

Gale Encyclopedia of Genetic Disorder / Gale Encyclopedia of Genetic Disorders, Two Volume Set - Volume 1 - A-L - I
.pdfdevelopmental delay, a coarse facial appearance, incompletely formed or absent fifth fingernails, and absent fifth fingers (distal phalanges). The cause of this disorder is unknown, and the severity of symptoms varies by individual.
Description
Coffin-Siris syndrome was first described in 1970 by Dr. Grange S. Coffin and Dr. Evelyn Siris. It may also be known as fifth digit syndrome. The cause of the disorder is unknown, and the combination of symptoms may vary by individual. All affected children have some form of mental retardation or developmental delay, and incompletely formed (hypoplastic) or absent fifth fingernails and tips of the fifth fingers (distal phalanges). There are some reports of fingers other than the fifth being affected, and affected toes and toenails. The face of a child with Coffin-Siris syndrome is usually described as coarse. This includes a flat nasal bridge, broad nose, wide mouth, thick lips, and in some cases, thick eyebrows, long eyelashes, palate malformations, a large tongue (macroglossia), and a small head (microcephaly). While some infants have an abnormal facial appearance, most of the facial features become more prominent as the child grows. Typically, there is sparse scalp hair in the infant and excessive growth of body hair (hirsutism). Reduced muscle tone (hypotonia), lax joints, delay in bone maturation, and short stature are commonly found. There are reports of frequent upper respiratory and ear infections. Occasionally, children with this disorder have cardiac or spinal abnormalities, hernias, vision or hearing problems, or delayed tooth development (dentition).
Infants with Coffin-Siris syndrome typically have sucking problems and feeding difficulties that may continue as they age. The extent of growth and mental retardation varies by individual. Mental retardation is usually reported as moderate. There are delays in motor activities such as rolling over, sitting up, and walking. Speech is usually delayed. Most children are more capable of responding to speech, rather than verbally expressing themselves.
Genetic profile
At present, the cause of Coffin-Siris syndrome is unknown. Most children reported with this disorder have a normal chromosome set (karyotype). There are a few cases in which a transfer of genetic material between chromosomes (translocation) has occurred. This may provide information about a specific chromosome site responsible for Coffin-Siris syndrome, but it has not been found in many individuals.
K E Y T E R M S
Consanguinity—A mating between two people who are related to one another by blood.
Hirsutism—The presence of coarse hair on the face, chest, upper back, or abdomen in a female as a result of excessive androgen production.
Hypoplasia—Incomplete or underdevelopment of a tissue or organ.
Hypotonia—Reduced or diminished muscle tone.
Karyotype—A standard arrangement of photographic or computer-generated images of chromosome pairs from a cell in ascending numerical order, from largest to smallest.
Phalanges—Long bones of the fingers and toes, divided by cartilage around the knuckles.
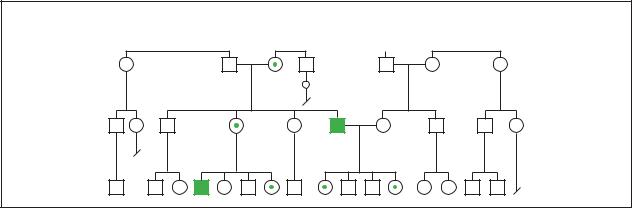
The majority of cases are sporadic, or random, in which the parents and siblings of an affected child are all healthy. However, there are some cases of affected siblings, and parental relatedness (consanguinity). CoffinSiris syndrome was originally thought to follow an autosomal recessive pattern of inheritance. This would mean that both healthy parents were carriers for the disorder, and the affected child inherited the affected gene from both parents. However, there are some reported cases that do not follow this pattern. An exact pattern of inheritance is unknown. The recurrence risk may be as high as 25%.
Demographics
At present, there are reports of more than 60 individuals affected with Coffin-Siris syndrome. It is more common in females, and the female to male ratio may be as high as a 3:1. There are cases of affected siblings, and parental relatedness. In general, cases are random, with affected children having healthy siblings and parents.
Signs and symptoms
At birth, infants with Coffin-Siris syndrome will have an absence or incomplete formation of the fifth fingernail and tip of the fifth finger (distal phalanx). This absence may also occur in the toes or in other fingers. Infants may have an abnormal facial appearance at birth. As the child grows, the facial abnormalities characteristic of Coffin-Siris syndrome become more apparent. Sparse scalp hair in an infant usually becomes more dense with age and excessive hair growth (hirsutism) develops.
syndrome Siris-Coffin
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
251 |

Cohen syndrome
Infants typically have sucking problems and feeding difficulties that may continue with age.
There is a delay in both gross and fine motor skills. Developments such as sitting up and walking may be delayed or not possible, depending upon the severity of the disorder. Speech is usually delayed and most children are better able to respond to language rather than express it. Some older children are able to form short sentences and answer simple questions. Mental retardation is usually moderate. Social adaptation is usually delayed.
Diagnosis
At present, the diagnosis of Coffin-Siris syndrome is based upon clinical findings. There are no laboratory tests that can confirm the disorder. The combination of symptoms such as coarse facial appearance, fifth finger appearance, and developmental delay would suggest Coffin-Siris syndrome. X ray of the hands to reveal the absence of the fifth finger bone is usually the best indicator of this syndrome. Neonatal ultrasounds for cardiac, kidney (renal), and other malformations that may be present with this disorder can also be informative.
Prenatal ultrasound may show intrauterine (occurring within the uterus) growth retardation, and can reveal the condition of the fifth finger. However, these symptoms alone cannot conclusively lead to a prenatal diagnosis of Coffin-Siris syndrome.
Due to the rarity, range of symptoms, and variability of Coffin-Siris syndrome, a definitive diagnosis may be difficult. It is important to exclude other disorders that may have similar symptoms. These include CoffinLowry syndrome, Cornelia de Lange syndrome, fetal hydantoin syndrome, trisomy 9p, and Brachymorphism- onychodysplasia-dysphalangism syndrome.
Treatment and management
The treatment or therapy required for children with Coffin-Siris syndrome is based on the particular symptoms of each individual. Some children may require surgery to repair malformations that may be seen with this disorder. This ranges from cleft palate repair to cardiac, renal, or other surgery. Speech therapy and special education may be considered depending upon the degree of mental retardation, developmental delay, and motor impairment.
Prognosis
Infants born with Coffin-Siris syndrome may experience a delay or absence of motor and mental activities, but with support can live into adulthood. The lifestyle of an individual with Coffin-Siris syndrome is dependent to
a large extent upon the degree of mental retardation and developmental delay.
Resources
PERIODICALS
Braun-Quentin C., et al. “Variant of Coffin-Siris Syndrome or Previously Undescribed Syndrome?” American Journal of Medical Genetics 64 (1996): 568-572.
Coffin G.S., and E. Siris. “Mental Retardation with Absent Fifth Fingernail and Terminal Phalanx.” American Journal of Diseases of Children 119 (1970): 433-439.
Dimaculangan D.P., et al. “Difficult Airway in a Patient with Coffin-Siris Syndrome.” Anesthesia and Analgesia 92 (2001): 554-555.
Fleck, B.J., et al. “Coffin-Siris Syndrome: Review and Presentation of New Cases From A Questionnaire Study.”
American Journal of Medical Genetics 99 (2001): 1-7. McPherson E.W., et al. “Apparently Balanced t(1;7)(q21.3;q34)
in an Infant With Coffin-Siris Syndrome.” American Journal of Medical Genetics 71 (1997): 430-433.
Rabe, P., et al. “Syndrome of Developmental Retardation, Facial and Skeletal Anomalies, and Hyperphosphatasia in Two Sisters: Nosology and Genetics of the Coffin-Siris Syndrome.” American Journal of Medical Genetics 41 (1991): 350-354.
ORGANIZATIONS
National Organization for Rare Disorders (NORD). PO Box 8923, New Fairfield, CT 06812-8923. (203) 746-6518 or (800) 999-6673. Fax: (203) 746-6481. http://www
.rarediseases.org .
WEBSITES
Coffin-Siris Syndrome.
http://members.aol.com/CoffinSiri/index.html .
Maureen Teresa Mahon, BSc, MFS
I Cohen syndrome
Definition
Cohen syndrome is a very rare genetic disorder characterized by infantile hypotonia (a weakening of the skeletal muscles), childhood obesity, and several malformations.
Description
Cohen syndrome was first described in 1973 by Dr. M. M. Cohen, Jr. in three children with distinct physical and developmental observations. Since then, over 100 cases have been reported throughout the world, offering the picture of an extremely rare disease with a wide range of clinical characteristics. The initial description given by
252 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

Cohen included obesity, mental retardation, low muscle tone, narrow hands and feet, and distinctive facial features with prominent upper central teeth. As of 2001, the underlying cause of the disease remains unknown.
Cohen syndrome has also been referred to as Pepper syndrome, Hypotonia-Obesity-Prominent Incisors syndrome, Obesity-Hypotonia syndrome, and Mirhosseini- Holmes-Walton syndrome.
Genetic profile
Research has suggested that the gene for Cohen syndrome lies between 8q21.3 and 8q22.1. This refers to a location on the long arm of chromosome 8 between positions 21.3 and 22.1 and is a rough estimate of where the gene may lie. This region was originally referred to as CHS1 but has since become known as COH1. The phrase ‘COH1 gene region’ is often used due to the fact that the exact location of the gene still remains to be discovered.
Chromosomes are the genetic material passed down from generation to generation that tell a person’s body how to work and how to grow. Each chromosome is composed of smaller pieces known as genes. A person inherits one set of 23 chromosomes from both the egg and the sperm of the parents. These chromosomes can then be matched into pairs, giving two copies of each chromosome and likewise two copies of each gene.
Cohen syndrome is an autosomal recessive disorder. Recessive means that both copies of the COH1 gene region must have a change or mutation for a person to be affected. An individual with only one changed COH1 gene region is not affected by the disease but can pass the disease on to a future child. These individuals are called carriers. If two carriers have a child there is a 25% chance with each pregnancy that the child will be affected. At this time prenatal diagnosis is not available.
Demographics
While Cohen syndrome affects all races and genders, several small samplings of affected populations have been studied around the world. Interestingly, it has been found that Cohen syndrome manifests in these populations in distinctly different ways, with certain clinical findings being familyor ethnic-specific.
For example, Cohen syndrome has been studied extensively in Finland. In the populations studied, individuals diagnosed with the syndrome typically have fewer white blood cells than normal (granulocytopenia), a specific eye abnormality called mottled retina, and mental retardation. As a rule, they do not have truncal obesity, a common characteristic of Cohen syndrome in
K E Y T E R M S
Astigmatism—A cause of poor eyesight, usually due to an error in the refraction of light within the eye.
Autism—A syndrome characterized by a lack of responsiveness to other people or outside stimulus, often in conjunction with a severe impairment of verbal and non-verbal communication skills.
Autosome—Chromosome not involved in specifying sex.
Coloboma of the iris—A birth defect leading to missing structures within the eye.
Granulocytopenia—A reduced number of white blood cells in the circulation.
Hypotonia—Reduced or diminished muscle tone.
Leucopenia—A decrease in white blood cells.
Microphthalmia—Small or underdeveloped eyes.
Mottled retina—Changes in the retina of the eye causing a loss of visual acuity.
Myopia—Nearsightedness. Difficulty seeing objects that are far away.
Neutropenia—A condition in which the number of leukocytes (a type of white or colorless blood cell) is abnormally low, mainly in neutrophils (a type of blood cell).
Philtrum—The center part of the face between the nose and lips that is usually depressed.
Retinal dystrophy—Degeneration of the retina, causing a decline in visual clarity.
other populations. Although the symptoms of Cohen syndrome are known to vary widely between affected individuals within the same family, affected people within the Finnish populations are very similar to each other in their presentation.
Due to the extreme rarity of the disease, the exact incidence of Cohen syndrome is not known. A relatively high frequency of the disease has also been noted in Israel. However, earlier reports suggesting a possible increase in the frequency of Cohen syndrome among Ashkenazi Jews no longer seems to be true.
Signs and symptoms
Four main areas are affected by Cohen syndrome: physical appearance, mental function, vision, and hema-
syndrome Cohen
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
253 |
Cohen syndrome
tology (blood function). The list of possible conditions is extensive however, and it is important to remember that each case is different. While a given characteristic may be common to the syndrome, not all affected individuals have been found to have it.
Physical appearance
When they are born, babies with Cohen syndrome usually look just like babies without the syndrome, although they are typically born at a low birth weight. As they grow, the various physical signs associated with the syndrome become increasingly obvious.
Narrow hands and feet with long slender fingers are a hallmark feature, found in approximately 89% of diagnosed individuals. Truncal obesity, or the abnormal deposition of fat around the mid-section of the body, has been observed in roughly 70% of patients. Most individuals with Cohen syndrome have large and rather noticeable front teeth, referred to as prominent upper central incisors. In general, the teeth are abnormal in shape and position. A majority of individuals with Cohen syndrome are also short, with many experiencing growth deficiency at all stages of life. Microcephaly (small head) is another common feature of the syndrome.
In addition, there are many other associated physical characteristics that occur less often. The palate (roof of the mouth) may be overly high, arched, and narrow. The mid-face can have an underdeveloped appearance and the area below the nose to the upper lip (philtrum) may be very short. The eyes can be down-slanting and thick hair and eyebrows may be observed.
Mental dysfunction
It is thought that every individual with Cohen syndrome experiences some level of developmental delay. Mental retardation can range from mild to severe. Even from infancy many are obviously behind in developmental milestones and are not able to sit up or roll over within the same time frame as their peers.
Most children with Cohen syndrome do learn to walk, although there have been a few reported cases of individuals who were wheelchair-bound. There is usually a noticeable delay, with affected children not learning to walk independently until much later than their peers (the normal average age for walking independently is 12 months).
Language deficiencies are also a common occurrence. Many affected individuals never learn to talk or have a vocabulary limited to a few singular words and two-word phrases. In general an IQ of less than 50 is considered average for Cohen syndrome.
Visual deficiencies
Vision is affected to varying degrees. Severe limitation in eyesight due to myopia is often observed. Several other dysfunctions and defects of the eyes causing low visual clarity have been reported including retinal dystrophy, strabismus, astigmatism, microphthalmia, and coloboma of the iris.
Hematologic abnormalities
Cohen syndrome can have a profound effect on the composition of the blood. Abnormally low counts of white blood cells, referred to as granulocytopenia, was once thought to be a standard symptom. It was hoped that it could help in early diagnosis because it can be tested for at birth. However, further studies have shown that not all affected individuals suffer from granulocytopenia. Some individuals have no blood disorders associated with their disease at all while others have various forms of white blood cell problems, such as a reduction in the number of white blood cells in the blood (leucopenia) or of neutrophils, which are specialized white blood cells (neutropenia).
Other deficiencies
Hypotonia, or low muscle tone, is found in 90-100% of the persons diagnosed with Cohen syndrome. Babies with hypotonia are described as “floppy” due to their lack of muscle strength. Although the observed hypotonia is not thought to be associated with any nervous system disorder, it does delay the overall development of the child, most notably in slowing the development of motor skills.
Social skills
Many studies have described Cohen syndrome patients as being outgoing and friendly with mild hyperactivity and severe attention deficits. There are a few reports of diagnosed individuals showing signs of autism, an extreme form of centering attention and interest on the self only.
Diagnosis
In 1972, Dr. Mirhosseini and others described two patients with symptoms similar to those observed in Cohen syndrome. These patients and a few subsequent cases were given a diagnosis of Mirhosseini-Holmes- Walton syndrome. Over the years, scientific opinion has come to consider Mirhosseini-Holmes-Walton syndrome and Cohen syndrome as different manifestations of the same disease.
Diagnosis of Cohen syndrome is difficult due to the varied nature of the symptoms. Most features of Cohen
254 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

syndrome are not evident in the newborn and many symptoms, such as truncal obesity and visual deficits are not easily observed until early childhood. In the past, the average age of diagnosis was approximately 6-8 years. However, as physicians become more aware of the disorder it is hoped that diagnosis will occur at earlier ages, offering affected individuals the opportunity for rapid intervention and treatment.
Incorrect diagnosis is not uncommon in patients with Cohen syndrome. Affected individuals may be misdiagnosed with Marfan syndrome, Sotos syndrome, hypothyroidism, Prader-Willi syndrome, or mental retardation of an unknown nature.
A correct and early diagnosis is important to ensure the favorable prognosis of the patient and so that the family can receive appropriate genetic counseling concerning the affected child or the risks involved in future pregnancies.
Treatment and management
Treatment of Cohen syndrome is focused on improving or alleviating symptoms as they arise. There is no cure for Cohen syndrome.
Early correction of vision problems, usually with glasses, often leads to general improvement of cognitive skills, an area of marked deficit in affected individuals.
As is the case for many disorders involving hypotonia and slowed development, physical and occupational therapy are invaluable tools. These treatment strategies are important at any age, but should be started as early as possible. There is no need to wait for a definitive diagnosis of Cohen syndrome as any child with hypotonia can benefit from physical and occupational therapy.
Prognosis
Varying symptoms lead to varying prognosis. Mental retardation can range from mild to severe. However, there is no way to predict the level of developmental delay a specific child will experience. Language deficiencies also vary a lot, with some children never learning to speak at all and others speaking full sentences. The hypotonia observed in infancy may persist and moderate obesity usually develops in mid-childhood.
As of 2001, there has been one reported case of a woman with Cohen syndrome giving birth. The child had some developmental delays but was thought not to have Cohen syndrome.
Resources
PERIODICALS
Kivitie-Kallio, S., J. Rajantie, E. Juvonen, and R. Norio.
“Granulocytopenia in Cohen syndrome.” British Journal
of Haematology 98 (1999): 308-311.
Young, I.D., and J. Moore. “Intrafamilial variation in Cohen syndrome.” Journal of Medical Genetics 24 (1987): 488-492.
ORGANIZATIONS
International Cohen Syndrome Support Group. 7 Woods Court,
Brackley, Northants, NN13-6HP. UK (012) 80–704515.
WEBSITES
NORD—National Organization for Rare Diseases, Inc.
http://www.rarediseases.org .
The Arc: A National Organization on Mental Retardation.
http://www.thearc.org .
Java O. Solis, MS
I Coloboma
Definition
Coloboma, also known as keyhole defect of the iris, is a congenital genetic disorder that affects the iris of the eye. Present at birth, coloboma implies the absence of tissue.
Description
A coloboma describes a condition wherein a portion of a structure of the eye is absent, usually the iris, retina, or the optic nerve. The disorder is often referred to as a keyhole defect of the iris because the shape of the coloboma appears as the shape of a keyhole or an upsidedown pear. There are many different types of colobomas, as described below.
Types of colobomas:
•Optic disc coloboma. This disorder occurs when the coloboma covers the optic nerve and may involve the macula, a structure in the eye that is responsible for visual acuity.
•Iris coloboma. This type of coloboma may be in one eye (unilateral) or in both eyes (bilateral). The pupil is often described as an upside-down pear shape when an individual has an iris coloboma.
•Retinal coloboma. In this disorder, a notch or cleft of the retina or part of the retina is missing. For example, 35% or more of the retina may be missing.
•Choroidal coloboma. This condition is similar to a retinal coloboma. The choroid is a structure in the eye that lies between the sclera and the retina.
•Morning glory syndrome. This condition, a type of optic nerve coloboma, affects the shape of the optic nerve. The syndrome is aptly named because it
Coloboma
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
255 |
Coloboma
K E Y T E R M S
Choroid—A vascular membrane that covers the back of the eye between the retina and the sclera and serves to nourish the retina and absorb scattered light.
Iris—The colored part of the eye, containing pigment and muscle cells that contract and dilate the pupil.
Macula—A small spot located in the back of the eye that provides central vision and allows people to see colors and fine visual details.
Optic nerve—A bundle of nerve fibers that carries visual messages from the retina in the form of electrical signals to the brain.
Pupil—The opening in the iris through which light enters the eye.
Retina—The light-sensitive layer of tissue in the back of the eye that receives and transmits visual signals to the brain through the optic nerve.
Sclera—The tough white membrane that forms the outer layer of the eyeball.
describes the appearance of the optic nerve, which looks like the inside of a morning glory flower.
Genetic profile
Colobomas may be isolated abnormalities in otherwise normal individuals or they may occur as part of a syndrome. As isolated findings, they are generally sporadic (not inherited). Some families, however, have shown an autosomal dominant inheritance pattern, meaning only one copy of the abnormal gene needs to be present for the disorder to occur. Some of the genetic disorders thought to contribute to coloboma include cateye syndrome, trisomy 13, trisomy 18, Sturge-Weber syndrome, and basal cell nevus syndrome.
Demographics
The condition occurs in about one in 10,000 births. Coloboma may be associated with hereditary or genetic conditions, trauma to the eye, or eye surgery.
Signs and symptoms
Chorioretinal colobomas are those that affect the choriod (light impermeable lining consisting primarily of blood vessels) and the retina (the photosensitive lining
The pupil in this eye is enlarged, extending to the lower edge of the cornea. Colobomas form because of a failure of the rudimentary eye to join the optic fissure during embryonic development. (Photo Researchers, Inc.)
inside the eye). The extent to which vision would be impaired depends on the size of the coloboma, and its impact on the optic nerve and macula. A coloboma can appear as a black indentation of varying depth at the edge of the pupil, and gives the pupil an odd or irregular shape. It may also appear as a split in the iris from the pupil to the edge of the iris.
Symptoms usually present as blurred or decreased vision, and an appearance of a hole or odd-shaped pupil in the individual’s eye. A smaller colboma, especially if it is not attached to the pupil, often causes a secondary image to focus on the back of the eye, producing blurred vision or decreased visual sharpness.
Diagnosis
A diagnosis is made by a physical exam and includes a detailed eye examination by an opthalmologist. The ophthalmologist will also ask the individual when the symptoms were first noticed, determine what part of the eye is affected, the size and shape of the dark area in the eye, and ask for reports of any changes in the individual’s vision.
Certain diagnostic tests are often used to diagnose coloboma. These include a visual acuity test, refraction test, and an in-depth history of symptoms.
Treatment and management
Colobomas may be accompanied by other problems that may be neurological or chromosomal in nature. In addition, some genetic syndromes also include coloboma as part of the disorder’s potential findings. More importantly, a specific combination of abnormalities identified by the acronym CHARGE must also be considered when a diagnosis of coloboma is made.
256 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

The medical condition known as CHARGE association is a very rare and serious condition. Individuals that have the condition will require attention from several specialists and treatment from an early age. Colobomas are usually one of the findings in individuals with CHARGE. The disorder includes these problems:
•(C)oloboma
•(H)eart defects
•(A)tresia of the choanae, which is a blockage of the nasal passages
•(R)etarded growth and development
•(G)enital hypoplasia, which occurs when the testes do not descend properly
•(E)ar abnormalities
While there is no specific treatment for coloboma, some treatments are available that can manage vision problems associated with the disorder. For example, physicians often recommend cosmetic contact lenses and sunglasses for individuals whose eyesight is adversely affected. Additional optical aids are often helpful such as eye patching. Since many indivduals with coloboma are highly sensitive to light, opthalmologists often recommend special lights or other personalized visual aids.
Prognosis
The effects of coloboma can be mild or severe, depending upon the extent and location of the gap or cleft. The gap itself is usually located at the bottom of the eye, but it may occur in the iris, choroid, macula or optic nerve.
A coloboma of the lens, particularly if it is large, may also include abnormalities of the iris and choroids, which increases the risk of retinal tearing. In severe cases of coloboma, the eye may be reduced in size. This condition is called microphthalmous, a disorder that can arise with or without coloboma.
The specific gene or genes responsible for coloboma have not yet been identified, but research continues throughout the United States, Scotland, and England.
Resources
ORGANIZATIONS
Royal National Institute for the Blind. PO Box 173, Peterborough PE2 6WS. http://www.rnib.org.uk .
WEBSITES
Coloboma. http://www.coloboma.org/whatis.html .
Medlineplus.
http://www.medline.adam.com/ency/article/003318.htm .
Bethanne Black
Coloboma-obesity-hypogenialism-mental
retardation syndrome see Coloboma
I Color blindness
Definition
Color blindness is an abnormal condition characterized by the inability to clearly distinguish different colors of the spectrum. The difficulties can be mild to severe. It is a misleading term because people with color blindness are not blind. Rather, they tend to see colors in a limited range of hues; a rare few may not see colors at all.
Description
Normal color vision requires the use of specialized receptor cells called cones, which are located in the retina of the eye. There are three types of cones, termed red, blue, and green, which enable people to see a wide spectrum of colors. An abnormality, or deficiency, of any of the types of cones will result in abnormal color vision.
There are three basic variants of color blindness. Red/green color blindness (deuteranopia) is the most common deficiency, affecting 8% of Caucasian males and 0.5% of Caucasian females. The prevalence varies with culture.
Blue color blindness (protanopia) is an inability to distinguish both blue and yellow, which are seen as white or gray. Protanopia is quite rare and has equal prevalence in males and females. It is common for young children to have blue/green confusion that becomes less pronounced in adulthood. Blue color deficiency often appears in people who have physical disorders such as liver disease or diabetes mellitus.
A total inability to distinguish colors (achromatopsia) is exceedingly rare. These affected individuals view the world in shades of gray. They frequently have poor visual acuity and are extremely sensitive to light (photophobia), which causes them to squint in ordinary light.
Genetic profile
Red/green and blue color blindness appear to be located on at least two different gene locations. The majority of affected individuals are males. Females are carriers but are not normally affected. This indicates that the X chromosome is one of the locations for color blindness. Male offspring of females who carry the altered gene have a fifty-fifty chance of being color-blind. The rare female that has red/green color blindness, or rarer
blindness Color
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
257 |
Color blindness
K E Y T E R M S
Achromatopsia—The inability to distinguish any colors.
Cones—Receptor cells that allow the perception of colors.
Deuteranopia—The inability or difficulty in distinguishing red/green colors.
Photophobia—An extreme sensitivity to light.
Protanopia—The inability or difficulty in distinguishing blue and yellow colors.
Retina—The light-sensitive layer of tissue in the back of the eye that receives and transmits visual signals to the brain through the optic nerve.
Rod—Photoreceptor that is highly sensitive to low levels of light and transmits images in shades of gray.
still, blue color blindness, indicates there is an involvement of another gene. As of 2001, the location of this gene has not been identified.
Achromatopsia, the complete inability to distinguish color, is an autosomal recessive disease of the retina. This means that both parents have one copy of the altered gene but do not have the disease. Each of their children has a 25% chance of not having the gene, a 50% chance of having one altered gene (and, like the parents, being unaffected), and a 25% risk of having both the altered gene and the condition. In 1997, the achromatopsia gene was located on chromosome 2.
Demographics
Researchers studying red/green color blindness in the United Kingdom reported an average prevalence of only 4.7% in one group. Only 1% of Eskimo males are color blind. Approximately 3% of boys from Saudi Arabia and 4% from India were found to have deficient color vision. Red/green color blindness may slightly increase an affected person’s chances of contracting leprosy. Pre-term infants exhibit an increased prevalence of blue color blindness. Achromatopsia has a prevalence of about one in 33,000 in the United States and affects males and females equally.
Color blindness is sometimes acquired. Chronic illnesses that can lead to color blindness include
Alzheimer disease, diabetes mellitus, glaucoma, leukemia, liver disease, chronic alcoholism, macular degeneration, multiple sclerosis, Parkinson disease, sickle cell anemia, and retinitis pigmentosa. Accidents
A common test used to detect color blindness. The number “hidden” in the image will not be visible to an individual with red/green color blindness. (Corbis)
or strokes that damage the retina or affect particular areas of the brain can lead to color blindness. Some medications such as antibiotics, barbiturates, anti-tuberculosis drugs, high blood pressure medications, and several medications used to treat nervous disorders and psychological problems may cause color blindness. Industrial or environmental chemicals such as carbon monoxide, carbon disulfide, fertilizers, styrene, and some containing lead can cause loss of color vision. Occasionally, changes can occur in the affected person’s capacity to see colors after age 60.
Signs and symptoms
The inability to correctly identify colors is the only sign of color blindness. It is important to note that people with red/green or blue varieties of color blindness use other cues such as color saturation and object shape or location to distinguish colors. They can often distinguish red or green if they can visually compare the colors. However, most have difficulty accurately identifying colors without any other references. Most people with any impairment in color vision learn colors, as do other young children. These individuals often reach adolescence before their visual deficiency is identified.
Diagnosis
There are several tests available to identify problems associated with color vision. The most commonly used is
258 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
Color Blindness
blindness Color
(Gale Group)
the American Optical/Hardy, Rand, and Ritter Pseudoisochromatic test. It is composed of several discs filled with colored dots of different sizes and colors. A person with normal color vision looking at a test item sees a number that is clearly located somewhere in the center of a circle of variously colored dots. A color-blind person is not able to distinguish the number.
The Ishihara test is comprised of eight plates that are similar to the American Optical Pseudoisochromatic test plates. The individual being tested looks for numbers among the various colored dots on each test plate. Some plates distinguish between red/green and blue color blindness. Individuals with normal color vision perceive one number. Those with red/green color deficiency see a different number. Those with blue color vision see yet a different number.
A third analytical tool is the Titmus II Vision Tester Color Perception test. The subject looks into a stereoscopic machine. The test stimulus most often used in professional offices contains six different designs or numbers on a black background, framed in a yellow border. Titmus II can test one eye at a time. However, its value is limited because it can only identify red/green deficiencies and is not highly accurate.
Treatment and management
There is no treatment or cure for color blindness. Most color vision deficient persons compensate well for their abnormality and usually rely on color cues and details that are not consciously evident to persons with typical color vision.
Inherited color blindness cannot be prevented. In the case of some types of acquired color deficiency, if the cause of the problem is removed, the condition may
improve with time. But for most people with acquired color blindness, the damage is usually permanent.
Prognosis
Color blindness that is inherited is present in both eyes and remains constant over an individual’s entire life. Some cases of acquired color vision loss are not severe, may appear in only one eye, and can last for only a short time. Other cases tend to be progressive, becoming worse with time.
Resources
BOOKS
Rosenthal, Odeda, and Robert H. Phillips. Coping with Color Blindness. Garden City Park, NY: Avery Publishing Group, 1997.
Sacks, Oliver. The Island of the Colorblind. New York, Knopf, 1997.
Wiggs, Janey L. Color Vision. In: Ophthalmology, edited by Myron Yanoff and Jay S. Duker. St. Louis, Mosby, 2000, pp. 8-10.
PERIODICALS
Arbour, N. C., et al. “Homozygosity Mapping of Achromatopsia to Chromosome 2 Using DNA Pooling.” Human Molecular Genetics 1997 May; 6(5): 689-694.
Dobson, V., et al. “Color Vision Measured with Pseudoisochromatic Plates at Five-and-a-Half Years in Eyes of Children from the CRYO-ROP Study.” Investigations in Ophthalmology and Visual Science 37 (12) (November 1996): 2467-2474.
Holroyd, E., Hall, D. M. “A Re-Appraisal of Screening for Colour Vision Impairments.” Child Care Health Developments 23 (5) (September 1997): 391-398.
Osuobeni, E. P. “Prevalence of Congenital Red-Green Color Vision Defects in Arab Boys from Riyadh, Saudi Arabia.” Ophthalmic Epidemiology 3 (3) (December 1996): 167-170.
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
259 |

Cone-rod dystrophy
ORGANIZATIONS
Achromatopsia Network. C/O Frances Futterman, PO Box 214, Berkeley, CA 94701-0214. http://www.achromat.org/ how_to_join.html .
American Academy of Ophthalmology. PO Box 7424, San Francisco, CA 94120-7424. (415) 561-8500. http://www
.eyenet.org .
International Colour Vision Society: Forschungsstelle fuer Experimentelle Ophthalmologie. Roentgenweg 11, Tuebingen, D-72076. Germany http://orlab.optom.unsw.edu
.au/ICVS .
National Society to Prevent Blindness. 500 East Remington Rd., Schaumburg, IL 60173. (708) 843-2020 or (800) 3312020. http://www.preventblindness.org .
WEBSITES
“Breaking the Code of Color.” Seeing, Hearing and Smelling
the World. http://www.hhmi.org/senses/b/b130.htm .
“Color Blindness.” Geocities.
http://www.geocities.com/Heartland/8833/coloreye.html .
“Medical Encyclopedia: Colorblind.” MEDLINEplus.
http://medlineplus.adam.com/ency/article/001002sym.htm .
University of Manchester.
http://www.umist.ac.uk/UMIST_OVS/welcome.html .
University of Nevada–Reno.
http://www.delamare.unr.edu/cb/ .
L. Fleming Fallon, Jr., MD, DrPH
I Cone-rod dystrophy
Definition
Cone-rod dystrophy (CRD) is a progressive retinal degenerative disease that causes deterioration of the cones and rods in the retina and frequently leads to blindness. Cone-rod dystrophy is also accompanied by amelogenesis imperfecta, an abnormality affecting the teeth.
Description
Cone-rod dystrophy is characterized by all of the following elements: skin pigmentation abnormality; involuntary, rhythmic movements of the eyes (nystagmus); degeneration of vision (optic atrophy); and sensitivity to light (photophobia).
Cone-rod dystrophy can be inherited as either an autosomal dominant or autosomal recessive trait. In its most common form, however, it is usually inherited as an autosomal recessive trait, which means that both parents have one copy of the cone-rod dystrophy gene but do not have the disease. Autosomal recessive cone-rod dystrophy (arCRD) is a genetically heterogeneous disease with
changes (mutations) in the ABCR gene. These mutations cause an abnormality in rod outer segment function that ultimately leads to dysfunction or death of the photoreceptor cells in the retina.
Genetic profile
The CRX gene has been shown to contain mutations that cause an autosomal dominant form of cone-rod dystrophy. This means that only one parent has to pass on the gene mutation in order for the child to be affected with the disease. This genetic form of CRD is clinically known as CORD2, or cone-rod dystrophy 2. Mutations in the CRX gene interfere in the development process of embryonic photoreceptor cells during the early stages of life. The result is abnormal photoreceptor cells with reduced function.
Demographics
Inherited retinal degeneration dystrophies have an incidence of approximately one in 4,000 people. Conerod dystrophy is an uncommon entity. The prevalence is estimated to be in the range of one in 10,000 to one in 100,000.
Signs and symptoms
The earliest symptom of CRD is loss of night vision that usually begins after the age of 20. The vision loss is progressive and unrelenting. Over the next decade, loss of all vision begins and by age 50, most people with cone-rod dystrophy have gone completely blind.
Cone-rod dystrophy is occasionally accompanied by amelogenesis imperfecta, which is characterized by abnormally shaped teeth and abnormalities in the tooth enamel.
Diagnosis
The earliest symptom of cone-rod dystrophy is decreased visual acuity. However, the diagnosis of conerod dystrophy is usually established with loss of the peripheral visual fields. Cone-rod dystrophy must be distinguished from retinitis pigmentosa (RP). In CRD, rods and cones are lost at approximately the same rate. It is further distinguished from RP by the absence of night blindness as a presenting symptom.
Treatment and management
As of 2001, there are no known treatments or cures for cone-rod dystrophy. It has been suggested, however, that people with cone-rod dystrophy may be able to slow the progression of their blindness by wearing sunglasses and avoiding bright light.
260 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |