.
24.Ricchetti M., Tekaia F., Dujon B. Continued Colonization of the Human Genome by Mitochondrial DNA. PLoS Biology 2004; September Issue.
25.Schmiedel J., Jackson S., Schefer J., Reichmann H. Mitochondrial Cytopathies. J Neurol 2003; 250: 267—277.
26.Shitara H., Kaneda H., Sato A. et al. Non-invasive visualization of sperm mitochondria behaviour in transgenic mice with introduced green fluorescent protein (GFP). FEBS Letters 2001; 500: 7—11.
27.Shoffner J.M., Wallace D.C. Oxidative phosphorylation diseases: disorders of two genomes. Adv Hum Genet 1990; 19: 267—330.
28.Shoubridge E.A. Mitochondrial encephalomyopathies. Curr Opin Neurol 1998; 11: 491—496.
29.Srivastava S., Moraes C.T. Manipulating mitochondrial DNA heteroplasmy by a mitochondrial targeted endonuclease. Human Mol Genet 2001; 10: 3093—3099.
30.Sue C., Schon E. Mitochondrial respiratory chain diseases and mutations in nuclear DNA: a promising Start? Brain Pathol 2000; 10: 3: 442—450.
31.Suomalainen A., Kaukonen J., Amati P. et al. An autosomal locus predisposing to deletions of mitochondrial DNA. Nature Genet 1995;
9:146—151.
32.Szibor M., Holtz J. Mitochondrial ageing. Basic Res Cardiol 2003; 98: 210—218.
33.Tarnopolsky M.A., Raha S. Mitochondrial myopathies: diagnosis, exercise intolerance, and treatment options. Med Sci Sports Exerc 2005; 37: 12: 2086—2093.
34.Taylor R.W., Wardell T.M., Connolly B.A. et al. Linked oligodeoxynucleotides show binding cooperativity and can selectively impair replication of deleted mitochondrial DNA templates. Nucleic Acids Res 2001; 29: 16: 3404—3412.
35.Tzagoloff A., Myers A.M. Genetics of mitochondria biogenesis. Ann Rev Biochem 1986; 55: 249—285.
36.Wallace D.C., Singh G., Lott M.T. et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988;
242:1427—1430.
37.Wallace D.C. Diseases of the mitochondrial DNA. Ann Rev Biochem 1992; 61: 1175—1212.
38.Wang Y., Michikawa Y., Mallidis C. et al. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proc Nat Acad Sci USA 2001; 27: 98: 7: 4022—4027.
39.Wredenberg A., Wibom R., Wilhelmsson H. et al. Increased mitochondrial mass in mitochondrial myopathy mice. PNAS 2002; 99: 23: 15066—15071.
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 2, 2007 |
69 |