

4.2. Два методи визначення можливості і напрямку
самодовільних процесів. Ентропія
Перший метод – метод факторів інтенсивності. Інтенсивними факторами можуть бути тиск, температура тощо. За цим методом самодовільні процеси можуть проходити в напрямку вирівнювання певного фактора інтенсивності. Рівновазі відповідає однакове значення цього фактора у всіх частинах системи.
Метод факторів інтенсивності є обмеженим. Він не придатний для визначення напрямку процесів в однорідних системах, де фактори інтенсивності однакові, наприклад, в хімічних реакціях.
Більш загальним є метод термодинамічних функцій. Він полягає в тому, що для конкретних умов існування певної термодинамічної системи підбирається термодинамічна функція стану системи, яка при протіканні самодовільного процесу збільшується (або зменшується) і в стані рівноваги досягає екстремального значення.
Клаузіус показав, що для ізольованих систем такою функцією може бути ентропія (S).
Ентропія системи – це функція стану системи, диференціал якої (dS)для елементарного рівноважного (оборотного) процесу дорівнює відношенню нескінченно малої кількості теплоти (Q), поглинутою системою, до абсолютної температури системи
![]() . (4.1)
. (4.1)
Для нерівноважного (необоротного, самодовільного) процесу
![]() . (4.2)
. (4.2)
Ентропія залежить від хімічної
природи речовин і температури. Згідно
з постулатом Планка, ентропія індивідуальної
кристалічної речовини при абсолютному
нулі дорівнює нулю: Sо
= 0. Із зростанням температури ентропія
збільшується, а при фазових перетвореннях
збільшується стрибкоподібно. Ентропію
речовини при стандартних умовах
позначають
![]() .
Для багатьох речовин стандартна ентропія
визначена і наводиться у довідниках
(табл. Д.2). Одиницею вимірювання ентропії
є Дж/(мольК).
.
Для багатьох речовин стандартна ентропія
визначена і наводиться у довідниках
(табл. Д.2). Одиницею вимірювання ентропії
є Дж/(мольК).
Для ізольованих систем, де відсутній обмін теплотою з навколишнім середовищем, Q = 0. Тоді з рівнянь (4.1-4.2) випливає, що для оборотних процесів dS= 0 (тобто ентропія стала), а для необоротних, самодовільних процесів dS 0 (тобто ентропія у ході процесу зростає).
Таким чином, визначивши зміну ентропії при процесі, можна робити висновок про напрямок процесу. Враховуючи, що ентропія – функція стану системи і dSє повним диференціалом ентропії, можна сформулювати правила визначення напрямку процесів:
якщо S 0 (ентропія зростає), то процес протікає самодовільно;
якщо S 0 (ентропія зменшується),то прямий процес самодовільно не проходить. Самодовільно протікає зворотний процес;
якщо S = 0 (ентропія не змінюється),то система знаходиться у стані рівноваги.
За допомогою поняття ентропії можна об'єднати перший і другий закони термодинаміки.
З другого закону термодинаміки випливає, що
![]() .
.
Звідси STdS. Підставимо це значення в рівняння першого закону - Q=dU+Wі одержимо:
ТdS dU + W
або
ТdS - dU W. (4.3)
З рівняння (4.3) видно, що максимальна робота має місце лише при оборотних процесах (теплота у цьому разі мінімальна).Для необоротних процесів, навпаки, теплота максимальна, а робота мінімальна.
4.3. Методи розрахунків ентропії речовин і зміни ентропії
процесів (реакцій)
Зміну ентропії при фазовому перетворенні речовини (Sф.п.) можна визначити за рівнянням
![]()
![]()
![]() , (4.1)
, (4.1)
де Нф.п.(Qф.п.) – теплота фазового перетворення, Дж/моль; Тф.п. – температура фазового перетворення, К.
Ентропію речовини при будь-якій температурі можна визначити з наступних міркувань:
![]() ;
;![]() ;
;
![]() ;
;![]() ;
;
![]() ;
;![]() . (4.5)
. (4.5)
Рівняння (4.5) можна розв'язати як за допомогою емпіричного ступеневого ряду
![]() , (4.6)
, (4.6)
так і через функції тепловмісту і приведеної енергії Гіббса
![]() , (4.7)
, (4.7)
де
![]() - приведена енергія Гіббса, Дж/(мольК);
- приведена енергія Гіббса, Дж/(мольК);![]() -тепловміст, кДж/моль.
-тепловміст, кДж/моль.
Приведена енергія Гіббса для багатьох речовин наводиться в довідниках (табл. Д.4).
Якщо в інтервалі температур від 298 до Т К має місце фазове перетворення, треба користуватись рівнянням (4.7), бо рівняння (4.6) у цьому випадку набуває вигляду
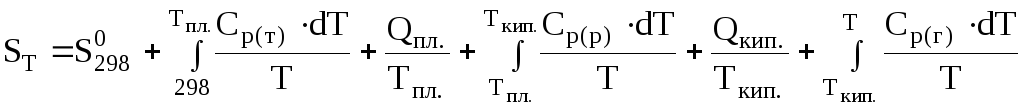 . (4.8)
. (4.8)
Як вже вказувалось, для визначення напрямку хімічної реакції в ізольованій системі треба розраховувати зміну ентропії реакції (S). Розглянемо цей випадок на прикладі одержання цинку пірометалургійним методом. У цьому випадку цинкову руду випалюють до оксиду цинку, останній змішують з коксом і нагрівають до 1370-1470 К. При цьому відбувається реакція
ZnO + C = Zn + CO,
зміна ентропії якої при сталій температурі буде дорівнювати
![]()
При визначенні SТ,і за допомогою емпіричного ступеневого ряду теплоємкості одержимо рівняння
![]() , (4.9)
, (4.9)
де
![]() - зміна стандартної ентропії реакції,
Дж/К;а,
в,
с
і с'
– зміна коефіцієнтів емпіричного
ступеневого ряду.
- зміна стандартної ентропії реакції,
Дж/К;а,
в,
с
і с'
– зміна коефіцієнтів емпіричного
ступеневого ряду.
Якщо ж визначити SТ,і через тепловміст і приведену енергію Гіббса, зміна ентропії реакції виражається рівнянням
![]() , (4.10)
, (4.10)
де
![]() - зміна тепловмісту у ході реакції, кДж;
- зміна тепловмісту у ході реакції, кДж;![]() - зміна приведеної енергії Гіббса, Дж/К.
- зміна приведеної енергії Гіббса, Дж/К.
Наведені рівняння визначення SТ (4.9-4.10) придатні як для оборотних, так і необоротних процесів. Це є наслідком властивості ентропії як функції стану системи, бо зміна функції стану від шляху процесу не залежить.