

108 METHODS FOR IN VITRO DNA AMPLIFICATION
Figure 4.6 |
Introducing extra |
functionalities by appropriately designed primers. |
(a) Incorporation |
of a 5 -terminal label (asterisk). |
(b)Incorporation of flanking restriction sites, useful for subsequent |
|
|
cloning. |
(c) Compensating for less than optimal length initial sequence information. |
|
|
This problem typically arises when one is trying to walk by from a known region of the |
|
|
|
||||||
genome into flanking unknown regions. Then one starts with a bit of known sequence at |
|
|
|||||||
the extreme edge of the charted region, and now the goal is to make a stab into the un- |
|
|
|||||||
known. There is still not a consensus on the best way to do this, but there have been a few |
|
|
|||||||
successes, and many failures. |
|
|
|
|
|
|
|
|
|
|
Consider the simple scheme shown in Figure 4.7 |
|
b. The known sequence is some- |
|
|||||
where within a restriction fragment of a length suitable for PCR. One can ligate arbi- |
|
|
|||||||
trary bits of known DNA sequence, |
called |
|
splints |
or |
adapters, |
onto the ends of |
the |
||
DNA |
fragment of interest. Now, in principle, |
the use of |
the one known primer and |
|
|
||||
one |
of the two ligated splint primers |
will allow selective amplification of |
one side |
of |
|
|
|||
the target. This amplification process will work as expected. However, the complica- |
|
|
|||||||
tion |
arises not from the target but |
from |
all |
the other |
molecules in the sample. They |
|
|
||
too |
are substrates for amplification |
using |
two |
copies of |
the splint primer |
(Fig. 4.7 |
|
c ). |
|

SINGLE-SIDED PCR |
109 |
Figure 4.7 |
Single-sided PCR. |
(a) General situation that requires this method; dashed |
line repre- |
sents the only known sequence in the target. |
(b)A potentially simple scheme. |
(c) Unwanted ampli- |
|
fication products that defeat the scheme in |
(b). |
|
|
Thus the desired product will be overwhelmed with undesirable side products. One must either separate these away (e.g., by using a capture tag on the known sequence primer) or find a way of preventing the undesired products from amplifying in the first
place (like suppression PCR, discussed later in this chapter). Sometimes it is useful to do both, as we will illustrate below.
In capture PCR, one can start with the very same set of primers shown for the unsuccessful example in Figure 4.7. However, the very first cycle of the amplification is
performed using |
only the sequence-specific primer with a capture |
tag, like |
biotin. |
|
Then, before any additional rounds of amplification are executed, the |
product |
of |
the |
|
first cycle is |
physically purified using streptavidin-coated magnetic |
beads |
or |
some |
other strepatvidin-coated surface (Fig. |
4.8). Now the splint primer is added, along with |
|
nonbiotinylated specific primer, and the |
PCR amplification is allowed to proceed nor- |
|
mally. When |
the procedure is successful, very pure desired product is achieved, since |
|
all of the |
potential side product precursors are removed before they are able to be |
|
amplified. |
|
|

110 METHODS FOR IN VITRO DNA AMPLIFICATION
Figure 4.8 Capture PCR, where the known sequence is used to purify the target DNA from a complex sample before fur-
ther rounds of amplification.
An alternative, general approach to single-sided PCR is to use a splint and primer
combination designed so that |
the primer will work only after the splint has been |
repli- |
cated once, and the splint can only be replicated by synthesis initiated at the known bit of |
||
sequence. Two versions of this are shown in Figure 4.9. In one case a splint is used that is |
||
dephosphorylated, so it cannot ligate to itself, and the double-stranded region is short and |
||
A T rich (Fig. 4.9 |
a ). After this is ligated to the target, the sample |
is heated to melt off |
the short splint strand. Next one cycle of DNA synthesis is allowed, using only the primer |
||
in the known target sequence. This copies the entire splint and produces a template for the |
||
second splint-specific primer. Both primers are now added, and ordinary PCR is allowed |
||
to proceed. However, only those molecules replicated during the first PCR cycle will be |
||
substrates for subsequent cycles. In the second version, called |
bubble PCR, the splint con- |
|
tains a noncomplementary segment of DNA which forms an interior loop (Fig. 4.9 |
b). As |
|
before, the first round of PCR uses only the sample-specific primer. This copies the mis- |
||
paired template strand faithfully so that when complementary primer is added to it, |
nor- |
|
mal PCR can ensue. These procedures are reported to work well. It is worth noting that |
||
they can easily be enhanced by |
attaching a capture tag to the target-specific primer in the |
|

SINGLE-SIDED PCR |
111 |
Figure 4.9 |
Two single-sided PCR schemes that use a linker that must be replicated before the |
|
|
complementary primer will function; dashed lines indicate known target sequence. |
(a) use of an ap- |
||
propriately designed dephosphorylated linker. |
(b)Bubble PCR. |
|
|
112 METHODS FOR IN VITRO DNA AMPLIFICATION
Figure 4.10 Inverse PCR, using primers designed to be extended outward on both sides of the known sequence (dashed lines).
first step. This will allow purification, as in capture PCR, before the reaction is allowed to continue.
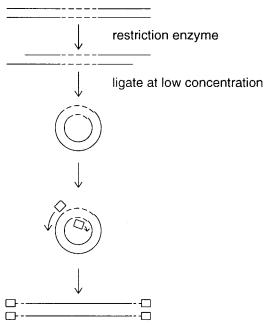
A third variation on the single-sided PCR theme is inverse PCR. This is extremely elegant in principle. Like many such novel tricks, it was developed independently and simultaneously by several different research groups. Inverse PCR is shown schematically in Figure 4.10. Here the target is cut with a restriction enzyme to generate ends that are easily ligatable. Then it is diluted to very low concentrations and exposed to DNA ligase. Under these condi-
tions the only products expected are DNA circles. To perform PCR, two primers are chosen within the known sequence, but they are oriented to face outward. Successful PCR with these primers should produce a linear product in which two, originally separate, segments of the
unknown sequence are now fused at a restriction site and lie in between the two bits of known sequence. In practice, this procedure has not seen widespread success. One difficulty is that it is not easy to obtain good yields of small double-stranded circles by ligation. A second problem is the topological difficulties inherent in replication of circular DNA. The polymerase complex must wind through the center of the DNA circle once for each turn of helix.
In principle, this latter problem could be removed by cleaving the known sequence with a restriction enzyme to linearize the target prior to amplification.
REDUCING COMPLEXITY WITH PCR
PCR allows any desired fraction of the genome to be selectively amplified if one has the primers that define that fraction. The complexity of a DNA sample was defined in
|
|
|
|
|
REDUCING COMPLEXITY WITH PCR |
113 |
|||||
Chapter 3. It is the total amount of different DNA sequence. For single-copy DNA the |
|
|
|
|
|||||||
complexity determines the rate of hybridization. Thus it can be very useful to selectively |
|
|
|
|
|||||||
reduce the complexity of a sample, since this speeds |
up subsequent attempts to analyze |
|
|
|
|
||||||
that sample by hybridization. The problem is that in general, one rarely has enough infor- |
|
|
|
|
|||||||
mation |
about the |
DNA |
sequences in a sample to choose a large but specific subset |
of it |
|
|
|
|
|||
for PCR amplification. |
|
|
|
|
|
|
|
|
|||
A powerful approach has been developed to use PCR to selectively reduce sample |
|
|
|
|
|||||||
complexity without any prior sequence knowledge |
at all. This approach promises to |
|
|
|
|
||||||
greatly facilitate genetic and physical mapping of new, uncharted genomes. It is based on |
|
|
|
|
|||||||
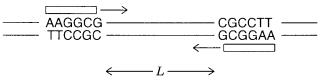
the use of short, random (arbitrary) primers. Consider the use of a |
|
|
single |
oligonucleotide |
|
||||||
primer |
of length |
|
n . As shown in Figure 4.11, this primer can produce DNA amplification |
|
|
|
|
||||
only if its complementary sequence exists as an inverted repeat, spaced within a distance |
|
|
|
|
|||||||
range amenable to efficient PCR. The inverted repeat requires that we specify a DNA se- |
|
|
|
|
|||||||
quence of 2 |
n |
bases. For a statistically random |
genome of |
|
N base |
pairs, |
the probability of |
|
|||
this occurring at any particular place is |
|
N 4 2n , which is quite small for almost any |
n |
large |
|||||||
enough to serve as an effective primer. However, any |
placement close enough for PCR |
|
|
|
|
||||||
will yield amplification products of the two primer sites. If |
|
L |
is the |
maximum |
practical |
|
|||||
PCR length, the probability that some observable PCR product will be seen is |
|
|
|
|
LN 4 2n . It |
||||||
is instructive to evaluate this expression for a mammalian genome with |
|
|
|
|
N 3 109 bp. |
||||||
For |
L 2000 the results are |
|
|
|
|
|
|
|
|
||
|
OLIGONUCLEOTIDE |
|
NUMBER OF |
|
TOTAL AMOUNT OF |
|
|
||||
|
|
|
LENGTH |
PCR PRODUCTS |
|
AMPLIFIED DNA |
|
|
|
||
|
|
|
8 |
|
1500 |
|
|
1.5 |
106 bp |
|
|
|
|
|
9 |
|
100 |
|
|
1.0 105 |
bp |
|
|
|
|
|
10 |
|
6 |
|
|
6.0 103 bp |
|
||
These results make it clear that by using single arbitrary short primers, we can |
sample |
|
|
|
|
||||||
useful discrete subsets of a genome. Each different choice of primer will presumably give |
|
|
|
|
|||||||
a largely nonoverlapping subset. The complexity of the reaction products can be |
con- |
|
|
|
|
||||||
trolled by the primer length to give simple or complex sets of DNA probes. This method |
|
|
|
|
|||||||
has been used, quite successfully, to search for new informative polymorphic |
genetic |
|
|
|
|
||||||
markers in plants. It has been called |
|
RAPD mapping, |
|
which is short for randomly ampli- |
|
||||||
fied polymorphic DNA. The idea is to amplify as large |
a number of bands as |
can |
be |
|
|
|
|
||||
clearly analyzed by a single electrophoretic lane and then |
to compare the patterns |
seen in |
|
|
|
|
|||||
a diverse set of individuals. |
|
|
|
|
|
|
|
|
|||
Figure 4.11 Sampling a DNA target (reducing complexity) with a single short oligonucleotide primer (RAPD method).

114 METHODS FOR IN VITRO DNA AMPLIFICATION
ADDITIONAL VARIANTS OF THE BASIC PCR REACTION
Here we will illustrate a number of variations on the basic PCR theme that increase the utility of the technique. The first of these is a convenient approach to the simultaneous
analysis of a number of different genomic targets. This approach is called |
multiplex PCR, |
and it is carried out in a reverse dot blot format. The procedure is illustrated schematically |
|
in Figure 4.12. Each target to be analyzed is flanked by two specific primers. One is ordi- |
|
nary; the other is tagged with a unique 20-bp overhanging DNA sequence. PCR can be |
|
carried out separately (or in one pot if conditions permit), and then the resulting products |
|
pooled. The tagged PCR products are next hybridized to a filter consisting of |
a set of |
spots, each of which contains the immobilized complementary sequence of one of |
the |
tags. The unique 20-bp duplex formed by each primer sequence will ensure that the corresponding PCR products become localized on the filter at a predetermined site. Thus the overall results of the multiplex PCR analysis will be viewed as positive or negative signals at specific locations on the filters. This approach, where amplified products are directed to a known site on a membrane for analysis, dramatically simplifies the interpretation of the results and makes it far easier to automate the whole process.
Figure 4.12 |
|
Inverse dot blot for the multiplex analysis of a |
|
||
set of |
pooled |
PCR |
reactions. |
(a) PCR with a set of |
different |
tagged |
primers, only |
one of which |
is shown. |
(b)Resulting |
|
dot blot.
|
|
ADDITIONAL VARIANTS |
OF |
THE |
BASIC |
PCR |
REACTION |
115 |
||||||||
For the approach shown in Figure 4.12, it would be far more useful to produce single- |
|
|
||||||||||||||
stranded product from the PCR reaction, since only one of the |
two strands of the ampli- |
|
|
|||||||||||||
fied target can bind to the filter; the other strand will |
actually act as a competitor. |
|
||||||||||||||
Similarly, for DNA sequencing, it is highly desirable to produce a single-stranded DNA |
|
|
||||||||||||||
product. Presence of the complementary strand presents an unwanted complication in the |
|
|
|
|
||||||||||||
sequencing reactions, and it can also act as a potential |
competitor. |
To |
produce |
single |
|
|||||||||||
strands in PCR, a very simple approach called |
|
|
|
|
|
asymmetric PCR |
can be |
used. Here ordi- |
||||||||
nary PCR is carried out for a few less than the usual number of cycles, say 20. Then one |
|
|
||||||||||||||
primer is depleted or eliminated, and the other is allowed to |
continue through an addi- |
|
|
|||||||||||||
tional 10 cycles of linear PCR. The result is a product that is |
almost |
entirely |
single |
|
||||||||||||
stranded. Clearly one can have whichever strand one wants by the appropriate choice of |
|
|
|
|||||||||||||
primer. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
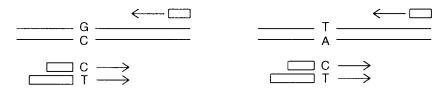
For diagnostic testing with PCR, one needs to distinguish different alleles at the DNA |
|
|
||||||||||||||
sequence level. A general approach for doing this is illustrated in Figure 4.13. It is called |
|
|||||||||||||||
allele-specific PCR. |
In the case shown |
in |
Figure |
4.13, we have a two-allele polymor- |
|
|||||||||||
phism. There is a single base difference possible, and we wish to know if a given individ- |
|
|
||||||||||||||
ual has two copies of one allele, or the other, or one copy of each. The goal is to distin- |
|
|
||||||||||||||
guish among these |
three alternatives in a single, definitive test. To do this, two primers are |
|
|
|||||||||||||
constructed that |
have 3 |
-ends specific |
for |
each |
of the |
alleles. A third general primer is |
|
|||||||||
used somewhere downstream where the two sequences are identical. The key element of |
|
|
|
|||||||||||||
allele-specific PCR is that because |
|
Taq |
|
polymerase |
does |
not |
have a |
3 |
-exonuclease, it |
|||||||
cannot use or degrade a primer with a mispaired 3 |
|
|
|
|
|
|
|
|
-terminus. Thus the allele-specific |
|||||||
primers will only amplify the allele to which they correspond precisely. Analysis of the |
|
|
||||||||||||||
results is simplified by using primers that are tagged either by having |
different lengths |
or |
|
|
||||||||||||
by having different colored fluorescent dyes. With length tagging, electrophoretic analysis |
|
|
||||||||||||||
will show different size bands for the two different alleles, and the heterozygote will be |
|
|
||||||||||||||
revealed as a doublet. With color tagging, the homozygotes will show, for example, red or |
|
|
|
|||||||||||||
green fluorescence, while the heterozygote will emit |
both |
red |
and |
green |
fluorescence, |
|
|
|||||||||
which our eye detects as yellow. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
Color can also be used to help monitor the quantitative progress of a PCR reaction. |
|
|||||||||||||||
Colored primers would not be useful unless their color were altered during the chain |
|
|
||||||||||||||
extension process. A similar problem holds for the potential |
use |
of |
colored |
|
dpppN’s. |
|
|
|||||||||
The best approach to date is a slightly complicated strategy, which takes advantage of |
|
|
||||||||||||||
the 5 -exonuclease activity of thermostable DNA polymerases (Heid et al. 1996). A |
|
|||||||||||||||
short oligonucleotide probe containing two different fluorescent labels is allowed to |
|
|
||||||||||||||
hybridize downstream from one of the PCR primers. The two |
dyes |
are |
close |
enough |
|
|
|
|||||||||
that the emission spectrum of the pair is altered by fluorescence |
|
resonance |
energy |
|
|
|||||||||||
transfer. As the |
primer is |
extended, the polymerase |
reaches |
this probe |
and |
degrades |
it. |
|
|
|||||||
Figure 4.13 Allele-specific PCR used in genetic analysis. Different primer lengths allow the results of amplification of the two different alleles to be distinguished.
116 |
METHODS FOR |
IN VITRO |
DNA AMPLIFICATION |
|
|
|
||||
A color change is produced because the |
nucleotide products of the digestion diffuse |
|||||||||
too far away from each other for energy transfer to be efficient. Thus each chain exten- |
||||||||||
sion results in |
the same incremental change in fluorescence. This procedure is called |
|||||||||
the TaqMan™ assay. A related spectroscopic trick, called molecular beacons, has re- |
||||||||||
cently been described in which a hybridization probe is designed as an oligonucleotide |
||||||||||
hairpin with different fluorescent dyes at its 3 |
|
- |
and 5 |
-ends (Kramer, 1996). In the |
||||||
hairpin these are close enough for efficient energy transfer. When the probe hybridizes |
||||||||||
to a longer target to form a duplex, its ends |
are |
now separated far apart |
in space, and |
|||||||
the energy transfer is eliminated. |
|
|
|
|
|
|
|
|||
Hairpins can also be used to produce selective PCR amplification. In suppression PCR, |
||||||||||
long GC-rich adapters are ligated onto the ends of a mixture |
of |
target |
fragments |
|||||||
(Diatchenko et al., 1996). When the ligation products are melted, the ends of the resulting |
||||||||||
single strands can form such stable hairpins |
that |
these |
ends become |
unaccessible |
for |
|||||
shorter primers complementary to the adapter sequences. However, if molecules in the |
||||||||||
target mixture contain a known internal target sequence, this can be used to initiate PCR. |
||||||||||
Chain extension from a primer complementary to the internal sequence will produce a |
||||||||||
product with only a single adapter. This will now allow conventional PCR amplification |
|
|||||||||
with one internal primer and one adapter primer. |
|
|
|
|
|
|
||||
A final PCR variant is called |
|
DNA shuffling |
(Stemmer, 1994). Here the goal is to en- |
|||||||
hance the properties of a target gene product by in vitro recombination. Suppose that a se- |
||||||||||
ries of mutant genes exist with different properties; the goal is to combine them in an op- |
||||||||||
timal way. The genes are randomly cleaved into fragments, pooled, and |
the |
resulting |
||||||||
mixture is subjected to PCR amplification using primers flanking the gene. Random as- |
||||||||||
sembly of overlapping fragments will lead |
to products that can be chain |
extended until |
||||||||
full length reassembled genes are produced. These then support exponential PCR amplifi- |
|
|||||||||
cation. The resulting populations of mutants are |
cloned and |
characterized |
by some kind |
|||||||
of screen or selection in order to concentrate those with the desirable properties. This new |
||||||||||
method appears to be extremely promising. A very interesting alternative method to shuf- |
|
|||||||||
fle DNA segments uses catalytic RNAs (Mikheeva and Jarrell, 1996). |
|
|
|
|||||||
TOTAL |
GENOME |
AMPLIFICATION |
METHODS |
|
|
|
|
|
|
|
A frequently encountered problem in biological research is insufficient amounts of sam- |
||||||||||
ple. If |
the sample is a cultured cell or |
microorganism, the simplest solution is to |
grow |
|||||||
more material. However, many samples of interest cannot be cultured. For example, many differentiated cells cannot be induced to divide without destroying or altering their phenotype. Sperm cells are incapable of division. Most microorganisms cannot be cultured by any known technique—we know of their existence only because we can see their cells or detect aspects of their DNA. Fossil samples and various clinical biopsies are other examples of rare materials with insufficient DNA for convenient analysis. Finally sorted chromosomes (Chapter 2) present the challenge of a very useful resource for which there is always more demand than supply.
In each of these cases mentioned above, one could use a particular set of primers to amplify any given known DNA region of interest. However, once this were done, the rest
of the sample would be lost for further analysis. Instead, what would be useful is an amplification method that first samples all of the DNA in the rare material. This can then be stockpiled for future experiments of a variety of types including more specific PCR, when

|
|
TOTAL GENOME AMPLIFICATION METHODS |
117 |
|||
needed. The issue is how to do this in such a way that the stockpile represents a complete, |
|
|
||||
or at least a relatively complete and even sampling of the original sample. The danger of |
|
|
||||
course is that the sample will consist of a set of regions with very different amplification |
|
|
||||
efficiencies with any particular set of primers. After PCR the stockpile |
will |
now be |
a |
|
||
highly distorted version of the original, and future experiments will all be plagued by this |
|
|
||||
distorted view. |
|
|
|
|
|
|
One approach to PCR sampling of an entire genome is the method of primer extension |
|
|
||||
preamplification (PEP). This was designed |
to be used on a single cell or |
single |
sperm. |
|
|
|
The detailed rationale for PEP will become apparent when genetics by single sperm PCR |
|
|
||||
is discussed in Chapter 7. PEP is illustrated schematically in Figure 4.14 |
|
|
a . A mixture of |
|||
all possible 4 |
n primers of length |
n is generated by automated oligonucleotide synthesis, |
|
|||
using at each step all four nucleotides rather than just a single one. This extremely com- |
|
|
||||
plex mixture is then used as a primer. Although the concentration of any one primer is |
|
|
||||
vanishingly small, there are always enough primers present that any particular DNA seg- |
|
|
||||
ment has a reasonable chance of amplification. Norman Arnheim and his coworkers have |
|
|
|
|||
reported reasonable |
success at using this |
approach with |
|
n |
15 (Arnheim and |
Ehrlich, |
1993). They use 50 cycles of amplification and estimate that at least 78% of the genome will be amplified to 30 or more copies by this method.
Figure 4.14 |
Methods for total genome PCR. |
(a) Primer extension preamplification (PEP). |
(b) |
Tagged random primer PCR (T-PCR).