

118 |
|
METHODS FOR IN VITRO DNA AMPLIFICATION |
|
|
|
|
|
||||
When we attempted to use the PEP method, we ran into the difficulty that a large num- |
|
||||||||||
ber of template-independent sequences were amplified in the reaction |
mixture. This |
is |
|
||||||||
shown |
in |
the results of Figure 4.15, which illustrates |
the |
pattern |
of hybridization |
seen |
|
||||
when |
PEP-amplified |
S. pombe |
chromosome |
1 |
is |
hybridized to |
an arrayed cosmid |
library |
|||
providing |
a fivefold |
coverage of the |
S. pombe |
genome. Almost |
all of |
the clones are de- |
|||||
tected with comparable intensities, even though only about 40% of them should contain |
|
|
|||||||||
material from chromosome 1. We reasoned that the complex set of long primers might al- |
|
|
|||||||||
low for very significant levels of primer dimers (Fig. 4.3 |
|
|
|
|
|
b) to be produced, and since the |
|||||
primers |
represented |
all possible DNA sequence, their |
dimers |
would |
also |
represent |
a |
|
|||
broad population of sequences. Thus, when used in hybridization, this mixture should detect almost everything, which, indeed, it seems to do.
Figure 4.15 |
Examples of ampli- |
||||
fication of |
S. pombe |
chromosome |
|||
I DNA. |
(a) |
A schematic of |
an ex- |
|
|
periment in which directly labeled |
|
|
|||
chromosome I or amplified chro- |
|
|
|||
mosome I is used as a hybridiza- |
|
||||
tion probe against an arrayed li- |
|
||||
brary |
of the |
entire |
S. |
pombe |
|
genome |
cloned into cosmids. |
|
(b) |
||
Actual hybridization results for la- |
|
||||
beled |
chromosome |
I |
|
(top), |
chro- |
mosome I labeled after T-PCR |
|
||||
amplification |
|
(center), |
chromo- |
||
some I labeled after PEP amplifi- |
|
||||
cation |
(bottom). |
(From |
Grothues |
||
et al., 1993.) |
|
|
|
||
|
|
|
APPLICATION OF PCR TO DETECT MOLECULES OTHER THAN DNA |
|
|
119 |
||||||||||
Three variations were introduced to circumvent the primer dimer |
problem in |
PEP. |
|
|
|
|
||||||||||
Together these constitute an approach we call |
|
|
|
|
T-PCR, |
for tagged random primer PCR |
||||||||||
(Grothues et al., 1993). Our primers consist of all 4 |
|
|
9 nonanucleotides. Their shorter length |
|
||||||||||||
and smaller complexity should be an |
advantage compared with the 4 |
|
|
|
|
|
15 compounds used |
|||||||||
in ordinary PEP. Each primer was equipped at its 5 |
|
|
|
-end |
with |
a constant 17 |
base |
se- |
||||||||
quence; this is the tag. Thus the actual primers used were |
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
GTTTTCCCAGTCACGACN |
|
9 |
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
where N is a mixture of A, T, G, and C. After a few rounds of PCR, the resulting mixture |
|
|
|
|
||||||||||||
was fractionated by gel filtration, and material small enough to be primer dimers was dis- |
|
|
|
|
|
|||||||||||
carded. Then the remaining mixture was used as a target for amplification |
with only |
the |
|
|
|
|
|
|||||||||
tag sequence as a primer (Fig. |
4.14 |
|
b). As shown |
in Figure |
4.15, |
this |
yielded |
reaction |
|
|
||||||
products that produced a pattern of hybridization with the |
|
|
|
|
S. pombe |
cosmid |
array |
almost |
||||||||
identical to that seen with directly labeled chromosome 1. Thus we feel that T-PCR offers |
|
|
|
|
|
|||||||||||
very good prospects for uniformly sampling a complex DNA sample. In our hands this |
|
|
|
|
|
|||||||||||
approach has been successful thus far with as little as 10 |
|
|
|
12 g DNA, which corresponds to |
|
|||||||||||
less than a single human cell. |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Quite a few variations on this approach have been |
developed by |
others. One |
example |
|
|
|
|
|||||||||
is degenerate oligonucleotide-primed PCR (DOP–PCR) described by Telenius et al. |
|
|
|
|
||||||||||||
(1992). Here primers are constructed like |
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
AAGTCGCGGCCGCN |
6ATG |
|
|
|
|
|
|||||
with a six |
base |
totally |
degenerate |
sequence |
flanked |
by a |
long |
5 |
|
|
|
|
unique |
sequence and |
||
a specific 3 to 6 base unique sequence. The 3 |
|
|
-sequence |
serves to select a subset of |
||||||||||||
potential PCR start points. The degenerate sequence acts to stabilize the primer-template |
|
|
|
|
||||||||||||
complex. The |
constant |
5 |
|
-sequence can be used for |
efficient amplification |
in subsequent |
|
|
|
|||||||
steps just as the tag sequence |
is used in T-PCR. It is not |
yet |
certain how to |
optimize |
|
|
|
|||||||||
whole genome PCR methods for particular applications. Issues that must be considered |
|
|
|
|
|
|||||||||||
include the overall efficiency of the amplification, the uniformity of the product distribu- |
|
|
|
|
|
|||||||||||
tion, and the fraction of the original target that is present in the final amplified product. A |
|
|
|
|
||||||||||||
recently published DOP–PCR protocol (Cheung and Nelson, 1996) looks particularly |
|
|
|
|
||||||||||||
promising. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
APPLICATION |
OF |
PCR |
TO |
DETECT |
MOLECULES |
OTHER |
THAN |
DNA |
|
|
|
|
|
|
|
|
A natural extension of PCR is its use to detect RNA. Two general approaches for doing |
|
|
|
|
|
|||||||||||
this are summarized in Figure 4.16. In one, which is specific |
for polyadenylated mRNA, |
|
|
|
|
|
||||||||||
an oligo-dT primer is used, with reverse transcriptase, to make a DNA copy of the RNA. |
|
|
|
|
|
|||||||||||
Then conventional PCR can be used to amplify that DNA. In the other approach, which is |
|
|
|
|
|
|||||||||||
more general, random (dN) |
|
n ’s are used to prime reverse transcriptase to make initial DNA |
|
|
||||||||||||
copies, and then ordinary PCR ensues. |
|
|
|
|
|
|
|
|
|
|
|
|||||
Less obvious is the use of PCR to detect antigens or other non-nucleic acid molecules. |
|
|
|
|
||||||||||||
We originally demonstrated |
the feasibility of this approach, which |
should have a |
broad |
|
|
|
|
|
||||||||

120 METHODS FOR IN VITRO DNA AMPLIFICATION
Figure 4.16 |
Two methods for PCR amplification of RNA. |
|
|
|
|
(a) |
Use of reverse transcriptase with an |
|
oligo-dT primer. |
(b)Use of reverse transcriptase with short random oligonucleotide primers. |
|||||||
range of applications, and it should be generalizable to almost any class of molecule. The |
|
|||||||
basic principle of |
what we call |
immuno-PCR |
|
(i-PCR) is shown in Figure 4.17. DNA is |
||||
used as the label to indirectly tag an antibody. Then the |
DNA |
is |
detected |
by ordinary |
||||
PCR. |
|
|
|
|
|
|
|
|
In the test case, shown in Figure 4.17, the antibody |
is allowed to detect an immobi- |
|||||||
lized antigen and to bind to it in the conventional way (Fig. 4.18). Then the sample is ex- |
||||||||
haustively washed to remove free antibody. Next a molecule is |
added that serves to cou- |
|
||||||
ple DNA to the bound antibody. That molecule is a chimeric protein fusion between the |
|
|||||||
protein streptavidin and two domains of staphylococcal protein A. The chimera was made |
|
|||||||
by conventional genetic engineering methods and expressed as |
a gene |
fusion in |
E. coli. |
|||||
After purification the chimeric protein is fully active. Its |
properties |
are |
summarized |
in |
||||
Table 4.1. The chimera is a tetramer. It is capable of binding |
four immunoglobulin G’s |
|||||||
and four biotins. |
|
|
|
|
|
|
|
|
After the chimera is bound to the immobilized antibody, any unbound excess material is |
|
|||||||
removed, and now biotinylated DNA is added (Fig. 4.18). In our hands, end-biotinylated |
|
|||||||
linearized pUC19 |
was used. This |
was prepared by filling in |
the |
ends of |
a |
restriction |
en- |
|
zyme-digested plasmid with biotinylated dpppU, but it could just as easily have been made by PCR with biotinylated primers. The biotinylated DNA binds to the immobilized chimera.
epitope
antibody
antigen
analyze PCR products
streptavidin-protein A chimera
Figure 4.17 Basic scheme for immuno-PCR: Detection of antigens with DNA-labeled antibodies.

APPLICATION OF PCR TO DETECT MOLECULES OTHER THAN DNA |
121 |
Figure 4.18 Detailed experimental flow chart for implementing immuno-PCR to detect an immobilized antigen.
TABLE 4.1 Streptavidin-Protein A Chimera
Expression vector: |
pTSAPA-2 |
Amino acid residues: |
289 per subunit |
Subunits: |
4 (subunit tetramer) |
Molecular mass: |
31.4 kDa per subunit |
|
126 kDa per molecule |
Biotin binding: |
4 per molecule |
|
1 per subunit |
IgG binding: |
4 per molecule |
|
1 per subunit |
|
(human IgG) |
|
|
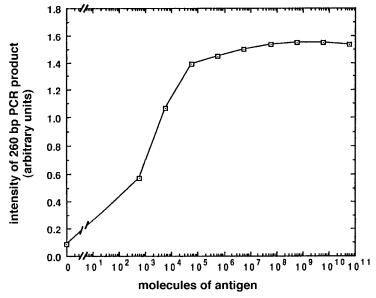
122 METHODS FOR IN VITRO DNA AMPLIFICATION
Figure 4.19 |
Result of detection of tenfold serial dilutions of an antigen, bovine serum albumin, |
by immuno-PCR. (Taken from Sano et al., 1992.) |
|
Excess unbound DNA is carefully removed by extensive washing. Now the entire sample is subjected to PCR using primers specific for the particular DNA tag. Typical results are shown for a set of serial dilutions of antigen in Figure 4.19. The results indicate that i-PCR promises to be an antigen-detection system of unparalleled sensitivity. Less than 600 molecules of antigen could be easily detected without any effort to optimize the system. This is
105 times |
more sensitive than conventional immunoassays. A major advantage of i-PCR is |
|
that the DNA molecule used is purely arbitrary. It can be |
changed at will to prevent the |
|
buildup of |
laboratory contaminants. It need not correspond |
(indeed it should not corre- |
spond) to any sequences found in the samples. Thus there should be no interference from sample DNA. Finally a number of different DNA labels could be detected simultaneously, which would open the way for multiplex PCR detection of several antigens simultaneously. Such an application has recently been reported (Hendrickson et al., 1995).
DNA AMPLIFICATION WITHOUT THERMAL CYCLING AND OTHER
ALTERNATIVES TO PCR
From a practical viewpoint it is difficult to fault PCR. If there is any step that is tedious, it is the need for stringent control at several different temperatures. It would be nice to eliminate
this requirement. From a commercial standpoint, existing PCR patents create quite a powerful band of protection around this technology and make the notion of potential competing technologies quite attractive as lucrative business ventures. Taken together, these considerations have fueled a number of attempts to create alternate DNA amplification procedures. Several of these have been shown to be practical. Some appear to be very attractive alternates
to PCR for certain applications. None yet have shown the generality or versatility of PCR. The degree of amplification achievable by these methods is quite impressive, but it is still considerably less than that seen with conventional PCR (Table 4.2).

|
|
|
OTHER ALTERNATIVES TO PCR |
123 |
|
TABLE 4.2 |
Comparison of Various In Vitro Nucleic Acid Amplification Procedures. |
|
|
||
|
|
|
|
|
|
|
Amplified |
Temperature |
Target-specific |
Amplification |
|
Method |
Species |
Used (°C) |
Probes Needed |
Extent |
|
PCR |
Target |
50–98 cycle |
2 or more |
10 |
QbR |
Probe |
37 isothermal |
1 |
10 |
LCR |
Probe |
50–98 cycle |
4 |
10 |
3SR |
Target |
42 isothermal |
2 |
10 |
SDA |
Target |
37 isothermal |
4 |
10 |
12
9
5
10
7
Source: Adapted from Abramson and Myers (1993).
Isothermal self-sustained sequence replication (3SR) is illustrated in Figure 4.20. In |
|
|||||||
this technique an RNA target is the preferred starting |
material. DNA targets can al- |
|
||||||
ways be copied by extending a primer |
containing |
a promoter |
site for an enzyme like |
|
||||
T7 RNA polymerase (and then that enzyme is used to generate an RNA copy of the |
|
|||||||
original DNA). The complementary DNA strand of the RNA is synthesized by Avian |
|
|||||||
myeloblastosis virus (AMV) reverse transcriptase (RT) using a primer that simultane- |
|
|||||||
ously introduces a promoter of T7 RNA polymerase. AMV RT contains an intrinsic |
|
|||||||
RNase H activity. This activity specifically degrades the RNA strand of an RNA-DNA |
|
|||||||
duplex. Thus, as AMV RT synthesizes the DNA complement, it degrades the RNA |
|
|||||||
template. The result |
is a single-stranded DNA complement |
of |
the original |
RNA. Now |
|
|||
a second primer, specific for the target |
sequence, is used to prime the |
RT to synthesize |
|
|||||
a double-stranded DNA (Fig. 4.20 |
|
a ). When this is completed, the resulting duplex now |
||||||
contains an intact promoter for T7 RNA polymerase so that enzyme can, rapidly, syn- |
|
|||||||
thesize many RNA copies. These RNAs are the complement of |
the original RNA tar- |
|
||||||
get (Fig. 4.20 |
b). |
|
|
|
|
|
|
|
Now, in a cyclical process, the RT makes DNA complements of the RNAs, degrading |
|
|||||||
them in the process by its RNaseH activity. RT then turns the single-stranded DNAs into |
|
|||||||
duplexes. These duplexes in turn serve |
as templates for |
T7 |
RNA |
polymerase to make |
|
|||
many more copies of single-stranded RNA. The key |
point is |
that all |
these |
reactions can |
|
|||
be carried on simultaneously at a constant temperature. A substantial level of amplifica- |
|
|||||||
tion is observed, and in principle, many of the same tricks and variations of PCR can be |
|
|||||||
implemented through the 3SR approach. Primer nesting does appear to be more difficult, |
|
|||||||
and it is not clear how well this technique will work in multiplexing. |
|
|
||||||
A method that is similar in spirit but rather different in detail is strand displacement |
|
|||||||
amplification (SDA). This is illustrated in Figure 4.21. It |
is based on the peculiarities of |
|
||||||
the restriction endonuclease |
Hinc |
II which recognizes |
the hexanucleotide sequence |
and |
||||
cleaves it, as shown below: |
|
|
|
|
|
|
|
|
|
|
GTTGAC |
|
GTT |
GAC |
|
||
|
|
CAACTG |
|
: CAA |
C T G |
|
||
The key feature of this enzyme exploited in SDA is the effect of alpha thio-substituted |
|
|||||||
phosphates on the enzyme. These can be introduced into DNA by the |
use of alpha-S- |
|
||||||
dpppA. When this is incorporated into the top strand of the recognition sequence, there is |
|
|||||||
no effect. However, in the bottom strand the thio derivatives inhibit cleavage (Fig. 4.21 |
a ). |
|||||||
How this peculiarity is used for isothermal amplification is illustrated in Figure 4.21 |
b. |
|||||||

124
Figur |
e |
4.20 |
|
Isothermal |
|
self-sustained |
|
se- |
||||
quence |
replication |
(3SR). |
(a) |
Synthesis |
of |
a |
||||||
double-stranded |
|
duple |
x from |
a |
|
starting |
RN |
A, |
||||
using |
|
one |
transcript-specific |
|
|
primer |
, |
TSP |
, |
|||
fused |
|
to a sequence |
containing |
|
a |
promoter |
|
|||||
site |
(solid |
bar) |
for |
T7 |
RN |
A |
polymerase |
|
||||
and |
a |
second |
|
transcript-specific |
|
|
primer |
|
B. |
|||
(b) Cyclical |
|
amplification |
|
by |
|
transcription |
|
|||||
and |
con |
v e rsion |
of |
the |
resulting |
|
RN |
A |
mole- |
|
||
cules to duple |
|
x DN |
As. |
|
|
|
|
|
|
|
||

OTHER ALTERNATIVES TO PCR |
125 |
Figure 4.21 |
Strand displacement amplification (SDA). A |
Hinc |
II site with alpha thio DNA deriva- |
|
|
|||
tives that block |
(solid circles) |
and do not block |
(open circles) |
cleavage. |
(b)Linear amplification by |
|
|
|
strand displacement from one |
Hinc |
II cleavage site. |
(c) Exponential amplification from two |
Hinc |
II |
|||
cleavage sites. |
|
|
|
|
|
|
|
|
A DNA polymerase I mutant with no 5 |
|
|
-exonuclease activity is used. This leads to |
||||
strand displacement. Consider first the effect of this enzyme on the target-primer com- |
|
|
|
|
|||
plex shown in the figure. The primer has a potential |
Hinc |
II |
site |
overhanging |
the tem- |
||
plate. The polymerase extends both the template and the primer, incorporating alpha-S- |
|
|
|
|
|||
A. The top strand of the resulting duplex can be |
cleaved |
by |
|
Hinc |
II; the |
bottom strand |
|
is resistant. This creates a target for |
the polymerase |
that can strand displace most of |
|
|
|
|
|
the top strand and continually make |
copies |
of it, |
resulting in linear amplification. |
|
|
|
|
126 |
METHODS FOR IN VITRO DNA AMPLIFICATION |
|
|
|
|
|
|
|
||||
If primers are established with overhanging |
|
|
Hinc |
|
II |
sites |
on both sides of a target |
|
||||
duplex, each strand can be made by linear amplification (Fig. 4.21 |
|
|
|
|
c ). Now, however, |
|
||||||
each newly synthesized strand can anneal with the original primer to form new com- |
|
|
||||||||||
plexes capable |
of further |
Hinc |
II cleavage and DNA polymerase-catalyzed |
amplifica- |
|
|||||||
tion. Thus the overall system will show exponential amplification in the presence of |
|
|
||||||||||
excess primers. |
|
|
|
|
|
|
|
|
|
|
|
|
|
In order to use SDA, one must have the |
desired target sequence |
flanked |
by |
|
Hinc |
II |
|||||
sites (or sites for any |
other restriction enzyme |
that might |
display |
similar |
properties). |
|
|
|||||
These sites can be introduced by using primers |
flanking the target sequence and tagged |
|
|
|||||||||
with |
additional |
5 |
-sequences containing the desired restriction |
enzyme |
cleavage |
sites. |
|
|||||
These flanking primers are used in a single cycle of conventional PCR; then SDA initiates |
|
|
||||||||||
spontaneously, and the amplification can be continued isothermally. |
|
|
|
|
|
|
|
|||||
|
Overall, SDA is a very clever procedure that combines a number of tricks in DNA en- |
|
|
|||||||||
zymology. It would appear to have some genuinely useful applications. However, SDA as |
|
|
|
|||||||||
originally described seems unlikely to become a generally used method because the re- |
|
|
||||||||||
sulting products have alpha-S-A, which is not |
always desirable, and the primers needed |
|
|
|||||||||
are rather complex and idiosyncratic. Recently variants on this scheme |
were |
developed |
|
|
||||||||
that have fewer restrictions. |
|
|
|
|
|
|
|
|
|
|||
|
Other modes of DNA polymerase-based amplification are still in their infancy, includ- |
|
|
|||||||||
ing rolling circle amplification (Fire and Xu, 1995) and protein-primed DNA amplification |
|
|
||||||||||
(Blanco et al., 1994). Alternate schemes for DNA amplification have been developed that |
|
|
||||||||||
avoid the use of DNA polymerase altogether. Foremost among these is the ligase chain re- |
|
|
|
|||||||||
action (LCR). This is illustrated in Figure 4.22. The target DNA is |
first |
denatured. Then |
|
|
||||||||
two oligonucleotides are annealed to one strand of the target. Unlike PCR, these two se- |
|
|
||||||||||
quences must |
be adjacent |
in the genome, and |
they |
correspond |
to the same DNA strand. |
|
|
|||||
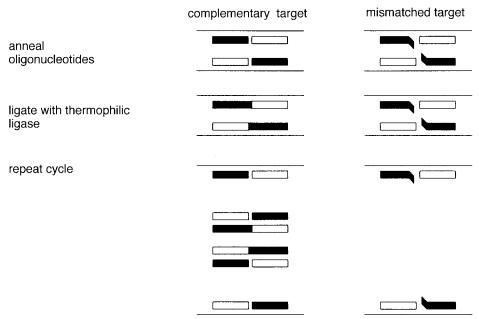
Figure 4.22 Example of allele-specific amplification using the ligase chain reaction (LCR).
|
|
|
|
|
|
|
FUTURE OF PCR |
127 |
If they match the target sequence exactly, DNA ligase will efficiently seal them together |
|
|
||||||
provided that a 5 |
-phosphate is present to form the phosphodiester bond between them. A |
|
||||||
complementary set of oligonucleotides can be used to form a ligation product directed by |
|
|
||||||
the other DNA strand. The overall result is to double the number of DNA strands. Both du- |
|
|
|
|||||
plexes are melted, more oligonucleotides anneal, and the process can be continued indefi- |
|
|
||||||
nitely. With continual thermal cycling, the result is exponential amplification of the target. |
|
|
||||||
This is most easily detected in automated systems by using a capture tag on one of the |
|
|
||||||
oligonucleotides to be ligated and a color-producing tag on the other. This procedure is ob- |
|
|
||||||
viously limited to small DNA target sequences, but it could form a powerful alternative to |
|
|
||||||
allele-specific PCR. Hybrid amplification procedures that combine LCR and polymerase |
|
|
||||||
extension reactions also appear to be very promising. |
|
|
|
|
|
|
||
The final amplification scheme we will discuss is carried out by the enzyme Q |
|
|
repli- |
|||||
case. This occurs strictly at the RNA level. Appropriate RNA targets can be made by sub- |
|
|
||||||
cloning DNA samples into vectors that |
embed the desired targets within Q |
|
|
sequences |
||||
and place them all downstream from a |
T7 RNA polymerase promoter so |
that |
an RNA |
|
|
|||
copy can be made to start the Q |
replication |
process (Q |
R). A much more general ap- |
|
||||
proach is to construct two separate RNA probes that can anneal to adjacent sequences on |
|
|
||||||
a target RNA. In the presence of T4 DNA ligase, the two probes will become covalently |
|
|
||||||
joined. Neither prone alone is a substrate for Q |
|
replicase. However, the ligation product |
|
|||||
is a substrate and is efficiently amplified (Fig. 4.23). |
|
|
|
|
|
|
||
Q has an unusual mode of replication. No primer is needed. No double-stranded in- |
|
|
||||||
termediate is formed. The enzyme recognizes specific secondary structure features and |
|
|
||||||
sequence elements on the template, |
and then makes a complementary copy of it. That |
|
|
|||||
copy dissociates from the template as it is made, and it folds into its own stable secondary |
|
|
||||||
structure which is a complement of that of the template. This structure also can serve as a |
|
|
||||||
template for replication. Thus the overall process continually produces both strands as |
|
|
||||||
targets, much in the manner of a dance in which the two partners |
move |
frenetically |
but |
|
|
|||
never stay in contact for an extended period. |
|
|
|
|
|
|
||
The usual mode of Q |
replication is very efficient. It is not uncommon to make 10 |
7 to |
||||||
108 copies of the original target. One can start from the single molecule level. However, |
|
|||||||
the system is not that easy to manipulate; Q |
replicase |
itself |
is a complex four-subunit |
|
||||
enzyme not that commonly available. The |
procedures needed to prepare the DNA target |
|
|
|
||||
for Q replication are somewhat |
elaborate, and there are considerable restrictions |
on |
|
|||||
what sorts of RNA insertions can be tolerated by the polymerase. For all these reasons the |
|
|
||||||
Q amplification system is most unlikely to replace PCR as a general tool |
for DNA |
|
||||||
analysis. It may, however, find unique niches for analyses where the idiosyncrasies of the |
|
|
||||||
system do not interfere, and where very high levels of amplification at constant tempera- |
|
|
||||||
ture are needed. |
|
|
|
|
|
|
|
|
FUTURE OF PCR |
|
|
|
|
|
|
|
|
In this chapter we have illustrated myriad variations and potential applications of PCR. In |
|
|
||||||
viewing these, it is important to keep in mind that PCR is a young technique. It is by no |
|
|||||||
means clear that today’s versions are |
the optimal ones or the most easily adaptable ones |
|
|
|||||
for the large-scale automation eventually needed for high-throughput genome analysis. |
|
|
||||||
Much additional thought needs to be |
given on how best to format PCR for widespread |
|
|
|||||
use and how to eliminate many of the |
current glitches and irreproducibility |
inherent in |
|
|
||||
such a high-gain amplification system. |
|
|
|
|
|
|
|
|