

Gale Encyclopedia of Genetic Disorder / Gale Encyclopedia of Genetic Disorders, Two Volume Set - Volume 1 - A-L - I
.pdfparent has this rearrangement, the risk for them to have a child with cri du chat is greater than 1%.
Demographics
It has been estimated that cri du chat syndrome occurs in one of every 50,000 live births. According to the 5p minus Society, approximately 50-60 children are born with cri du chat syndrome in the United States each year. It can occur in all races and in both sexes.
Signs and symptoms
An abnormal larynx causes the unusual cat-like cry made by infants that is a hallmark feature of the syndrome. As children with cri du chat get older, the cat-like cry becomes less noticeable. This can make the diagnosis more difficult in older patients. In addition to the cat-like cry, individuals with cri du chat also have unusual facial features. These facial differences can be very subtle or more obvious. Microcephaly (small head size) is common. During infancy, many patients with cri du chat do not gain weight or grow normally. Approximately 30% of infants with cri du chat have a congenital heart defect. Hypotonia (poor muscle tone) is also common, leading to problems with eating, and slow normal development. Mental retardation is present in all patients with cri du chat but the degree of mental retardation varies between patients.
Diagnosis
During infancy the diagnosis of cri du chat syndrome is strongly suspected if the characteristic cat-like cry is heard. If a child has this unusual cry or other features seen in cri du chat syndrome, chromosome testing should be performed. Chromosome analysis provides the definitive diagnosis of cri du chat syndrome and can be performed from a blood test. Chromosome analysis, also called “karyotyping”, involves staining the chromosomes and examining them under a microscope. In some cases the deletion of material from chromosome 5 can be easily seen. In other cases, further testing must be performed. FISH (fluorescence in-situ hybridization) is a special technique that detects very small deletions. The majority of the deletions that cause cri du chat syndrome can be identified using the FISH technique.
Cri du chat syndrome can be detected before birth if the mother undergoes amniocentesis testing or chorionic villus sampling (CVS). This testing would only be recommended if the mother or father is known to have a chromosome rearrangement, or if they already have a child with cri du chat syndrome.
Treatment and management
Currently, there is no cure for cri du chat syndrome. Treatment consists of supportive care and developmental therapy.
Prognosis
Individuals with cri du chat have a 10% mortality during infancy due to complications associated with congenital heart defects, hypotonia, and feeding difficulties. Once these problems are controlled, most individuals with cri du chat syndrome have a normal lifespan. The degree of mental retardation can be severe. However, a recent study suggested that the severity is somewhat affected by the amount of therapy received.
Resources
BOOKS
Gardner, R., J. McKinlay, and Grant R. Sutherland. Chromosome Abnormalities and Genetic Counseling. New York: Oxford University Press, 1996.
Jones, Kenneth. Smith’s Recognizable Patterns of Human Malformation, 5th Edition. Philadelphia: W.B. Saunders Company, 1997.
Rimoin, David, Michael Connor, and Reed Pyeritz. Emery and Rimoin’s Principles and Practice of Medical Genetics,
Third Edition. New York: Churchill Livingstone, 1996.
PERIODICALS
Van Buggenhout, G. J. C. M., et al. “Cri du Chat Syndrome: Changing Phenotype in Older Patients.” American Journal of Medical Genetics 90 (2000): 203-215.
ORGANIZATIONS
5pSociety. 7108 Katella Ave. #502, Stanton, CA 90680. (888) 970-0777. http://www.fivepminus.org .
Alliance of Genetic Support Groups. 4301 Connecticut Ave. NW, Suite 404, Washington, DC 20008. (202) 966-5557. Fax: (202) 966-8553. http://www.geneticalliance.org .
Cri du Chat Society. Dept. of Human Genetics, Box 33, MCV Station, Richmond VA 23298. (804) 786-9632.
Cri du Chat Syndrome Support Group. http://www.criduchat
.u-net.com .
National Organization for Rare Disorders (NORD). PO Box 8923, New Fairfield, CT 06812-8923. (203) 746-6518 or (800) 999-6673. Fax: (203) 746-6481. http://www
.rarediseases.org .
WEBSITES
OMIM—Online Mendelian Inheritance in Man.
http://www.ncbi.nlm.nih.gov/Omim/ .
Holly Ann Ishmael, MS, CGC
Crouzon craniofacial dysostosis see
Crouzon syndrome
syndrome chat du Cri
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
291 |

Crouzon syndrome
I Crouzon syndrome
Definition
Crouzon syndrome is a genetic condition that causes early closure of the bones in the skull. This event is called craniosynostosis and causes the skull to be formed differently in affected individuals. Because of the craniosynostosis, individuals affected with Crouzon syndrome will have the characteristic facial features described below.
Description
Other features of Crouzon syndrome include wideset and prominent eyes. Individuals with this syndrome may also have a condition called strabismus, which means the eyes have difficulty focusing on objects. Other facial features may include an underdeveloped upper jaw, which causes tooth abnormalities. Individuals with Crouzon syndrome often have a beak-shaped nose and hearing loss. A skin condition, called acanthosis nigricans, occurs in approximately 5% of individuals with Crouzon syndrome. It is important to note that there is a wide range of severity in Crouzon syndrome. No two individuals with the condition will necessarily have all the listed features.
It is rare for individuals with Crouzon syndrome to have learning delays or mental impairments. Affected individuals often undergo several corrective surgeries, increasing the need for continual medical care throughout their lives. This can be very stressful and difficult for individuals and their families. Additionally, since people with Crouzon syndrome have significant facial differences, it may be difficult for them (and their parents) to feel accepted by society. There may be psychological implications, ranging from the affected person feeling bad for “looking different” to the parents having trouble bonding to their child for similar reasons. The psychological impact may be less if there are others in the family with Crouzon syndrome. Having more than one family member with this syndrome may help those affected feel less isolated and give them a stronger support system.
Genetic profile
Crouzon syndrome is caused by mutations in the FGFR2 (location 10q25.3-q26) and FGFR3 (location 4p16.3) genes. Crouzon syndrome is inherited in an autosomal dominant manner. An affected individual has one copy of the FGFR mutation and has a 50% chance to pass it on to each of his or her children, regardless of that child’s gender. As of 1997, about 75% of affected people
have a family history of Crouzon syndrome, which is typically a parent with the condition. In the remaining 25%, the genetic mutation occurs as a new event in the affected individual, and there is no one in their family with the disease. These new mutations are thought to occur because of advancing paternal age, i.e. the age of the patient’s father is a factor. Additionally, there is no increased recurrence risk for Crouzon syndrome above the general population risk when there is no family history of the condition.
FGFR2 and FGFR3 are responsible for the proper growth, movement, and creation of specific cells in the body, known as fibroblasts. Fibroblasts are often part of the bony structures in the body (such as the skull), so problems in fibroblast growth and movement would naturally lead to skull/bone problems. As of 1998, about 95% of patients have an FGFR2 mutation, and 5% have an FGFR3 mutation. However, nearly all of the affected individuals that also have acanthosis nigricans have one common FGFR3 mutation.
Demographics
As of 2000, Crouzon syndrome occurs in about one per 25,000 live births. It affects all ethnic groups equally.
Signs and symptoms
There commonly is bilateral (two-sided) coronal craniosynostosis in Crouzon syndrome. A cloverleaf skull may be present if the sagittal (long suture going from front to back of the head) and/or lambdoidal (short suture at very back of the head) sutures are involved. This causes the skull shape to be taller than usual, often described as “tower-shaped.” The pattern looks like a cloverleaf because the skull is taller, and the sides of the skull and face bulge slightly from right to left. Additionally, the eye orbits are very shallow, causing the eyes to protrude significantly. This eye finding is always present in the condition. Strabismus may be present and eyes may be wide-set, making vision poor. Some individuals may have unexplained difficulties with their vision. The nose can be narrow and beak-shaped, forcing the individual to breathe through their mouth as a result.
The upper jaw may not be formed properly and can cause dentition problems, most commonly a missing tooth. The palate (upper ridge of the mouth) may be high and narrow, causing crowding of the existing teeth. Occasionally, clefting (improper closure) of the lip and palate may occur. Mild to moderate conductive hearing loss (due to abnormal ear structure formation) may occur in a proportion of cases.
Intellectual development is typically within normal limits. Only rare cases have been reported with signifi-
292 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
cant mental deficiency. In about 30% of patients, hydrocephalus can occur. Hydrocephalus is an accumulation of fluid in the brain and skull, and this may progress or worsen with time. This typically shows up as a general enlarging of the skull. Sometimes the fluid can put increased pressure on various structures of the brain, limiting their growth and development. Hydrocephalus may be an explanation for the few reported cases of Crouzon syndrome with learning problems. Occasionally, seizures may occur in the condition.
Individuals with Crouzon syndrome may be shorter than the normal expected height. This seems to affect females with the condition more than males.
Diagnosis
Historically, Crouzon syndrome has been diagnosed after careful physical examination and further studies. A diagnosis of Crouzon syndrome can be made through observing several of the following features. The abnormally shaped head is typically seen right away, in the newborn period. It may sometimes be seen in the prenatal period with an ultrasound examination. X-ray or physical examination of the skull can diagnose craniosynostosis. Once craniosynostosis is seen, it is important to determine whether it occurred because of abnormal biology of the cranial suture, possibly caused by an FGFR mutation. This is known as primary craniosynostosis and would make Crouzon syndrome a possibility. Craniosynostosis may also be caused by abnormal outside forces (known as secondary craniosynostosis) such as decreased brain growth or abnormal fetal head positioning. This may have occurred in the prenatal period, and in these cases the abnormal head shape may correct itself with time. The next step is to determine the type of craniosynostosis. A cloverleaf skull makes Crouzon syndrome a possibility, but it is also seen more commonly in other genetic craniosynostosis syndromes.
Some babies with Crouzon syndrome have breathing problems in the newborn period, due to narrowed nasal passages. Protruding eyes are a hallmark feature for the condition, and can be seen almost immediately after birth. The lack of abnormalities in the extremities (hands and feet) are also considered part of the diagnosis of Crouzon syndrome versus another type of craniosynostosis.
As of 2001, molecular (DNA-based) genetic testing to diagnose Crouzon syndrome is available at a few laboratories. This testing is specific for the condition, separating it from other craniosynostosis syndrome possibilities. A blood or other type of sample (such as fetal cells from amniotic fluid) from the affected individual is provided, and the FGFR2 gene is analyzed.
Abnormal results occur when a mutation in the sequence of the FGFR2 DNA is identified from genetic analysis. This means that the mutation caused the symptoms in the individual, confirming the diagnosis of Crouzon syndrome. As mentioned earlier, not every person with Crouzon syndrome will have an FGFR2 mutation. Therefore, one could conceivably go through genetic testing and have no mutation found. This could mean that the person’s symptoms are not caused by Crouzon syndrome.
As of 2001, only a little more than 50% of the mutations that cause Crouzon syndrome are known. Therefore, a negative result could also mean that the patient has a genetic mutation that is unable to be found by current technology. Once a mutation is found in a family, it is much easier (and less time-consuming) to test others in the same family. For people with the features of Crouzon syndrome and acanthosis nigricans, there is DNA-based testing to determine if they have the common FGFR3 mutation.
Prenatal testing is available for both FGFR2 and FGFR3 mutations, done via amniocentesis or chorionic villus sampling (CVS). This is only offered when there is a parent with a known mutation. However, knowing prenatally that an individual has a mutation tells nothing about the extent of the disease. The only way to determine the severity of Crouzon syndrome is by seeing the individual after birth, not by molecular testing. A prenatal ultrasound can sometimes make a possible diagnosis of a syndrome involving craniosynostosis, but it is not as accurate as direct DNA testing. Additionally, a cloverleaf skull seen on a prenatal ultrasound usually implies a more severe outcome for the baby than other types of craniosynostosis.
Treatment and management
Treatment of individuals with Crouzon syndrome often involves the coordinated efforts of several medical specialists in a team setting. The specialists may include a pediatrician, plastic surgeon, neurosurgeon, geneticist, genetic counselor, dentist, social worker, audiologist, speech pathologist, psychologist, and otolaryngologist.
Craniosynostosis is typically repaired through a series of operations. There is a major surgery performed as early as the first three months of life, followed by several others that may extend over the lifespan. Each series of operations is tailored to the individual, but it is rare for the correction to be “perfect” despite the interventions. Because the skull is continually growing in the early part of life, timing of these surgeries is critical for proper brain formation and better results. Surgeries after the skull has stopped growing rarely yield good results.
syndrome Crouzon
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
293 |

Crouzon syndrome
K E Y T E R M S
Acanthosis nigricans—A skin condition characterized by darkly pigmented areas of velvety wartlike growths. Acanthosis nigricans usually affects the skin of the armpits, neck, and groin.
Amniocentesis—A procedure performed at 16-18 weeks of pregnancy in which a needle is inserted through a woman’s abdomen into her uterus to draw out a small sample of the amniotic fluid from around the baby. Either the fluid itself or cells from the fluid can be used for a variety of tests to obtain information about genetic disorders and other medical conditions in the fetus.
Chorionic villus sampling (CVS)—A procedure used for prenatal diagnosis at 10-12 weeks gestation. Under ultrasound guidance a needle is inserted either through the mother’s vagina or abdominal wall and a sample of cells is collected from around the fetus. These cells are then tested for chromosome abnormalities or other genetic diseases.
Coronal suture—Skull suture that lies behind the forehead area, across the head from left side to the right side.
Craniosynostosis—Premature, delayed, or otherwise abnormal closure of the sutures of the skull.
Mutation—A permanent change in the genetic material that may alter a trait or characteristic of an individual, or manifest as disease, and can be transmitted to offspring.
Otolaryngologist—Physician who specializes in the care of the ear, nose, and throat and their associated structures.
Strabismus—An improper muscle balance of the ocular musles resulting in crossed or divergent eyes.
Suture—”Seam” that joins two surfaces together.
Surgeries performed before various portions of the facial region have stopped growing also have a poor prognosis, and will require additional follow-up procedures.
For individuals with hydrocephalus, sometimes a shunt, or tube, needs to be placed in order to allow the fluid to drain from the affected area(s) of the brain.
For babies with respiratory distress, oxygen and ventilation are often provided. Occasionally, a tracheostomy
(opening in the windpipe) is created to help the individual breathe.
Because their eyes protrude so significantly, people with Crouzon syndrome sometimes have trouble closing their eyes. Surgical eye closure may be necessary, which allows the eye and its various structures (such as the cornea) to remain protected.
Occasionally, surgeries to correct structural ear abnormalities (resulting in hearing loss) are necessary.
Prognosis
The most problematic complication in Crouzon syndrome is the craniosynostosis. Prognosis primarily depends upon the severity and extent of this skull abnormality. Consequently, the success of corrective surgeries often determines prognosis.
Resources
BOOKS
Charkins, Hope. Children with Facial Difference: A Parent’s Guide. Bethesda, MD: Woodbine House, 1996.
ORGANIZATIONS
AboutFace USA. PO Box 458, Crystal Lake, IL 60014. (312) 337-0742 or (888) 486-1209. aboutface2000@aol.com.http://www.aboutface2000.org .
American Cleft Palate-Craniofacial Association. 104 South Estes Dr., Suite 204, Chapel Hill, NC 27514. (919) 9939044. Fax: (919) 933-9604. http://www.cleftline.org .
Children’s Craniofacial Association. PO Box 280297, Dallas, TX 75243-4522. (972) 994-9902 or (800) 535-3643. contactcca@ccakids.com. http://www.ccakids.com .
Crouzon Support Network. PO Box 1272, Edmonds, WA 98020. penny@crouzon.org. http://www.crouzon.org .
Crouzon’s/Meniere’s Parent Support Network. 3757 North Catherine Dr., Prescott Valley, AZ 86314-8320. (800) 8424681. katy@northlink.com.
WEBSITES
“Craniofacial Anomalies.” Columbia Presbyterian Medical Center Neurological Institute. http://cpmcnet.columbia
.edu/dept/nsg/PNS/Craniofacial.html .
Deepti Babu, MS
Cryptophthalmos syndactyly syndrome see
Fraser syndrome
Cutis-gyrata syndrome of Beare and Stevenson see Beare-Stevenson cutis gyrata syndrome
Cystathionine beta-synthetase see
Homocystinuria
294 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

I Cystic fibrosis
Definition
Cystic fibrosis (CF) is an inherited disease that affects the lungs, digestive system, sweat glands, and male fertility. Its name derives from the fibrous scar tissue that develops in the pancreas, one of the principal organs affected by the disease.
Description
Cystic fibrosis affects the body’s ability to move salt and water in and out of cells. This defect causes the lungs and pancreas to secrete thick mucus, blocking passageways and preventing proper function.
CF affects approximately 30,000 children and young adults in the United States, and about 3,000 babies are born with CF every year. CF primarily affects people of white northern European descent; rates are much lower in non-white populations.
Many of the symptoms of CF can be treated with drugs or nutritional supplements. Close attention to and prompt treatment of respiratory and digestive complications have dramatically increased the expected life span of a person with CF. Several decades ago most children with CF died by age two years; today, about half of all people with CF live past age 31. That median age is expected to grow as new treatments are developed, and it is estimated that a person born in 1998 with CF has a median expected life span of 40 years.
Genetic profile
Cystic fibrosis is a genetic disease, meaning it is caused by a defect in the person’s genes. Genes, found in the nucleus of all the body’s cells, control cell function by serving as the blueprint for the production of proteins. Proteins carry out a wide variety of functions within cells. The gene that, when defective, causes CF is called the CFTR gene, which stands for cystic fibrosis transmembrane conductance regulator. A simple change in this gene leads to all the consequences of CF. There are over 500 known changes in the CFTR gene that can cause CF. However, 70% of all people with an abnormal CFTR gene have the same defect, known as delta-F508.
Genes can be thought of as long strings of chemical words, each made of chemical letters, called nucleotides. Just as a sentence can be changed by rearranging its letters, genes can be mutated, or changed, by changes in the sequence of their nucleotide letters. The gene changes in CF are called point mutations, meaning that the gene is mutated only at one small spot along its length. In other
words, the delta-F508 mutation is a loss of one “letter” out of thousands within the CFTR gene. As a result, the CFTR protein made from its blueprint is made incorrectly, and cannot perform its function properly.
The CFTR protein helps to produce mucus. Mucus is a complex mixture of salts, water, sugars, and proteins that cleanses, lubricates, and protects many passageways in the body, including those in the lungs and pancreas. The role of the CFTR protein is to allow chloride ions to exit the mucus-producing cells. When the chloride ions leave these cells, water follows, thinning the mucus. In this way, the CFTR protein helps to keep mucus from becoming thick and sluggish, thus allowing the mucus to be moved steadily along the passageways to aid in cleansing.
In CF, the CFTR protein does not allow chloride ions out of the mucus-producing cells. With less chloride leaving, less water leaves, and the mucus becomes thick and sticky. It can no longer move freely through the passageways, so they become clogged. In the pancreas, clogged passageways prevent secretion of digestive enzymes into the intestine, causing serious impairment of digestion— especially of fat—which may lead to malnutrition. Mucus in the lungs may plug the airways, preventing good air exchange and, ultimately, leading to emphysema. The mucus is also a rich source of nutrients for bacteria, leading to frequent infections.
To understand the inheritance pattern of CF, it is important to realize that genes actually have two functions. First, as noted above, they serve as the blueprint for the production of proteins. Second, they are the material of inheritance: parents pass on characteristics to their children by combining the genes in egg and sperm to make a new individual.
Each person actually has two copies of each gene, including the CFTR gene, in each of his or her body cells. During sperm and egg production, however, these two copies separate, so that each sperm or egg contains only one copy of each gene. When sperm and egg unite, the newly created cell once again has two copies of each gene.
The two gene copies may be the same or they may be slightly different. For the CFTR gene, for instance, a person may have two normal copies, or one normal and one mutated copy, or two mutated copies. A person with two mutated copies will develop cystic fibrosis. A person with one mutated copy is said to be a carrier. A carrier will not have symptoms of CF, but can pass on the mutated CFTR gene to his or her children.
When two carriers have children, they have a one in four chance of having a child with CF each time they conceive. They have a two in four chance of having a
fibrosis Cystic
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
295 |

Cystic fibrosis
K E Y T E R M S
Carrier—A person who possesses a gene for an abnormal trait without showing signs of the disorder. The person may pass the abnormal gene on to offspring.
CFTR—Cystic fibrosis transmembrane conductance regulator. The protein responsible for regulating chloride movement across cells in some tissues. When a person has two defective copies of the CFTR gene, cystic fibrosis is the result.
Emphysema—A chronic lung disease that begins with breathlessness during exertion and progresses to shortness of breath at all times, caused by destructive changes in the lungs.
Mucociliary escalator—The coordinated action of tiny projections on the surfaces of cells lining the respiratory tract, which moves mucus up and out of the lungs.
Mucolytic—An agent that dissolves or destroys mucin, the chief component of mucus.
Pancreatic insufficiency—Reduction or absence of pancreatic secretions into the digestive system due to scarring and blockage of the pancreatic duct.
child who is a carrier, and a one in four chance of having a child with two normal CFTR genes.
Approximately one in every 25 Americans of northern European descent is a carrier of the mutated CF gene, while only one in 17,000 African-Americans and one in 30,000 Asian-Americans are carriers. Since carriers are symptom-free, very few people will know whether or not they are carriers, unless there is a family history of the disease. Two white Americans with no family history of CF have a one in 2,500 chance of having a child with CF.
It may seem puzzling that a mutated gene with such harmful consequences would remain so common; one might guess that the high mortality of CF would quickly lead to loss of the mutated gene from the population. Some researchers now believe the reason for the persistence of the CF gene is that carriers, those with only one copy of the gene, are protected from the full effects of cholera, a microorganism that infects the intestine, causing intense diarrhea and eventual death by dehydration. It is believed that having one copy of the CF gene is enough to prevent the full effects of cholera infection, while not enough to cause the symptoms of CF. This so-called “heterozygote advantage” is seen in some other genetic disorders, including sickle-cell anemia.
Signs and symptoms
The most severe effects of cystic fibrosis are seen in two body systems: the gastrointestinal (digestive) system and the respiratory tract, from the nose to the lungs. CF also affects the sweat glands and male fertility. Symptoms develop gradually, with gastrointestinal symptoms often the first to appear.
Gastrointestinal system
Ten to fifteen percent of babies who inherit CF have meconium ileus at birth. Meconium is the first dark stool that a baby passes after birth; ileus is an obstruction of the digestive tract. The meconium of a newborn with meconium ileus is thickened and sticky, due to the presence of thickened mucus from the intestinal glands. Meconium ileus causes abdominal swelling and vomiting, and often requires surgery immediately after birth. Presence of meconium ileus is considered highly indicative of CF. Borderline cases may be misdiagnosed, however, and attributed instead to a “milk allergy.”
Other abdominal symptoms are caused by the inability of the pancreas to supply digestive enzymes to the intestine. During normal digestion, as food passes from the stomach into the small intestine, it is mixed with pancreatic secretions, which help to break down the nutrients for absorption. While the intestines themselves also provide some digestive enzymes, the pancreas is the major source of enzymes for the digestion of all types of foods, especially fats and proteins.
In CF, thick mucus blocks the pancreatic duct, which is eventually closed off completely by scar tissue formation, leading to a condition known as pancreatic insufficiency. Without pancreatic enzymes, large amounts of undigested food pass into the large intestine. Bacterial action on this rich food source can cause gas and abdominal swelling. The large amount of fat remaining in the feces makes it bulky, oily, and foul-smelling.
Because nutrients are only poorly digested and absorbed, the person with CF is often ravenously hungry, underweight, and shorter than expected for his age. When CF is not treated for a longer period, a child may develop symptoms of malnutrition, including anemia, bloating, and, paradoxically, appetite loss.
Diabetes becomes increasingly likely as a person with CF ages. Scarring of the pancreas slowly destroys those pancreatic cells which produce insulin, producing type I, or insulin-dependent, diabetes.
Gallstones affect approximately 10% of adults with CF. Liver problems are less common, but can be caused by the build-up of fat within the liver. Complications of liver enlargement may include internal hemorrhaging,
296 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

abdominal fluid (ascites), spleen enlargement, and liver failure.
Other gastrointestinal symptoms can include a prolapsed rectum, in which part of the rectal lining protrudes through the anus; intestinal obstruction; and rarely, intussusception, in which part of the intestinal tube slips over an adjoining part, cutting off blood supply.
Somewhat fewer than 10% of people with CF do not have gastrointestinal symptoms. Most of these people do not have the delta-F508 mutation, but rather a different one, which presumably allows at least some of their CFTR proteins to function normally in the pancreas.
Respiratory tract
The respiratory tract includes the nose, the throat, the trachea (or windpipe), the bronchi (which branch off from the trachea within each lung), the smaller bronchioles, and the blind sacs called alveoli, in which gas exchange takes place between air and blood.
Swelling of the sinuses within the nose is common in people with CF. This usually shows up on x ray, and may aid the diagnosis of CF. However, this swelling, called pansinusitis, rarely causes problems, and does not usually require treatment.
Nasal polyps, or growths, affect about one in five people with CF. These growths are not cancerous, and do not require removal unless they become annoying. While nasal polyps appear in older people without CF, especially those with allergies, they are rare in children without CF.
The lungs are the site of the most life-threatening effects of CF. The production of a thick, sticky mucus increases the likelihood of infection, decreases the ability to protect against infection, causes inflammation and swelling, decreases the functional capacity of the lungs, and may lead to emphysema. People with CF will live with chronic populations of bacteria in their lungs, and lung infection is the major cause of death for those with CF.
The bronchioles and bronchi normally produce a thin, clear mucus, which traps foreign particles including bacteria and viruses. Tiny hair-like projections called cilia on the surface of these passageways slowly sweep the mucus along, out of the lungs and up the trachea to the back of the throat, where it may be swallowed or coughed up. This “mucociliary escalator” is one of the principal defenses against lung infection.
The thickened mucus of CF prevents easy movement out of the lungs, and increases the irritation and inflammation of lung tissue. This inflammation swells the passageways, partially closing them down, further
Accumulation of mucus in the smaller passageways of the lungs can plug them up, decreasing functional lung volume. As the air is exhaled, much of it becomes trapped in the small pores of the lungs. This leads to expansion of the lung and swollen appearance seen in the left lung above. (Custom Medical Stock Photo, Inc.)
hampering the movement of mucus. A person with CF is likely to cough more frequently and more vigorously as the lungs attempt to clean themselves out.
At the same time, infection becomes more likely since the mucus is a rich source of nutrients. Bronchitis, bronchiolitis, and pneumonia are frequent in CF. The most common infecting organisms are the bacteria
Staphylococcus aureus, Haemophilus influenzae, and
Pseudomonas aeruginosa. A small percentage of people with CF have infections caused by Burkholderia cepacia, a bacterium which is resistant to most current antibiotics (Burkholderia cepacia was formerly known as
Pseudomonas cepacia). The fungus Aspergillus fumigatus may infect older children and adults.
The body’s response to infection is to increase mucus production; white blood cells fighting the infection thicken the mucus even further as they break down and release their cell contents. These white blood cells also provoke more inflammation, continuing the downward spiral that marks untreated CF.
As mucus accumulates, it can plug up the smaller passageways in the lungs, decreasing functional lung volume. Getting enough air can become difficult; tiredness, shortness of breath, and intolerance of exercise become more common. Because air passes obstructions more easily during inhalation than during exhalation, over time, air becomes trapped in the smallest chambers of the lungs, the alveoli. As millions of alveoli gradually expand, the chest takes on the enlarged, barrel-shaped appearance typical of emphysema.
fibrosis Cystic
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
297 |
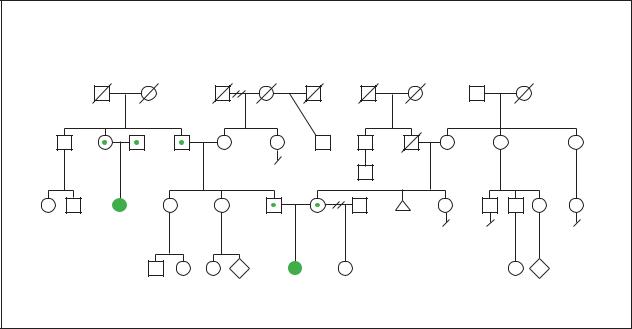
fibrosis |
|
Cystic Fibrosis |
|
|
||
|
Autosomal Recessive |
|
|
|||
|
Family history significant for CF |
|
||||
Cystic |
|
|
||||
Irish |
Italian |
|
|
German |
English |
|
|
|
|
|
|
|
Parkinsons |
|
|
|
|
|
|
disease |
|
|
|
|
3 |
Heart |
|
|
|
|
|
attack |
|
|
|
2 |
|
|
|
|
|
|
|
32y |
38y |
|
|
|
|
|
F508/ |
F508/ |
|
|
|
|
|
Negative |
Negative |
|
|
|
|
|
P |
|
|
|
P |
|
|
3mos |
11y |
|
|
|
|
|
F508/ F508 |
|
|
|
|
|
|
Sweat test 112 |
|
|
|
|
(Gale Group)
For unknown reasons, recurrent respiratory infections lead to “digital clubbing,” in which the last joint of the fingers and toes becomes slightly enlarged.
Sweat glands
The CFTR protein helps to regulate the amount of salt in sweat. People with CF have sweat that is much saltier than normal, and measuring the saltiness of a person’s sweat is the most important diagnostic test for CF. Parents may notice that their infants taste salty when they kiss them. Excess salt loss is not usually a problem except during prolonged exercise or heat. While most older children and adults with CF compensate for this extra salt loss by eating more salty foods, infants and young children are in danger of suffering its effects (such as heat prostration), especially during summer. Heat prostration is marked by lethargy, weakness, and loss of appetite, and should be treated as an emergency condition.
Fertility
Ninety-eight percent of men with CF are sterile, due to complete obstruction or absence of the vas deferens, the tube carrying sperm out of the testes. While boys and men with CF form normal sperm and have normal levels of sex hormones, sperm are unable to leave the testes, and fertilization is not possible. Most women with CF are fertile, though they often have more trouble getting pregnant than women without CF. In both boys and girls, puberty
is often delayed, most likely due to the effects of poor nutrition or chronic lung infection. Women with good lung health usually have no problems with pregnancy, while those with ongoing lung infection often do poorly.
Diagnosis
The decision to test a child for cystic fibrosis may be triggered by concerns about recurring gastrointestinal or respiratory symptoms, or salty sweat. A child born with meconium ileus will be tested before leaving the hospital. Families with a history of CF may wish to have all children tested, especially if there is a child who already has the disease. Some hospitals now require routine screening of newborns for CF.
Sweat test
The sweat test is both the easiest and most accurate test for CF. In this test, a small amount of the drug pilocarpine is placed on the skin. A very small electrical current is then applied to the area, which drives the pilocarpine into the skin. The drug stimulates sweating in the treated area. The sweat is absorbed onto a piece of filter paper, and is then analyzed for its salt content. A person with CF will have salt concentrations that are one-and-one- half to two times greater than normal. The test can be done on persons of any age, including newborns, and its results can be determined within an hour. Virtually every person who has CF will test positively on it, and virtually everyone who does not will test negatively.
298 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
Genetic testing
The discovery of the CFTR gene in 1989 allowed the development of an accurate genetic test for CF. Genes from a small blood or tissue sample are analyzed for specific mutations; presence of two copies of the mutated gene confirms the diagnosis of CF in all but a very few cases. However, since there are so many different possible mutations, and since testing for all of them would be too expensive and time-consuming, a negative gene test cannot rule out the possibility of CF.
Couples planning a family may decide to have themselves tested if one or both have a family history of CF. Prenatal genetic testing is possible through amniocentesis. Many couples who already have one child with CF decide to undergo prenatal screening in subsequent pregnancies. Siblings in these families are also usually tested, both to determine if they will develop CF, and to determine if they are carriers, to aid in their own family planning. If the sibling has no symptoms, determining his or her carrier status is often delayed until the teen years or later, when he or she is closer to needing the information to make decisions.
Newborn screening
Some states now require screening of newborns for CF, using a test known as the IRT test. This is a blood test which measures the level of immunoreactive trypsinogen, which is generally higher in babies with CF than those without it. This test gives many false positive results immediately after birth, and so requires a second test several weeks later. A second positive result is usually followed by a sweat test.
Treatment and management
There is no cure for cystic fibrosis. Treatment has advanced considerably in the past several decades, increasing both the life span and the quality of life for most people affected by CF. Early diagnosis is important to prevent malnutrition and infection from weakening the young child. With proper management, many people with CF engage in the full range of school and sports activities.
Nutrition
People with CF usually require high-calorie diets and vitamin supplements. Height, weight, and growth of a person with CF are monitored regularly. Most people with CF need to take pancreatic enzymes to supplement or replace the inadequate secretions of the pancreas. Tablets containing pancreatic enzymes are taken with every meal; depending on the size of the tablet and the
meal, as many as 20 tablets may be needed. Because of incomplete absorption even with pancreatic enzymes, a person with CF needs to take in about 30% more food than a person without CF. Low-fat diets are not recommended except in special circumstances, since fat is a source of both essential fatty acids and abundant calories.
Some people with CF cannot absorb enough nutrients from the foods they eat, even with specialized diets and enzymes. For these people, tube feeding is an option. Nutrients can be introduced directly into the stomach through a tube inserted either through the nose (a nasogastric tube) or through the abdominal wall (a gastrostomy tube). A jejunostomy tube, inserted into the small intestine, is also an option. Tube feeding can provide nutrition at any time, including at night while the person is sleeping, allowing constant intake of high-quality nutrients. The feeding tube may be removed during the day, allowing normal meals to be taken.
Respiratory health
The key to maintaining respiratory health in a person with CF is regular monitoring and early treatment. Lung function tests are done frequently to track changes in functional lung volume and respiratory effort. Sputum samples are analyzed to determine the types of bacteria present in the lungs. Chest x rays are usually taken at least once a year. Lung scans, using a radioactive gas, can show closed off areas not seen on the x ray. Circulation in the lungs may be monitored by injection of a radioactive substance into the bloodstream.
People with CF live with chronic bacterial colonization; that is, their lungs are constantly host to several species of bacteria. Good general health, especially good nutrition, can keep the immune system healthy, which decreases the frequency with which these colonies begin an infection, or attack on the lung tissue. Exercise is another important way to maintain health, and people with CF are encouraged to maintain a program of regular exercise.
In addition, clearing mucus from the lungs helps to prevent infection, and mucus control is an important aspect of CF management. Bronchial drainage is used to allow gravity to aid the mucociliary escalator. For this technique, the person with CF lies on a tilted surface with head downward, alternately on the stomach, back, or side, depending on the section of lung to be drained. An assistant thumps the rib cage to help loosen the secretions. A device called a “flutter” offers another way to loosen secretions: it consists of a stainless steel ball in a tube. When a person exhales through it, the ball vibrates, sending vibrations back through the air in the lungs. Some special breathing techniques may also help clear the lungs.
fibrosis Cystic
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
299 |

fibrosis |
|
|
Cystic Fibrosis |
|
|||
|
|
Autosomal Recessive |
|
||||
|
|
No family history |
|
||||
Cystic |
|
|
|
||||
Eastern European |
Eastern European |
|
Dutch |
English |
|||
Jewish |
|
Jewish |
|
|
|||
|
|
|
|
||||
|
|
|
|
|
|
? |
|
|
|
Bipolar |
HIV |
|
|
d.30y |
|
|
|
|
|
Childbirth |
|||
|
|
disease |
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
Brain |
|
41y |
|
37y |
|
|
|
tumor at |
|
|
|
|
|
|
|
6y now, |
|
|
|
|
|
|
|
17y |
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Mild |
7wks |
9y |
3y |
1y |
|
|
|
learning |
|
|
|
F508/N1303k |
|
|
|
disabilities |
|
|
|
Sweat test 98 |
|
(Gale Group)
Several drugs are available to prevent the airways from becoming clogged with mucus. Bronchodilators can help open up the airways; steroids reduce inflammation; and mucolytics loosen secretions. Acetylcysteine (Mucomyst) has been used as a mucolytic for many years but is not prescribed frequently now, while DNase (Pulmozyme) is a newer product gaining in popularity. DNase breaks down the DNA from dead white blood cells and bacteria found in thick mucus.
People with CF may pick up bacteria from other CF patients. This is especially true of Burkholderia cepacia, which is not usually found in people without CF. While the ideal recommendation from a health standpoint might be to avoid contact with others who have CF, this is not usually practical (since CF clinics are a major site of care), nor does it meet the psychological and social needs of many people with CF. At a minimum, CF centers recommend avoiding prolonged close contact between people with CF, and scrupulous hygiene, including frequent hand washing. Some CF clinics schedule appointments on different days for those with and without B. cepacia colonies.
Some doctors choose to prescribe antibiotics only during infection, while others prefer long-term antibiotic treatment against S. aureus. The choice of antibiotic depends on the particular organism or organisms found. Some antibiotics are given as aerosols directly into the lungs. Antibiotic treatment may be prolonged and aggressive.
Supplemental oxygen may be needed as lung disease progresses. Respiratory failure may develop, requiring temporary use of a ventilator to perform the work of breathing.
Lung transplantation is another option for people with CF, although the number of people who receive them is still much lower than those who want them. Transplantation is not a cure, however, and has been likened to trading one disease for another. Long-term immunosuppression is required, increasing the likelihood of other types of infection. About 50% of adults and more than 80% of children who receive lung transplants live longer than two years. Some CF patients whose livers have been damaged by fibrosis also undergo liver transplants.
Long-term use of ibuprofen has been shown to help some people with CF; presumably by reducing inflammation in the lungs. Close medical supervision is necessary, however, since the effective dose is high and not everyone benefits. Ibuprofen at the required doses interferes with kidney function, and together with aminoglycoside antibiotics, may cause kidney failure.
A number of experimental treatments are currently the subject of much research. Some evidence indicates that aminoglycoside antibiotics may help overcome the genetic defect in some CF mutations, allowing the protein to be made normally. While promising, these results would apply to only about 5% of those with CF.
300 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |