

Gale Encyclopedia of Genetic Disorder / Gale Encyclopedia of Genetic Disorders, Two Volume Set - Volume 1 - A-L - I
.pdfBiotinidasedeficiency |
|
Biotinidase Deficiency |
|
|||
|
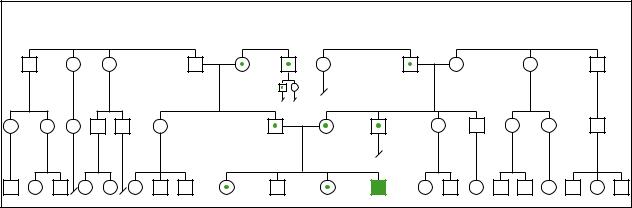
Autosomal Recessive |
|
||||
|
|
|
|
|
|
|
71y |
73y |
|
|
|
d.72y |
70y |
|
Breast cancer |
2 |
Alzheimer |
|
||
|
at 67y |
|
|
disease |
|
|
|
|
|
|
|
||
2 |
|
|
|
|
|
|
44y |
42y |
40y |
39y |
38y |
|
35y |
Deaf |
|
|
d.2y 11y |
9y |
6y |
|
from birth |
|
Seizures |
|
Diagnosed by |
|
|
now 18y |
|
|
|
|
newborn |
|
|
|
|
|
|
screen |
|
(Gale Group)
symptoms usually do not appear immediately at birth, biotinidase deficiency is also referred to as late-onset or juvenile multiple carboxylase deficiency. A related disorder, early-onset or neonatal multiple carboxylase deficiency, is caused by the lack of a different enzyme, holocarboxylase synthetase, and, as the name suggests, results in symptoms in the newborn period.
Genetic profile
Inheritance pattern
Biotinidase deficiency is an autosomal recessive disorder affecting both males and females. In individuals with this disorder, both copies of the biotinidase gene are defective. Both parents of an affected child have one abnormal copy of the gene, but usually do not show symptoms because they also have one normal copy. The normal copy provides approximately 50% of the usual enzyme activity, a level adequate for the body’s needs. Individuals with one abnormal copy of the gene and 50% enzyme activity are said to be carriers or heterozygotes. As is typical of autosomal recessive inheritance, their risk for having another child with the disorder is 25% in each subsequent pregnancy.
Gene location
The gene for biotinidase is located on the short arm of chromosome 3 (3p25). As of 1999, at least 40 differ-
ent mutations in this gene had been identified in individuals with biotinidase deficiency. The fact that there are a number of different types of mutations helps explain why symptoms are variable from one individual to another. However, the presence of variability even within a family suggests there may be other, as yet unknown, factors that affect the severity of the disease.
Demographics
Individuals with biotinidase deficiency have been described in various ethnic groups worldwide. In the general population, the incidence of the disease is estimated at about one in 60,000 individuals and one in every 123 individuals is a carrier.
Signs and symptoms
The onset of symptoms is typically between three and six months of age but varies widely from one week to several years. The most common clinical features are hair loss (alopecia), skin rash (dermatitis), seizures (convulsions), decreased muscle tone (hypotonia), difficulty walking (ataxia), breathing problems, redness of the eyes (conjunctivitis), hearing and vision loss, and developmental delay. Children with biotinidase deficiency are prone to fungal and bacterial infections, suggesting that
158 |
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
the immune system is also affected. Symptoms are highly variable among affected individuals even, within a single family.
Biotinidase deficiency is classified as either partial or profound. If there is at least 10% enzyme activity, the deficiency is considered partial and is usually associated with minimal to mild symptoms. Profound biotinidase deficiency, defined as less than 10% of normal activity, is characterized by many of the symptoms mentioned above, and can, if left untreated, result in coma and death.
Diagnosis
Children with profound biotinidase deficiency may show general signs such as vomiting, seizures, and low muscle tone, all of which can be associated with a number of different disorders. Diagnosis can be difficult because of the many different enzyme deficiencies (inborn errors of metabolism) with similar symptoms and test results. For example, abnormally high amounts of certain acidic products in the blood and urine can be typical of a number of different metabolic disorders including biotinidase deficiency. Accurate diagnosis is made by measuring the activity of the enzyme in blood or skin cells. A number of states and countries test for this disorder at birth as part of a comprehensive newborn screening program. Infants whose tests indicate they have biotinidase deficiency can be started on treatment before symptoms appear. With regular treatment these infants usually remain symptom-free.
Carrier testing
Most carriers can be detected by measuring biotinidase activity in their blood. Fifty percent of normal enzyme activity is characteristic of carriers. Specific DNA tests can usually detect the particular gene mutation in any affected individual or carrier.
Prenatal diagnosis
If a couple has had one child with biotinidase deficiency, they can be offered prenatal testing in future pregnancies. Prenatal testing is accomplished by measuring biotinidase activity in amniotic fluid cells obtained by amniocentesis around the sixteenth week of pregnancy. Alternatively, if specific gene mutations have been identified in the parents, fetal DNA from amniotic fluid cells can be studied to test for these same mutations in the fetus. Carrier couples who are considering prenatal diagnosis should discuss the risks and benefits of this type of testing with a geneticist or genetic counselor.
Treatment and management
Treatment of the profound form of biotinidase deficiency consists of giving large doses of biotin orally. Partial deficiencies are usually treated with lower doses. The biotin must be in a free form; that is, not attached to other molecules as would be the case with the biotin found in food. Properly treated, biotinidase deficiency is not a life-threatening condition, but biotin treatment must continue throughout life. No treatment is needed before birth because the developing fetus is provided with sufficient free biotin from the mother.
Prognosis
Daily treatment with free biotin usually results in rapid improvement of the skin condition, hair regrowth, and a lessening or cessation of seizure activity. Many children whose development has been affected by biotinidase deficiency have shown some improvement after treatment. Hearing and vision losses are less reversible. Children who are diagnosed at birth through newborn screening programs rarely develop symptoms if they are started on biotin replacement therapy immediately.
Resources
BOOKS
Wolf, Barry. “Disorders of Biotin Metabolism.” In Metabolic and Molecular Bases of Inherited Disease, edited by C.R. Scriver, et al. New York: McGraw-Hill, 2001.
PERIODICALS
Blanton, S. H., et al. “Fine Mapping of the Human Biotinidase Gene and Haplotype Analysis of Five Common Mutations.” Human Heredity 50 (March-April 2000): 102-11.
Norrgard, K. J., et al. “Mutations Causing Profound Biotinidase Deficiency in Children Ascertained by Newborn Screening in the United States Occur at Different Frequencies Than in Symptomatic Children.” Pediatric Research 46 (July 1999): 20-27.
ORGANIZATIONS
National Organization for Rare Disorders (NORD). PO Box 8923, New Fairfield, CT 06812-8923. (203) 746-6518 or (800) 999-6673. Fax: (203) 746-6481. http://www
.rarediseases.org .
WEBSITES
“Biotinidase.” Online Mendelian Inheritance in Man.
http://www.ncbi.nlm.nih.gov/entrez/dispomim
.cgi?id=253260 (May 24, 2001).
Thibodeau, D. L., and B. Wolf. “Biotinidase Deficiency. A Booklet for Families and Professionals.” http://views
.vcu.edu/biotin .
Tyler for Life Foundation Home Page.http://www.tylerforlife.com/biotinidase.htm .
Sallie Boineau Freeman, PhD
deficiency Biotinidase
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
159 |

Bipolar disorder
I Bipolar disorder
Definition
Bipolar disorder is characterized by mood swings, which are unpredictable and range from mania (elevated and irritable mood) to depression (a mood characterized by loss of interest and sadness). The disorder causes significant difficulties or impairment in social, occupational, and general functioning capabilities.
Description
Bipolar Type II (BT II) disorder is a psychological disorder characterized by fluctuation of cycles (time periods) of mania and depression. The manic cycle or phase is commonly associated with irritability, decreased need for sleep (sleep disruption), euphoria (an exaggerated false self-perception of feeling good), social extroversion (excessive friendliness), and feeling more important than one truly is (grandiosity). The depressive episode or cycle is correlated with a broad spectrum of symptoms. Most patients in depressive cycles exhibit common symptoms, which include fatigue, impaired concentration/decision making, and altered sleep and appetite patterns. This cycle can further progress to the level where patients feel excessively shameful and guilty. In totality, the symptoms for the depressive cycle can lead to thoughts of death or dying. The disorder is also called Manic-Depressive Psychosis, and Major Affective Disorder.
Genetic profile
There is significant evidence that correlates BT II with genetic causes. Studies have shown monozygotic twins (identical twins) have an 80% concordance rate (presence of the same disorder in twins). Additionally, studies have demonstrated that the disorder is transmitted to children (progeny) by autosomal dominant inheritance. This means that either affected parent has a 50% chance of having a child (regardless if the child is male or female) with the disorder.
Further studies concerning the genetic correlations have revealed specific chromosomes (the structure that contains genes) that contain mutated genes. Susceptible genes are located in specific regions of chromosomes 13, 18, and 21. The building blocks of genes, called nucleotides, are normally arranged in a specific order and quantity. If these nucleotides are repeated in a redundant fashion a genetic abnormality usually results. Recent evidence suggests a special type of nucleotide sequence (CAG/CTG repeats) is observed in patients with BT II on chromosome 18. However, the presence of this sequence
does not worsen the disorder or change the age of onset. It is currently thought that expression of BT II involves multiple mutated genes. Further research is ongoing to determine precise mechanisms and to develop genetic markers (gene tags) for predicting which individuals are at higher risk.
Demographics
Manic-depression is a common psychological disorder that is difficult to diagnose (detect). It is estimated that about three million people in the United States are affected. Community oriented studies suggest that the lifetime prevalence (number of cases in terms of time) is approximately 0.5%. The disorder is more common in women than in men. Women have been observed at increased risk of developing subsequent episodes in the immediate period after giving birth. After treatment, most patients with BT II return to fully functional levels. Approximately 15% of patients do not display functioning due to persistent mood changes, which continues to cause occupation and interpersonal difficulties.
Signs and symptoms
The following signs and symptoms are indicative of bipolar disorder:
1.Presence or history of major depressive episodes:
•Feeling sad or empty
•Decreased interest in pleasure and daily activities
•Weight changes (gain or loss)
•Sleep changes (difficulty falling asleep or waking up)
•Thinking and moving in an agitated or slowed manner
•Feeling loss of energy or fatigued for most of the day
•Feeling worthless or having unnecessary guilt for nearly every day
•Decreased ability to think, concentrate, or indecisiveness nearly every day
•Recurrent thoughts of death or suicide (without a plan or attempts)
2.Presence or history of at least one hypomaniac episode (persistent elevated or irritable mood lasting throughout at least four days). The criteria includes three or more of the following:
•Grandiosity
•Decreased requirement for sleep (patient feels rested after only three hours of sleep)
160 |
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |

•Pressure or overly talkative
•Racing thoughts (flight of ideas)
•Irrelevant distractibility (attention). The patient is easily distracted to something that is unimportant.
•Increase in goal-directed activities
•Excessive involvement with risky pleasurable activities (sexual indiscretions, buying sprees, or foolish monetary investments)
3.There is an uncharacteristic change in functioning
4.Mood and functioning changes are detected by others
5.Lacks severity since impairment is not pronounced
6.There has never been a manic or mixed episode. A mixed episode is characterized by a period of time, usually about one week in which the patient exhibits diagnostic criteria for both major depressive and manic episodes nearly every day. The criteria for manic and hypomanic episodes are identical.
7.The symptoms are severe to cause problems in occupation, social, and relationship functioning.
8.The symptoms are not associated with another medical condition, which can present with criteria similar to a manic episode.
For BT II to be chronic, criteria for the depressive
episode should be met continuously for at least two years. Patients with concurrent catatonic features also exhibit disturbances with movement (immobility, peculiar or excessive motor activity). The features of BT II with melancholia often include near complete absence of the capacity for pleasure. Patients with BT II and atypical features usually present with mood reactivity (mood improves with positive event) and two or more of the following: increased appetite or significant weight gain; difficulty waking up from sleep; heavy, almost paralyzed feeling in the arms or legs; long term sensitivity to interpersonal rejection. BT II with postpartum onset usually occurs within four weeks after childbirth. Manic-depres- sion with a seasonal pattern is also related to seasonal change, age, gender, and latitude. The prevalence of the seasonal specifier increases with higher latitudes, young persons, winter months, and female gender. Rapid cycler’s are those who exhibit the criteria for BT II and have at least four episodes of a mood disturbance in the previous 12 months.
Diagnosis
The diagnosis of BT II is based on the specific criteria described in the Signs and Symptoms section. BT II should be distinguished from Unipolar (major) depression. Patients who exhibit BT II often present with signs
KEY TERMS
Nucleotides—Building blocks of genes, which are arranged in specific order and quantity.
of eating more (hyperphagia), sleeping more (hypersomnia), very low energy levels, overweight, and worsening of mood during evening hours. The BT II affected person also tends to deny or minimize poor judgement and acting differently when compared to others. Close friends, family members, and roommates are often very helpful in assisting the clinician make the correct diagnosis. Unipolar (major) depression usually presents with anxiety, difficulty sleeping, and loss of appetite, loss of weight and feeling worse during morning hours, which improves as the day goes on.
Complications
Suicide is the major complication of BT II. This is related to time. The longer the depression the more serious a threat, especially when there are secondary reinforcements, which promote such aggression. Alcoholics and patients with chronic (long-term) medical diseases are particularly prone to planning and implementing a suicide attempt. There are four major groups that are likely to carry out a suicide attempt. They include:
•Individuals who are overwhelmed by problems in living. They tend to be acts related to aggression and impulsive behaviors, not significant depressive episodes.
•Individuals who are attempting to control others.
•High-risk groups who are chronically ill with another medical disease.
•Patients with other severe types of psychotic illness, delusions, and paranoia.
Treatment and management
Treatment of BT II is focused along three categories: standard medications, psychosocial interventions, and newly discovered medications (gabapentin augmentation).
Standard medications
Standard treatments include medications such as lithium carbonate and sodium valproate. With lithium carbonate, beneficial effects usually appear one to two weeks after administration with oral doses. The response rate with lithium is encouraging since 70-80% of patients with acute manic attacks show improvement of symptoms. Side effects from lithium treatment include gas-
disorder Bipolar
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
161 |
Bipolar disorder
trointestinal discomfort, diarrhea, baldness, skin eruptions, and fluid retention. Lithium is primarily useful as a prophylactic (prevention) medication from future attacks. Another medication, haloperidol can be given initially and gradually reduced for lithium replacement and maintenance.
Valproic acid is a second line medication intended for patients who respond poorly to or cannot tolerate side effects. Valproic acid seems to be more efficient than lithium for treating BT II patients with the rapid cycling variety (more than four episodes a year).
Recent reports indicate a new medication, gabapentin (an anti-manic medication), is efficient for treating acute phase (sudden onset) BT II. This chemical seems to be particularly useful when combined with other psychotropics (medications commonly used to treat mental illnesses). Very recent evidence suggests that gabapentin can potentially induce aggressive and disruptive behavior in children treated with this drug for seizures (abrupt and abnormal jerking of muscles due to abnormal firing of nerve impulses from the brain).
Psychosocial interventions
Psychosocial interventions include both patient education and psychotherapy. It is important for patients to receive social support and illness management skills. Family and friends must be aware of the high rates of social dysfunction and marital discord. Involvement in national support groups is advisable (National Depressive and Manic-Depressive Association).
Psychoeducation usually focuses on:
•Assessment of what parameters will have an impact on the outcome of patient’s disease.
•Implementing the boundaries and requirements of treatment.
•Implementation of a personal cost-benefit analysis concerning specific treatment directions.
•Implementing a follow-up program.
•Implementing future directions, which may include adjustment or change interventions.
Genetic counseling should be a part of family education programs since the predisposition of this disorder has been genetically proven to increase among firstdegree relatives.
Prognosis
Overall the long-term outcome for BT II patients is variable. Patients must maintain strict compliance with medications. Psychotherapy and education can assist the patient and family members with pertinent information concerning relapses, noncompliance with prescription
medications, and specific adjustments necessary for the welfare of the affected individual. Patients taking psychotropic medications must understand the importance of regular dosing as prescribed and the necessity for constant psychiatric follow up visits. In comparison to major depression (Unipolar), BT II depression is usually associated with longer depression, more severe depressive symptoms, more relapses (having active symptoms return after a period of remission) and experience more incapacitation and hospitalization. Some studies have shown that early onset BT II is associated with more recurrences, but not necessarily worse outcomes.
Resources
BOOKS
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, fourth edition. Washington, DC, American Psychiatric Association, 1994.
Maxmen, J. S., and M. G. Ward. Essential Psychopathology and Its Treatment. New York, NY: W. W. Norton & Company: 1995: 206-283.
Muench, K. H. Genetic Medicine. New York, NY: Elsevier Science Publishing Co., Inc., 1988: 48-49.
PERIODICALS
Benazzi, F. “Early-versus late-onset bipolar II disorder.”
Journal of Psychiatry and Neuroscience 25 (2000): 53-56. Callahan, A. M., and M. S. Bauer. “Psychosocial interventions for bipolar disorder.” The Psychiatric Clinics of North
America 22 (1999): 675-686.
Parikh, S. V., J. B. Vincent, and J. L. Kennedy. “Clinical characteristics of bipolar disorder subjects with large CAG/CTG repeat DNA.” Journal of Affective Disorders 55 (1999): 221-224.
Sanchez, L., O. Hagino, E. Weller, and R. Weller. “Bipolarity in children.” The Psychiatric Clinics of North America 22 (1999): 629-639.
Schaffer, C. B., and L. C. Schaffer. “Open maintenance treatment of bipolar disorder spectrum patients who responded to gabapentin augmentation in the acute phase of treatment.” Journal of Affective Disorders 55 (1999): 237-240.
ORGANIZATIONS
National Depressive and Manic-Depressive Association. 730 N. Franklin, Suite 501, Chicago, IL 60610-7204. (800) 8263632 or (312) 642-7243. http://www.ndmda.org .
WEBSITES
American Psychological Association.
http://helping.apa.org/ .
National Mental Health Organization.
http://www.nmha.org .
Laith Farid Gulli, MD
Bloch-Sulzberger syndrome see
Incontinentia pigmenti
162 |
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |

I Bloom syndrome
Definition
Bloom syndrome is a rare inherited disorder characterized primarily by short stature and a predisposition to various types of cancer. It is always associated with a decreased stability in the chromosomes that can be seen by cytogenetic laboratory techniques.
Description
Bloom syndrome (BS) was first described by D. Bloom in 1954. The clinical symptoms of BS include small body size, sun-sensitive skin that is prone to a reddish rash, patchy spots on the skin that are either lighter or darker than the expected skin color, severe immune deficiency, and an enormous predisposition to various types of cancer. The hallmark of the disorder is genetic instability that manifests itself in chromosomes that tend to exchange material with one another.
Genetic profile
BS is inherited in an autosomal recessive manner. The gene responsible for this disorder is known as BLM and it is located on chromosome 15, in band q26.1. Changes or mutations in the BLM gene lead to decreased stability in the chromosomes. Chromosomes of people with BS will show an increased amount of gaps, breaks, and structural rearrangements.
The most characteristic chromosomal abnormality in BS involves the tendency for deoxyribonucleic acid (DNA) strands to exchange material, most likely during replication. DNA is the molecule that encodes the genetic information and determines the structure, function, and behavior of a cell. The exchange of DNA may occur between a chromatid of each of the two homologues of a chromosome pair, forming a unique structure called a quadriradial, or between the two sister chromatids of one chromosome, known as sister-chromatid exchange (SCE).
The BLM gene produces the BLM protein. The BLM protein is a member of the helicase family and is thus capable of unwinding DNA and RNA. This unwinding process provides single stranded templates for replication, repair, recombination, and transcription. Additionally, the BLM protein may function in a postreplication recombination process that resolves errors generated during replication. Mutations (changes) prevent the BLM gene from making BLM protein. Without adequate amounts of this protein, errors are likely to occur in these important processes and these errors are less likely to be repaired.
KEY TERMS
Carcinoma—Any cancer that arises in the epithelium, the tissue that lines the external and internal organs of the body.
Chromatid—Each of the two strands formed by replication of a chromosome. Chromatids are held together by the centromere until the centromere divides and separates the two chromatids into a single chromosome.
Erythema—Redness of the skin due to dilatation of capillaries.
Fecal blood testing—Examination of the stool for any evidence of blood, which may be a sign of cancers in the digestive tract.
Homologues—Chromosomes or chromosome parts identical with respect to their construction and genetic content (i.e. the two chromosome #1s are homologous, as are the two #2s, #3s, etc...).
Leukemia—Cancer of the blood forming organs which results in an overproduction of white blood cells.
Lymphoma—A malignant tumor of the lymph nodes.
Sigmoidoscopy—The visual examination of the inside of the rectum and sigmoid colon, using a lighted, flexible tube connected to an eyepiece or video screen for viewing.
Telangiectatic—A localized collection of distended blood capillary vessels.
As of 2001, it is known that mutations in the BLM gene lead to the symptoms of BS. However, the precise relationship between these mutations and the symptoms seen in BS is still unknown.
Additionally, the DNA of individuals affected with BS is much more prone to spontaneous mutations, perhaps because the inadequate amount of BLM hinders the correction of these errors.
Demographics
BS is a very rare condition, thought to affect a very small proportion of the general population (approximately 1/6,330,000). However, in the Ashkenazi Jewish population, approximately 1/60,000 people are affected with BS. Approximately 1/100 people of this ethnic group are carriers of a mutation in the BLM gene. These carriers do not have BS but are capable of passing it on to
syndrome Bloom
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
163 |
Bloomsyndrome |
Bloom Syndrome |
|
(Gale Group)
their children if the other parent is also a carrier. If both parents are carriers, each pregnancy will have a 25% chance of being affected with the disorder. Carriers, or individuals with only one copy of the abnormal gene, do not appear to have an increased risk for cancer or other symptoms associated with BS. They have near normal or normal genetic stability.
Signs and symptoms
There are two characteristic signs that are seen in nearly all individuals with BS. The first is an overall small body size, which is usually noted at birth and continues throughout the person’s lifetime. The growth deficiency is often accompanied by a small brain and head. The head may be dolichocephalic as well, meaning that is it elongated from the front to the back of the head. The average height for an adult with BS is 147.5 cm for males and 138.6 cm for females.
The second characteristic that is very common in individuals with this disorder is an enormous predisposition to cancer. Both benign (non-cancerous) and malignant (cancerous) tumors arise at an early age and with great frequency in a wide variety of body locations and cell types. Thirty-seven percent of patients have malignant tumors. The mean age at diagnosis of a cancer is 24 years with a range of 2–46 years. Lymphomas and leukemias are common and generally appear before the age of 25. Carcinomas are common as well, usually appearing after the age of 20, most often in the colon, skin, breast, or cervix. Cancer is the most common cause of death for individuals with BS. Radiation treatment or chemotherapy can lead to further complications in these patients due to the increased sensitivity to exposures that may damage their fragile chromosomes.
There are additional features that may or may not be present in individuals with BS and they vary in severity
from person to person. In some cases of BS, the person may have some unique facial features, including a narrow, triangular face shape, a prominent nose, a small jaw, and protuberant ears. The voice may be high pitched and somewhat squeaky in tone.
Infants may experience repeated respiratory tract infections, ear infections, and vomiting and diarrhea that can lead to a life-threatening loss of body water (dehydration). Additionally, after the first significant exposure to sunlight, an infant may develop a reddish “butterfly rash” on the cheeks and nose described as erythematous or telangiectatic. The severity of the rash can vary from a faint blush during the summertime to a severely disfiguring, flaming red lesion. Rarely, other areas of the body that are exposed to sunlight can show a similar rash. In childhood, the skin may begin to appear “patchy” showing some spots with less pigment than the rest of the skin (hypopigmentation) and some with more pigment than the rest of the skin (hyperpigmentation).
Men diagnosed with this disorder may have abnormally small testes and might be unable to produce sperm, making them infertile. Women can have early menopause and often have reduced fertility.
Individuals with BS have a higher incidence of diabetes mellitus when compared to the general population. The average age of onset of diabetes is 25 years, earlier than the usual age of onset of type II diabetes and later than that of type I. Additionally, this disorder can lead to a compromised immune system, resulting in an increased susceptibility to bacterial infections. Infections of the respiratory tract and ears are seen most commonly.
Intelligence in individuals with BS seems to be average to low average. When they exist, limitations in intellectual abilities range from minimal to severe. Even when intelligence is normal in these individuals, there tends to be a poorly defined and unexplained learning disability
164 |
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
that is often accompanied by a short attention span. BS is often accompanied by a persistent optimistic attitude.
Diagnosis
BS can be suspected by the doctor but is generally confirmed by a cytogenetic study known as sister chromatid exchange (SCE) analysis. This disorder is the only one that features an increased risk of SCE. This analysis is indicated in any child or adult with unexplained growth deficiency regardless of whether or not other features of the BS are present.
SCE analysis involves taking a blood sample, treating it with a special process in the laboratory, and examining the chromosomes. In individuals with BS, the chromosomes will show an approximately 10-fold increased rate of sister chromatid exchange. Most likely, unique chromosome structures called quadriradials will also be visible in a higher frequency than expected. SCE and quadiradials are present in untreated cells from individuals without BS, although much less frequently.
In addition to examining the chromosomes, it is also possible to look for specific changes in the BLM gene. This type of evaluation is generally used only for those who may be carriers of the gene mutation rather than those who are suspected to have the disorder. Carriers cannot be identified by SCE analysis because they do not show an increased rate of SCE.
Carrier testing is available for the Ashkenazi Jewish population. In these individuals, there is one particular mutation in the BLM gene that is responsible for most cases of BS. A blood sample can be tested for the presence of this mutation. Almost all Ashkenazi Jewish carriers of the BS gene can be identified in this manner. The great majority of carriers of the mutation causing BS are of Ashkenazi Jewish descent and, thus, this test is designed for that high-risk population. The test is not accurate for people from other ethnic populations in whom the specific changes of the BLM gene are not so well understood.
Prenatal diagnosis is available for carrier couples with previously identified mutations in the BLM gene.
It is thought that BS is highly underdiagnosed. Many affected individuals are treated for a symptom or are mistakenly considered to have another rare disorder.
Treatment and management
There is no treatment for BS—the underlying genetic defect cannot be repaired. However, early diagnosis and management can increase the life span of these individuals.
Babies and young children with BS are often poor eaters. Thus, nutritious food and multivitamins may help improve growth. Treatment with growth hormone has been attempted in several cases but has been generally unsuccessful. Further investigation into this possibility has been limited due to reports that cancer has developed in conjunction with growth hormone treatment.
The reddish skin lesions can be controlled by avoiding the sun, wearing a hat or bonnet, and by using a sunscreen. Avoidance of sun exposure is most critical in the first few years of life, since the severity of the skin lesion appears to be established at that time.
Cancer surveillance is of utmost importance in BS. After the age of 20, annual sigmoidoscopy and fecal blood testing are recommended, as well as breast selfexaminations and pap smears for women. It is suggested that the individual be followed closely by a specialist or clinic knowledgeable about BS so that any subtle symptoms of carcinomas can be treated. Early surgical removal of these tumors provides the best chance of a cure. Individuals may wish to store their bone marrow early in life in case a later treatment diminishes their existing bone marrow. Unfortunately, early diagnosis of leukemia is not known to improve the chances of curative therapy; thus, surveillance of the blood and blood-form- ing tissues in children with BS is not recommended as a part of the cancer surveillance.
Additionally, individuals with this disorder are instructed to avoid x rays, chemotherapeutic drugs and other environmental exposures that may damage their unusually fragile chromosomes. Due to the immunodeficiencies often associated with BS, it is important to treat any bacterial infections promptly.
Prognosis
The mean age at death is 23 years with a range from 1–48 years. Cancer is the most common cause of fatalities in individuals with BS and is thought to be responsible for approximately 80% of deaths. Chronic respiratory infection is the next most common cause of death.
Resources
PERIODICALS
Gennery, A. R., et al. “Immunodeficiency Associated With DNA Repair Defects.” Clinical and Experimental Immunology 121 (2000): 1-7.
German, James. “Bloom’s Syndrome.” Dermatologic Clinics 13 (January 1995): 7-18.
Meyn, M. S. “Chromosome Instability Syndromes: Lessons for Carcinogenesis.” Current Topics in Microbiology and Immunology 221 (1997): 71-148.
Nakura, J., et al. “Helicases and Aging.” Cellular and Molecular Life Sciences 57 (2000): 716-730.
syndrome Bloom
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
165 |

Blue rubber bleb nevus syndrome
Rong, Suo-Bao, Jouni, Valiaho, and Mauno Vihinen. “Structural Basis of Bloom Syndrome (BS) Causing Mutations in the BLM Helicase Domain.” Molecular Medicine 6 (2000): 155-164.
Watt, Paul M., and Ian D. Hickson. “Genome Stability: Failure to Unwind Causes Cancer.” Current Biology 6 (1996): 265-267.
Woods, C. Geoffrey. “DNA Repair Disorders.” Archives of Disease in Childhood 78 (1998): 178-184.
WEBSITES
“Bloom Syndrome.” OMIM—Online Mendelian Inheritance in
Man. National Center for Biotechnology Information.
http://www3.ncbi.nlm.nih.gov/omim/ .
“Bloom Syndrome.” Pediatric Database. PEDBASE.
http://www.icondata.com/health/pedbase/index.htm.
“Bloom Syndrome.” University of Pittsburgh, Department of
Human Genetics. Genetics Education and Counseling
Program. http://www.pitt.edu/~edugene/ .
Mary E. Freivogel, MS
I Blue rubber bleb nevus syndrome
Definition
Blue rubber bleb nevus syndrome (BRBNS) is a rare disorder characterized by hemangiomas of the skin and gastrointestinal (GI) tract. Hemangiomas are benign or noncancerous tumors of newly formed blood vessels and skin. This syndrome derives its name from these distinctive rubber-like skin lesions.
Description
In 1860 G. G. Gascoyen first reported the association of cutaneous or skin nevi and intestinal lesions with GI bleeding. William Bean in 1958 first used the term BRBNS to describe the rubber-like tumors. Because of his description, BRBNS is sometimes called Bean syndrome. Besides the skin and GI tract, nevi are found on all internal organs and even the brain. Nevi are birthmarks of the skin that are probably hereditary because they are not caused by external factors.
Genetic profile
To date, the gene that causes BRBNS has not been identified. The fact that it has not been discovered does not imply the gene does not exist. Some cases of BRBNS are familial and support an autosomal dominant form of inheritance, meaning that only one copy of the non-
working gene is required to manifest the condition. An affected parent has a 50% chance of passing the disorder to his or her offspring. However, most cases are sporadic without a familial tendency.
Demographics
Less than 180 cases have been reported worldwide. BRBNS affects all races, both sexes, and may be present at birth. The effects on life expectancy are unknown because so few cases exist.
Signs and symptoms
The distinctive blue skin blebs are the hallmark of BRBNS and are not cancerous. Blebs are nevi that measure more than 5 mm around. Composed of skin and large dilated blood vessels, the nevi do not disappear and are found on internal organs such as the stomach, liver, spleen, heart, bone, muscle, bladder, and vulva. They are easily compressible and refill after compression. Occasionally, the nevi are painful. Ranging in size from millimeters to several centimeters, the nevi can number from a few to hundreds. As the patient ages, they can increase in size and number. In rare cases, large lesions can cause skeletal deformities that may lead to amputation.
Nevi are usually present at birth. Sometimes, however, they may not appear until ages two or three.
Patients with BRBNS develop an extreme paleness or pallor of the skin. This paleness results because anemia, a low blood count, decreases the amount of oxygen available to the surface skin. Often they complain of fatigue that results from low iron stores and the anemia.
Chronic or acute bleeding in the GI tract may be detected when blood is present in the stool. Chronic bleeding causes anemia, pallor, fatigue, and low iron stores. Iron supplements will help to increase the blood count. Acute bleeding in the GI tract happens quickly and can rapidly decrease a normal blood count. Immediate blood transfusion or surgery to remove the bleeding nevus can correct this condition.
Diagnosis
The first key to diagnosis of this condition is the appearance of the skin nevi. If they do not have the distinct rubbery texture, blue color, and refill after they have been compressed, another diagnosis should be considered. Endoscopy is required to examine the GI tract for nevi. If they are present, then the diagnosis is confirmed. However, lack of nevi in the GI tract does not completely rule out BRBNS, since they may not develop until adolescence.
166 |
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |

During an endoscopy a viewing instrument attached to a flexible tube is passed through the mouth to the small intestine. Or, the tube can be inserted through the rectum to the colon. The doctor can then examine the GI tract for nevi.
A patient will require blood tests to assess anemia and iron deficiency as well as a stool test for the presence of blood. Although nevi may be found on the brain, few patients have neurological signs such as seizures or partial paralysis.
Treatment and management
Treatment of BRBNS will depend upon the severity, number, size, and location of the nevi. Skin lesions that are life-threatening can be safely removed by surgery, or laser therapy. The severity of bleeding from GI lesions will determine how they are treated. Surgery can remove single lesions; however, the number may be too great to excise them all. Treatment methods that are less invasive than surgery use endoscopy to tie off bleeding nevi.
Patients who have neurological signs should have a magnetic resonance image (MRI) of the brain to discover the extent of nevi. Seizures can usually be controlled by medications. Physical therapy may improve paralysis.
Prognosis
Although BRBNS is a chronic, progressive disease it does not appear to be fatal. If the GI bleeding and anemia are treated, the patient will usually cope well. If a patient expresses concerns about his or her physical appearance psychological counseling should be considered.
Resources
BOOKS
Fry, L. An Atlas of Dermatology. New York: Parthenon
Publications, 1997.
Helm, K. Atlas of Differential Diagnosis in Dermatology. New
York: Churchill Livingston, 1997.
PERIODICALS
Ertem, D., et al. “Blue Rubber Bleb Nevus Syndrome.” Pediatrics 107, no. 2 (February 2001): 418-20.
Fernandes, C., et al. “Blue Rubber Bleb Naevus: Case Report and Literature Review.” European Journal of Gastroenterology and Hepatology 11, no. 4 (April 1999): 455-7.
Kim, S. J. “Blue Rubber Bleb Nevus Syndrome With Central Nervous System Involvement.” Pediatric Neurology 22, no. 5 (May 2000): 410-2.
ORGANIZATIONS
Nevus Network, The Congenital Nevus Support Group. PO Box 1981, Woodbridge, VA 22193. (703) 492-0253.http://www.nevus.org .
KEY TERMS
Anemia—A blood condition in which the level of hemoglobin or the number of red blood cells falls below normal values. Common symptoms include paleness, fatigue, and shortness of breath.
Cutaneous—Of, pertaining to, or affecting the skin.
Endoscopy—A slender, tubular optical instrument used as a viewing system for examining an inner part of the body and, with an attached instrument, for biopsy or surgery.
Nevus—Any anomaly of the skin present at birth, including moles and various types of birthmarks.
Nevus Outreach, Inc. 1616 Alpha St., Lansing, MI 48910. (517) 487-2306. http://www.nevus.org .
WEBSITES
“Blue Rubber Bleb Nevus Syndrome.” University of Texas South-
western Medical Center. http://www2.utsouthwestern
.edu/brbns/ .
Fenske, Neil, and Basil Cherpelis. “Blue Rubber Bleb Nevus
Syndrome” In Dermatology/Diseases of the Vessels.
E-Medicine http://emedicine.com/derm/topic56.htm .
Suzanne M. Carter, MS, CGC
Brachmann-de Lange syndrome see
Cornelia de Lange syndrome
I Brachydactyly
Definition
Brachydactyly (BD) refers to shortening of the fingers or toes due to underdevelopment of the bones in the hands or feet.
Description
The word brachydactyly comes from the Greek terms brachy, meaning “short,” and daktylos, meaning “digit.” This term is used to describe the hands and feet of people who have shortened digits (fingers or toes). The digits themselves may be shorter than normal, or they may appear small because of shortening of the other bones in the hands or feet. This shortening occurs when one or more of the hand or foot bones fail to develop or grow normally.
Brachydactyly
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
167 |