

11.7. Комбинирование равновесий
Константа равновесия
реакции Kpоднозначно связана с изменением
стандартной энергии ГиббсаGоуравнением (11.23). Порядок и знак величиныGопозволяют
качественно предвидеть положение
равновесия. Большая положительная
величинаGоозначает, что конечные продукты имеют
значительно больший потенциал, чем
исходные вещества, и равновесие сильно
смещено в сторону исходных веществ,
константа равновесия много меньше
единицы, выход продуктов мал. Например,
для реакции![]() N2+
N2+![]() О2LNОпри2675 КGо= 64,5 кДж,Kp= 3,5.10–3и выход NО равен
2,88%.
О2LNОпри2675 КGо= 64,5 кДж,Kp= 3,5.10–3и выход NО равен
2,88%.
Большая отрицательная
величина Gоуказывает на то, что равновесие сильно
смещено в сторону образования продуктов
реакции, их выход значительный, а
константа равновесия велика.Так, для
реакцииСО+![]() О2LСО2при
1396КGо= –
158 кДж, константа равновесияKp= 8,2.105, а содержание СО и О2в равновесной смеси составляют сотые
доли процента.
О2LСО2при
1396КGо= –
158 кДж, константа равновесияKp= 8,2.105, а содержание СО и О2в равновесной смеси составляют сотые
доли процента.
Так как энергия Гиббса Gявляется функцией состояния системы, то ее измененияGв каком-либо процессе не зависят от пути процесса. ПоэтомуGреакции, протекающей в несколько стадий, представляет сумму измененийGвсех стадий. Это дает возможность комбинирования равновесий и расчета констант равновесия для тех реакций, которые экспериментально не изучались.
Для такого расчета, аналогичного расчетам тепловых эффектов реакций по закону Гесса, необходимо, чтобы конечные продукты одной реакции (первая стадия суммарной реакции) находились в том же состоянии, в каком они являются исходными веществами для другой реакции (вторая стадия). Это достигается использованием стандартных изменений энергии Гиббса Gо, для которых принимается, что каждый из компонентов находится при давлении, равном 1 атм, твердые и жидкие вещества являются чистыми фазами, а газообразные вещества – идеальными газами.
Рассмотрим, например, расчет константы равновесия реакции
2Н2+ О2L2Н2О(г) (1)
при 1396 К по данным о Gореакций (при той же температуре):
СО+ Н2ОLСО2+ Н2Gо= 9570 Дж (2)
2СО + О2L2СО2Gо= –315800 Дж (3)
Как видно из записей, уравнение реакции (1) можно получить, вычитая из уравнения (3) удвоенное выражение реакции (2). Тогда
![]() =
=![]() – 2
– 2![]() = – 315800 – 2. 9570 = – 334940 Дж,
= – 315800 – 2. 9570 = – 334940 Дж,
откуда
lnKp= –![]() /RT= 334940/8,31.1396 = 28,87
/RT= 334940/8,31.1396 = 28,87
и константа равновесия реакции (1) Kp= 3,5.1012.
При непосредственном использовании величин Kpвсе действия сложения и вычитания величинGопри комбинировании равновесий заменяются действиями умножения и деления величинKp.
11.8. Химическое равновесие и тепловой закон Нернста
Константа химического равновесия однозначно связана со стандартным изменением энергии Гиббса при реакции и расчет Kpпри любых температурах можно свести к расчету величинGо. Согласно уравнению Гиббса – Гельмгольца
Gо= –Т ![]() dT+IT. (11.40)
dT+IT. (11.40)
Однако, по уравнению (11.40) можно рассчитать Gо(а, следовательно иKр) при любой температуре при условии, что величинаGоопределена хотя бы при одной температуре, так как значение постоянной интегрированияIнельзя вычислить заранее – будучи независимой от температуры, она различна для разных реакций.
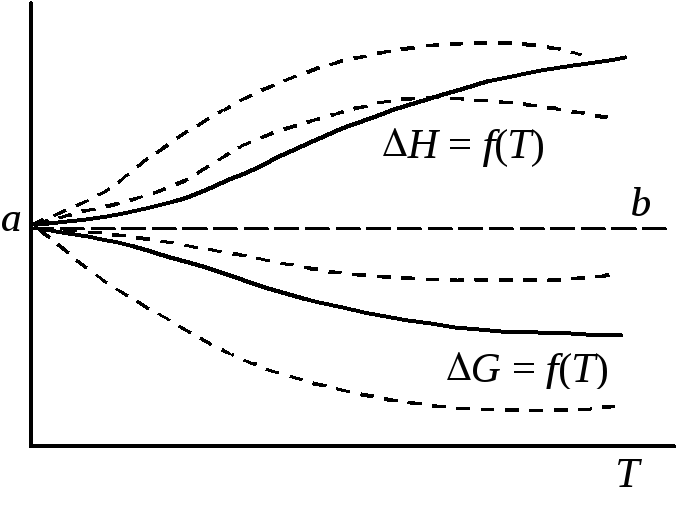
Величины Нпри разных температурах можно вычислить по экспериментальным данным о тепловом эффекте реакции при какой-либо температуре (например, начиная с очень низких температур) и теплоемкостях реагирующих веществ. ПосколькуG=H–TS, то приТ0GН. Но при других температурах каждому значениюНсоответствует множество значенийGов зависимости от от величиныI (рис.11.2, пунктирные линии). Так какGоявляется однозначной функцией температуры, то из всего набора зависимостейGо=f(T) следует выбрать лишь одну кривую, соответствующую строго определенному значению постояннойI. Решить эту задачу с помощью первого и второго законов термодинамики нельзя.
Нернст (1906) высказал предположение, что в конденсированных системах при температуре абсолютного нуля кривые температурной зависимости Но=f(Т) иGо=f(Т) имеют общую касательную, т.е.
![]() =
=
![]() . (11.41)
. (11.41)
Рис. 11.2. Зависимость
H
и
G
от
температуры
Из уравнения Гиббса – Гельмгольца G=Н+Т(G/Т)pследует, что приT= 0 производнаяG/Тпревращается в неопределенность, так как
(G/Т)T
0=![]() . (11.42)
. (11.42)
Взяв для раскрытия неопределенностей отношение пределов производных числителя и знаменателя по температуре, найдем в соответствии с уравнением (11.41), что величина
(G/Т)T0=![]() = 0. (11.43)
= 0. (11.43)
Поэтому уравнение (11.41) может быть окончательно записано в виде
lim (H/T)T0 = lim (G/T)T0= 0. (11.44)
Уравнение (11.44) представляют первоначальную форму так называемого теплового закона (тепловой теоремы)Нернста, илитретьего закона термодинамики в формулировке Нернста. Из уравнения следует, что правильна та криваяG=f(Т), которая горизонтально достигает оси ординат. Поэтому кривыеН=f(Т) иGо=f(Т) не просто соединяются в точке абсолютного нуля, но касаются друг друга уже при приближении к ней; в соответствии с уравнением (11.43) они имеют общую горизонтальную касательнуюab.
Уравнение (11.44) также означает, что
![]() ср
=
ср
=![]() S= 0, (11.45)
S= 0, (11.45)
т.е. теплоемкость конденсированной системы при Т0 не должна меняться при реакции, а также что при абсолютном нуле и вблизи него процесс в конденсированных системах протекает без изменения энтропии.
Тепловая теорема Нерста дает возможность определить постоянную интегрирования I. Если температурную зависимость теплового эффекта в температурном интервале от 0 до Т представить уравнением Кирхгофа
HT
= H0
+
![]() , (11.46)
, (11.46)
то уравнение (11.40) можно записать как
GT
= H0
– T
![]()
![]() + IT. (11.47)
+ IT. (11.47)
Интегрирование уравнения (11.47) в пределах температур от Т0 к Т дает:
 (11.48)
(11.48)
или
 . (11.49)
. (11.49)
Это уравнение совпадает с уравнением (11.47), если принять, что Т0 0 и
![]() . (11.50)
. (11.50)
Но из теплового закона Нернста вытекает, что для конденсированных систем эта величина равняется нулю, следовательно, постоянная интегрирования в уравнении (11.47) I= 0.
Тепловой закон не относится непосредственно к газам. Однако Нернст показал, что с помощью специального приема этим законом можно воспользоваться и для расчета термодинамических характеристик газовых и гетерогенных систем. Для этого надо провести процесс с помощью цикла, который включает конденсацию исходных веществ, реакцию между ними в конденсированной фазе и обратную возгонку продуктов реакции.
Рассмотрим некоторую реакцию в газовой фазе
іАі L kBk
и представим для нее цикл:

 іАі
L
kBk
(газовая
фаза)
іАі
L
kBk
(газовая
фаза)
испарениеконденсация
іАі L kBk (конденсированная фаза)
Вещества взяты при произвольных давлениях пара РіиРk, а после окончания реакции устанавливаются равновесные давленияріирк.Общее изменение энергии Гиббса для реакции в газовой фазе
GT
= RT
![]() –RT
–RT
![]() = RT
= RT
![]() +RT
+RT
![]() =
=
= – RT
lnKp+
RT
![]() . (11.51)
. (11.51)
Зависимость давлениянасыщенного пара от температуры над конденсированными фазами описывается уравнением Клаузиуса – Клапейрона (5.43). Интегрирования этого уравнения дает:
lnР=![]() +jилиRT
lnР=
+jилиRT
lnР=![]() +RTj(11.52)
+RTj(11.52)
где постоянная интегрирования jназываетсяистинной химической постоянной.
Зависимость теплоты испарения (конденсации) определяется уравнением Кирхгофа:
= 0
+
![]() . (11.53)
. (11.53)
Подстановка этого уравнения в (11.52) дает:
RT lnР
= 0
![]()
![]() +RT j. (11.54)
+RT j. (11.54)
Подставим эти значения в правую часть уравнения (11.51), а в левую его часть – уравнения (11.54):
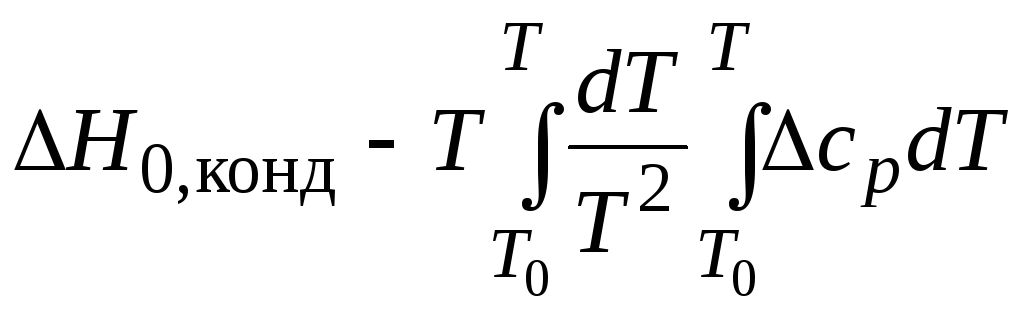 =–
RT lnKp
+ k0,k
– i0,i
–
=–
RT lnKp
+ k0,k
– i0,i
–

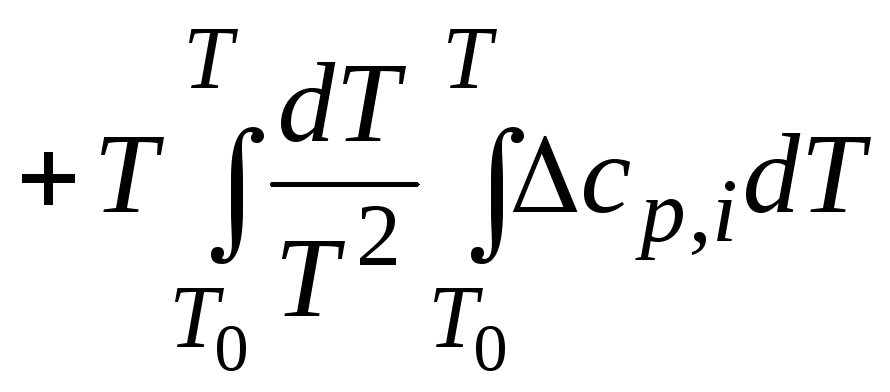 +RT
j. (11.55)
+RT
j. (11.55)
Различие между тепловыми эффектами реакции между веществами в конденсированном состоянии и в газовой фазе определяется, по закону Гесса, различием теплот испарения исходных веществ и продуктов реакции:
Hгаз = Hконд – (k0,k – и0,и). (11.56)
Итак,
lnKp
=![]() . (11.57)
. (11.57)
Это уравнение можно использовать для расчетов констант равновесий реакций в газовой фазе по термическим данным.