

Gale Encyclopedia of Genetic Disorder / Gale Encyclopedia of Genetic Disorders, Two Volume Set - Volume 1 - A-L - I
.pdfBrachydactyly
BD is usually isolated, meaning that it is not associated with any other medical problems. BD may occur along with other physical differences or health problems, often as part of a “syndrome.”
BD occurs in a variety of patterns, depending upon which hand or foot bones are affected and how severely they are shortened. It is important to know some basic information about the bone structure of the hands and feet in order to understand the various patterns of BD. Beyond the wrist and ankle, each hand and foot contains 19 tube-shaped (tubular) bones in a specific arrangement. For purposes of orientation, the fingers and toes are numbered from one (thumb or great toe) to five (little finger or little toe). When a fist is made, the bones in the hand that extend from the wrist to the knuckles are called metacarpals. There are five metacarpals, one for the thumb (first metacarpal) and each finger. Each thumb and finger contains several bones called phalanges. A single one of these bones is called a phalanx. The phalanges are arranged end to end and are separated by joints. The thumb has two phalanges and each finger has three phalanges. The phalanges within a particular finger are named according to their location. The phalanges closest to the metacarpals are called the “proximal” phalanges, those in the middle of the fingers are called the “middle” phalanges, and those at the ends of the fingers are called the “distal” or “terminal” phalanges. The thumbs have only proximal and distal phalanges.
The foot bones are very similar to the hand bones. Like the metacarpals, there are five metatarsal bones that extend from the ankle to each of the toes. The bones in the toes are also called phalanges. There are two phalanges in the great toe and three phalanges in each of the other toes.
BD can involve any of the phalanges, metacarpals, and metatarsals in many different combinations. The shortening of these bones may range from mild to severe. Sometimes certain bones are completely absent. Shortening of the bones may occur in one, several, or all of the digits. For a particular finger or toe, the entire digit may be short or only a particular phalanx may be underdeveloped. When BD involves the distal phalanges, the fingernails or toenails may be small or absent. A digit may also be of normal length but appear short due to shortening of its corresponding metacarpal or metatarsal bone. Reduced length of a metacarpal bone is often easiest to appreciate when the hand is held in a fist.
BD can also occur with other abnormalities of the hands and feet. When a phalanx is abnormally shaped, the finger or toe may be bent to one side (clinodactyly). Sometimes the digits have webbing between them (syndactyly). The phalanges may also be fused together at
their ends (symphalangism). This makes it difficult to bend a digit at the joint where the phalanges are fused.
BD frequently occurs in characteristic patterns that can be inherited through families. These patterns are classified as particular types of BD, depending upon which bones and which digits of the hands and/or feet are shortened. There are several classification systems used to describe these different types of BD. The system that is used most frequently was developed by Dr. Julia Bell in 1951 and is called the “Bell Classification.”
There are five main types of BD in the Bell Classification, which are designated types A through E. Their major features are as follows:
•In type A, the middle phalanges of one, several, or all of the fingers and/or toes are shortened. This form of BD is further divided into types A1, A2, and A3. In type A1, the middle phalanges of all digits and the proximal phalanges of the thumbs and great toes are shortened. People with this form of BD generally have hands and feet that appear small with relatively equal shortening of all digits. In type A2, the middle phalanges of the index finger and second toe are shortened and often abnormally shaped. In type A3, the middle phalanx of the fifth finger is shortened and this finger often bends toward the fourth finger. Several other forms of BD type A have also been described.
•In type B, the distal phalanges and nails of the fingers and/or toes are small or absent. The middle phalanges may also be shortened, and the tips of the thumbs and/or great toes may be broad or have a “duplicated” (double) appearance. In this type of BD, the digits typically look as though their tips have been amputated.
•In type C, the middle phalanges of all of the fingers may be shortened, but the fourth finger is least affected and is often the longest finger. The index and middle fingers may be bent toward the fourth finger. The first metacarpal bone can also be short, making the thumb appear small.
•In type D, the distal phalanges of the thumbs and/or great toes are shortened and broad.
•In type E, the metacarpals and/or metatarsals are shortened. The fourth and fifth metacarpals and metatarsals are most commonly shortened, but any of them may be affected.
Genetic profile
Many different genetic signals are required for normal formation of the hand and foot bones. BD is usually caused by abnormalities in these genetic blueprints. Sometimes BD can be caused by exposure to drugs or medications taken during pregnancy. Problems with
168 |
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |

blood flow to the hands or feet during fetal life may also cause BD.
The types of BD in the Bell Classification are inherited in families from one generation to the next. Their pattern of inheritance is called autosomal dominant. This means that they are caused by abnormalities in only one copy of a gene from a particular gene pair. In fact, one form of BD (type A1) was the first human condition that was recognized to have this type of inheritance pattern. Autosomal dominant forms of BD can be inherited by a child of either sex from a parent of either sex. The gene change causing BD may also occur in a particular person for the very first time within a family. Each child born to a person having autosomal dominant BD has a 50% chance of also having BD. However, the degree of hand or foot abnormalities can be very different between people with the same type of BD, and even among members of the same family.
Until recently, nothing was known about the genes that cause BD. This has changed with the identification of the genes that cause two forms of autosomal dominant BD (types B and C) in the past several years. The gene causing BD type C was the first to be identified in 1997. The name of this gene is the “Cartilage Derived Morphogenetic Protein 1” gene, abbreviated as CDMP1. This gene is located on the long arm of chromosome 20 (at location 20q11.2) and provides an important genetic signal to the developing bones of the limbs. Most people with BD type C have abnormalities in one of their two copies of this gene.
The gene causing BD type B was identified in 2000. This gene is called ROR2 and is located on the long arm of chromosome 9. Like CDMP1, ROR2 also provides an important genetic blueprint for the normal development of bones. BD type B is caused by alterations in one copy of this gene.
One interesting feature of the CDMP1 and ROR2 genes is that they can also cause other medical conditions with bone problems that are much more severe than BD. This happens when both copies of either gene are altered in the same person. The genes for other types of autosomal dominant BD have not yet been discovered.
Demographics
BD occurs in people of many different racial and ethnic backgrounds. It is difficult to determine the overall frequency of BD in the general population because many people who have BD never seek medical attention for their shortened digits. Types A3 and D are the most common forms of BD, but their frequencies vary widely between groups of people from different backgrounds. For example, type A3 has been found in fewer than 1% of
KEY TERMS
Clinodactyly—An abnormal inward curving of the fingers or toes.
Digit—A finger or toe. Plural–digits.
Metacarpal—A hand bone extending from the wrist to a finger or thumb.
Metatarsal—A foot bone extending from the ankle to a toe.
Phalanges—Long bones of the fingers and toes, divided by cartilage around the knuckles.
Symphalangism—Fusion of phalanges at their ends.
Syndactyly—Webbing or fusion between the fingers or toes.
Americans, compared to 21% of Japanese people. Because isolated forms of BD are generally inherited as autosomal dominant traits, they should affect males and females in equal numbers. However, several types of BD may be more common in females.
Signs and symptoms
BD is often evident at birth, but may also develop or become more obvious during childhood. It usually does not cause pain or other physical symptoms. In fact, many people who have BD consider it to be a normal family trait rather than a medical condition. When BD does cause problems, they are usually related to the size, appearance, or function of the hands or feet. The altered appearance of the hands or feet may make persons with BD feel self-conscious. Shortening of the digits may also make it difficult to find comfortable shoes or gloves. In its severe forms, BD may affect a person’s ability to grip objects or participate in certain jobs or leisure activities. Hand function may be especially affected when BD is associated with clinodactyly, syndactyly, or symphalangism. When BD is associated with significant deformities of the feet, walking may be difficult or painful.
In some cases, BD occurs in combination with other physical changes or medical problems. For instance, people with autosomal dominant forms of BD are often shorter than expected and may have other alterations of the skeleton besides short digits. Some people with BD type E also have hypertension (high blood pressure). BD may also be present as one finding in a number of different genetic conditions (syndromes).
Brachydactyly
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
169 |

Branchiootorenal syndrome
Diagnosis
The diagnosis of BD is made when a person has shortening of the digits due to lack of normal growth and development of one or more bones in the hands or feet. When the bones are significantly shortened, this is easily noticed in the appearance of the hands and feet. When the shortening is mild, it may only be apparent on x rays. Some people may not realize that they have BD until told by a physician who has carefully examined their hands and feet.
X rays of the hands and feet are used to look at the bones in detail. A special analysis of the hand x rays called a “metacarpophalangeal profile” is often performed for people with BD. This involves measuring the length of each hand and finger bone. These measurements are then compared to the normal range of sizes for each bone. The metacarpophalangeal profile is used to identify particular patterns of BD. X rays may also reveal other bone changes that help to pinpoint a specific type of BD or another genetic condition. If a person has short stature or other bone changes, a series of x rays of the entire skeleton (skeletal survey) may be recommended.
Since BD is often inherited, detailed information about a person’s relatives can be very important in evaluating someone with BD. A geneticist may wish to examine other family members or obtain x rays of their hands and feet. Because BD can occur in a variety of genetic conditions, a geneticist evaluating someone with BD will usually review his or her medical history and perform a detailed physical examination. The presence of other physical differences or medical problems may indicate that the brachydactyly is part of another condition rather than an isolated finding.
Laboratory tests are usually not helpful in diagnosing BD when it is an isolated finding. Although the genes for BD types B and C are known, testing of these genes is not routinely available or usually necessary. If a person with BD has signs or symptoms of another underlying condition, certain laboratory tests may be recommended. These tests may identify other associated medical problems or help to pinpoint a specific diagnosis.
Treatment and management
Many people who have BD are perfectly healthy and do not require any specific treatment for their hands and feet. When use of the hands is impaired, physical therapy or hand exercises may improve grip strength or flexibility. Evaluation by an orthopedist or physical therapist may also be helpful for people who have trouble walking comfortably due to bone changes in the feet. Surgery can be used to lengthen the hand or foot bones in some severe forms of BD. Surgery may also be helpful for people who have significant clinodactyly, syndactyly, or sympha-
langism. For most people with BD, however, surgery is not needed. If BD is associated with other medical problems, such as hypertension, specific treatments for these problems may be indicated.
Prognosis
Isolated BD generally has an excellent prognosis. When BD is associated with other health problems or is part of another condition, the overall prognosis depends upon the nature of the associated condition.
Resources
BOOKS
Temtamy, Samia A., and Victor A. McKusick. The Genetics of Hand Malformations. New York: Alan R. Liss, 1978.
Winter, Robin M., Richard J. Schroer, and Leslie C. Meyer. “Hands and Feet.” In Human Malformations. Vol. 2, edited by Roger E. Stevenson, Judith G. Hall, and Richard M. Goodman. NewYork: Oxford University Press, 1993, pp. 828–43.
PERIODICALS
Armour, C. M., D. E. Bulman, and A. G. W. Hunter. “Clinical and Radiological Assessment of a Family with Mild Brachydactyly Type A1: The Usefulness of Metacarpophalangeal Profiles.” Journal of Medical Genetics 37 (April 2000): 292–296.
Oldridge, M., et al. “Dominant Mutations in ROR2, Encoding an Orphan Receptor Tyrosine Kinase, Cause Brachydactyly Type B.” Nature Genetics 24 (March 2000): 275–78.
Polinkovsky, A., et al. “Mutations in CDMP1 Cause Autosomal Dominant Brachydactyly Type C.” Nature Genetics 17 (September 1997): 18–19.
WEBSITES
Online Mendelian Inheritance in Man (OMIM).http://www.ncbi.nlm.nih.gov/Omim/ .
David B. Everman, MD
I Branchiootorenal syndrome
Definition
Branchiootorenal (BOR) syndrome is an autosomal dominant condition characterized by ear abnormalities, hearing loss, cysts in the neck, and kidney problems.
Description
The name branciootorenal syndrome describes the body systems most commonly affected by this genetic disorder. The term “branchio” refers to the abnormalities of the neck found in individuals with this syndrome.
170 |
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |

Cysts (lump or swelling that can be filled with fluid) and fistulas (abnormal passage from the throat to the skin) in the neck occur frequently. The term “oto” refers to the ear disorders associated with the syndrome. For example, the outer ear can be unusual in appearance. Hearing loss is also common. Finally, the term “renal” stands for the kidney problems commonly seen in patients with this condition. These can be very mild or very severe, as can any of the symptoms associated with this disorder.
Dr. M. Melnick first described branchiootorenal (BOR) syndrome in 1975. Another name for BOR syndrome is Melnick-Fraser syndrome. Individuals with BOR syndrome typically have physical differences that are present at birth (congenital). These birth defects are caused by a change (mutation) in a gene.
Genetic profile
Scientists recently discovered that mutations in the EYA1 gene cause BOR syndrome. The EYA1 gene is located on chromosome 8. The exact function of the EYA1 gene is unknown, but mutations in this gene disrupt normal development, producing the physical differences common to BOR syndrome. A mutation in this gene can affect the normal development of the ear, kidney, and the branchial arches. The branchial arches are tissues that develop very early in pregnancy and are involved in the formation of the face and neck.
BOR syndrome is inherited in a dominant manner. This means that only one gene in the pair must be mutated in order for the individual to be affected. If a person has a mutation in one of their EYA1 genes, the disorder is typically present. The characteristics of the syndrome can be extremely variable in severity.
A mutation in the EYA1 gene may be inherited from a parent with BOR syndrome. A mutation can also occur by chance, in an individual without a family history of BOR syndrome. If a child inherits an abnormal gene from a parent, the signs of the disorder can be very different between the parent and the child. This is called variable expressivity. For example, a parent who has a very mild form of BOR syndrome can have a severely affected child. The reverse situation can also occur.
Once an individual has a mutation in the EYA1 gene, there is a 50/50 chance with each pregnancy that the gene will be passed on. This means that there is a 50/50 chance of having a child with BOR syndrome. Male and female children have the same risk. It does not matter if the gene is inherited from the mother or the father.
Demographics
BOR syndrome occurs in one of every 40,000 live births. BOR syndrome is seen in all ethnic groups and
KEY TERMS
Autosomal dominant—A pattern of genetic inheritance where only one abnormal gene is needed to display the trait or disease.
Bilateral—Relating to or affecting both sides of the body or both of a pair of organs.
Cleft palate—A congenital malformation in which there is an abnormal opening in the roof of the mouth that allows the nasal passages and the mouth to be improperly connected.
Congenital—Refers to a disorder which is present at birth.
Cyst—An abnormal sac or closed cavity filled with liquid or semisolid matter.
Deoxyribonucleic acid (DNA)—The genetic material in cells that holds the inherited instructions for growth, development, and cellular functioning.
Ear tags—Excess pieces of skin on the outside of the ear.
Fistula—An abnormal passage or communication between two different organs or surfaces.
Gene—A building block of inheritance, which contains the instructions for the production of a particular protein, and is made up of a molecular sequence found on a section of DNA. Each gene is found on a precise location on a chromosome.
Gustatory lacrimation—Abnormal development of the tear ducts causing tears when chewing.
Lacrimal ducts—Tear ducts.
Microtia—Small or underdeveloped ears.
Mutation—A permanent change in the genetic material that may alter a trait or characteristic of an individual, or manifest as disease, and can be transmitted to offspring.
Preauricular pits—Small pits in the skin on the outside of the ear.
Renal agenesis—Absence or failure of one or both kidneys to develop normally.
Renal hypoplasia—Abnormally small kidneys.
Unilateral—Refers to one side of the body or only one organ in a pair.
Variable expressivity—Differences in the symptoms of a disorder between family members with the same genetic disease.
syndrome Branchiootorenal
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
171 |
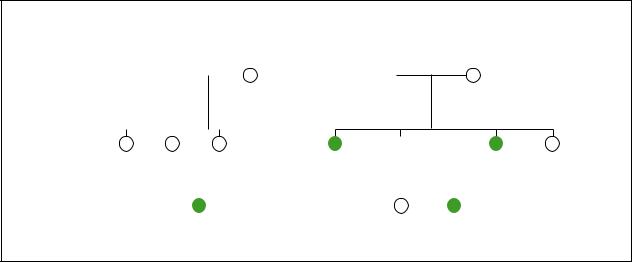
Branchiootorenal syndrome
Branchiootorenal Syndrome
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hearing loss |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
Cleft palate |
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
Bifid uvula |
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hearing loss |
|
|
|
|
Hearing loss |
|||||||||||
|
|
|
|
|
|
|
|
|
|
Polycystic kidneys |
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
Hearing loss |
|
Hearing loss |
Hearing loss |
||||||||||||||||||||
|
|
Branchial cleft cyst |
|
Cleft palate |
Kidney problem |
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|
One kidney missing |
|
|
|
|
|
|
|
|
|
||||||||
(Gale Group)
cultures. It also affects males and females equally. One study suggested that 2% of individuals with severe hearing loss have BOR syndrome.
Signs and symptoms
The characteristics associated with BOR syndrome are highly variable. Some individuals with BOR syndrome have many physical deformations. Other individuals with BOR syndrome have a few minor physical differences. The birth defects can occur on only one side of the face (unilateral) or be present on both sides (bilateral).
Abnormal development of the ears is the most common characteristic of BOR syndrome. The ears may be smaller than normal (microtia) and may have an unusual shape. Ear tags (excess pieces of skin) may be seen on the cheek next to the ear. Preauricular pits (small pits in the skin on the outside of the ear) are found in 75% of patients with BOR syndrome. Hearing loss is present in 85% of individuals with BOR syndrome and this loss may be mild or severe.
The most distinctive finding in individuals with BOR syndrome is the presence of cysts or fistulas in the neck region due to abnormal development of the branchial arches. These cysts and fistulas can be filled with or discharge fluid.
Approximately two-thirds of individuals with BOR syndrome also have kidney abnormalities. These abnormalities can be very mild and cause no health problems, or they can be very severe and life threatening. The kidneys can be smaller than normal (renal hypoplasia),
abnormally shaped, malfunctioning, or totally absent (renal agenesis).
Other less common characteristics associated with BOR syndrome include cleft palate, facial nerve paralysis, and abnormalities of the tear ducts. The tear ducts (lacrimal ducts) may be absent or abnormal. Some patients with BOR syndrome uncontrollably develop tears while chewing (gustatory lacrimation).
Diagnosis
The diagnosis of BOR syndrome is made when an individual has the common characteristics associated with the condition. An individual does not need to have all three components of the disorder in order to be diagnosed with the condition.
There is no readily available genetic test that can diagnose BOR syndrome. Some laboratories are performing DNA testing for mutations in the EYA1 gene, however, this testing is currently being offered on a research basis only. Individuals interested in this type of testing should discuss it with their doctor.
Treatment and management
Once a child is diagnosed with BOR syndrome, additional tests should be performed. A hearing evaluation is necessary to determine if there is hearing loss. If hearing loss is evident, the child should be referred to a hearing specialist. Hearing tests may need to be performed on a regular basis. Speech therapy may also be helpful. An ultrasound of the kidney may be necessary, due to the increased risk for birth defects in these areas.
172 |
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |

Finally, minor surgery may be required to correct the branchial cysts and fistulas commonly found in BOR syndrome.
Prognosis
The prognosis for individuals with BOR syndrome is very good. Individuals with BOR syndrome typically have a normal life span and normal intelligence.
Resources
BOOKS
Jones, Kenneth Lyons. “Melnick-Fraser Syndrome.” In Smith’s Recognizable Patterns of Human Malformation. 5th edition. Philadelphia: W.B. Saunders, 1997.
PERIODICALS
Chen, Achih, et al. “Phenotypic Manifestations of Branchiootorenal Syndrome.” American Journal of Medical Genetics 58 (1995): 365-370.
ORGANIZATIONS
Alliance of Genetic Support Groups. 4301 Connecticut Ave. NW, Suite 404, Washington, DC 20008. (202) 966-5557. Fax: (202) 966-8553. http://www.geneticalliance.org .
National Kidney Foundation. 30 East 33rd St., New York, NY 10016. (800) 622-9010. http://www.kidney.org .
National Organization for Rare Disorders (NORD). PO Box 8923, New Fairfield, CT 06812-8923. (203) 746-6518 or (800) 999-6673. Fax: (203) 746-6481. http://www
.rarediseases.org .
Research Registry for Hereditary Hearing Loss. 555 N. 30th St., Omaha, NE 68131. (800) 320-1171. http://www.boystown
.org/btnrh/deafgene.reg/waardsx.htm
WEBSITES
“Branchio-Oto-Renal (BOR) Syndrome.” Boystown Research
Registry. www.odc.state.or.us/tadoc/hloss3.htm .
“Brachiootorenal Dysplasia.” OMIM—Online Mendelian
Inheritance in Man. www.ncbi.nlm.nih.gov/htbin-post/
Omim/dispmim?113650 .
“Brachiootorenal Syndrome.” GeneClinics. www.geneclinics
.org/profiles/bor/details.html .
Holly Ann Ishmael, MS
I Breast cancer
Definition
Breast cancer is a disease in which abnormal breast cells begin to grow uncontrollably, forming tumors. It often shows up as a breast lump, breast thickening, or skin change.
Description
The breasts are areas of tissue located on the front chest wall, and are essentially part of the skin. They are like “specialized sweat glands” in their structure and function, in that they can produce and secrete fluids, like milk. They are made of ductal tissue, supporting connective tissue, and fat. The breasts naturally drain fluid through the lymph channels to the axillary lymph nodes, located in the armpit areas. Within the breasts are intricate structures of ducts and lobules, which are channels and areas that create and transport milk during lactation.
Excluding skin cancers, breast cancer is the most common cancer among women and the leading cause of death in women in their middle years of life (as of 2000). Male breast cancer, though rare, accounts for less than 1% of all breast cancers. Both genetic and environmental factors are thought to cause breast cancer. Of all breast cancer diagnoses, only approximately 5-10% are caused by hereditary factors like specific alterations in breast cancer susceptibility genes, or by a genetic cancer syndrome. In these instances, individuals may have a strong family history of cancer and the cancers may be diagnosed at an earlier age than usual.
Breast cancers vary in their type and size, and this can be determined by a breast biopsy. Breast cancer may commonly be detected by a mammogram, a physician’s clinical breast examination (CBE), or a patient’s own breast selfexamination (BSE). Breast cancer, if it is the first cancer diagnosed, may sometimes metastasize (spread) to other organs, such as the liver, bone, lungs, skin, or brain. The breasts may also be the site of metastasis from other primary cancers.
Breast cancer may present as a lump or other change within the breast. As with other types of cancer, the initial diagnosis may be unexpected. Each cancer has a unique prognosis, and this will affect the patient’s concern. If an individual has a very strong family history of breast cancer, the diagnosis may be somewhat expected, but no less emotionally taxing. Treatment and management of the cancer may be extremely exhausting, painful, and stressful for the patient and his or her family.
Genetic profile
Cells in breast tissue normally divide and grow, according to controls and instructions of various genes. If these genes have changes within them, the instructions for cellular growth and division may go awry. Abnormal, uncontrolled cell growth may occur, causing breast cancer. Therefore, all breast cancers are genetic because they all result from changes within genes. However, most breast cancers occur later in life after years of exposure to various environmental factors that can cause alter-
cancer Breast
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
173 |

Breast cancer
KEY TERMS
Alteration—Change or mutation in a gene, specifically in the DNA that codes for the gene.
Benign—A non-cancerous tumor that does not spread and is not life-threatening.
Bilateral breast cancer—Cancer of both breasts, caused by two separate cancer processes.
Bile—A substance produced by the liver, and concentrated and stored in the gallbladder. Bile contains a number of different substances, including bile salts, cholesterol, and bilirubin.
Breast biopsy—Small sample of tissue taken from the breast and studied, to diagnose and determine the exact type of breast cancer.
Breast self-exam (BSE)—Examination by an individual of their own breasts.
CA-125 (Carbohydrate antigen 125)—A protein that is sometimes high when ovarian cancer is present. A blood sample can determine the level of CA-125 present.
Clinical breast exam (CBE)—Examination of the breasts, performed by a physician or nurse.
Malignant—A tumor growth that spreads to another part of the body, usually cancerous.
Mammogram—A procedure in which both breasts are compressed/flattened and exposed to low doses of x rays, in an attempt to visualize the inner breast tissue.
Metastasis—The spreading of cancer from the original site to other locations in the body.
Multifocal breast cancer—Multiple primary cancers in the same breast.
Primary cancer—The first or original cancer site, before any metastasis.
Tumor—An abnormal growth of cells. Tumors may be benign (noncancerous) or malignant (cancerous).
ations (such as the body’s own hormones, asbestos exposure, or smoking).
A small proportion of breast cancers is caused by inherited genetic alterations. In 1994 a breast cancer susceptibility gene, known as BRCA1 (location 17q21), was identified. The discovery of BRCA2 (location 13q12) followed shortly in 1995. Women with alterations in these genes have an increased risk for breast and ovarian cancer, and men have an increased risk for prostate cancer. Men with a BRCA2 alteration have an increased risk for breast cancer. Slightly increased risks for colon
and pancreatic cancers (in men and women) are associated with BRCA2 alterations.
BRCA1 and BRCA2 alterations are inherited in an autosomal dominant manner; an individual has one copy of a BRCA alteration and has a 50% chance of passing it on to each of his or her children, regardless of that child’s gender. Nearly all individuals with BRCA alterations have a family history of the alteration, usually a parent. In turn, they also may have a very strong family history of breast, ovarian, prostate, colon, and/or pancreatic cancers. Aside from BRCA1 and BRCA2, there likely are other breast cancer susceptibility genes that are still unknown (such as BRCA3). Additionally, there may be other genes that convey increased risks solely for other cancers, such as ovarian cancer.
BRCA1 and BRCA2 are thought to function as “tumor-suppressor genes,” meaning that their normal role is to prevent tumors from forming. Specifically, they control cellular growth and division, all the while preventing the over-growth that may lead to cancer. Alterations in tumor-suppressor genes, such as BRCA1 and BRCA2, would naturally lead to an increased risk of developing cancer. However, this risk is not 100%.
There are rare, genetic cancer syndromes that may include breast cancer. As a group, these comprise less than 1% of all breast cancer diagnoses. In these instances, an individual may have other health problems (unrelated to cancer) and a family history of a wide variety of cancers and symptoms. These health problems can initially appear unrelated, but may be caused by alterations in a specific gene. As an example, Cowden syndrome typically involves early-onset thyroid and breast cancers, as well as specific tissue growths on the face, limbs, and mouth. An individual with Cowden syndrome may have all or some of these symptoms. It is now known that alterations in the PTEN gene cause Cowden syndrome. Other known cancer syndromes are caused by specific alterations in different genes. These genes are responsible for the various symptoms and cancers in an individual.
Demographics
On average, a North American woman faces a lifetime risk of approximately one in nine (11%) to develop breast cancer. Most cases of breast cancer occur in women past the age of 50, and more commonly in individuals of North American descent.
As of 2000, the prevalence of BRCA alterations in the general population is estimated to be between 1/500 and 1/1,000. However, there are specific alterations that are commonly found in certain ethnic groups. In the Ashkenazi (Eastern European) Jewish population, two specific BRCA1 alterations and one BRCA2 alteration
174 |
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
are commonly seen and range in prevalence from 0.1% to 1.0% in this group. As a result, hereditary forms of breast and ovarian cancer are more predominant in people of Ashkenazi Jewish ethnicity. A common BRCA1 alteration has been found in the Dutch population; a specific BRCA2 alteration exists in about 0.6% of people from Iceland. Additionally, common alterations have been identified in both BRCA1 and BRCA2 in French Canadians, and a BRCA1 alteration has often been seen in West Africans.
Signs and symptoms
Various symptoms may bring someone to medical attention in order to investigate the possibility of breast cancer. These may include a breast lump that persists, as opposed to one that only appears at certain times of a woman’s menstrual cycle (which is more common). Other signs include changes from the normal breast shape, pain, itchiness, fluid leaking from the nipple (especially if a woman is not pregnant), a turned-in nipple, fatigue, or unexplained weight loss. Sometimes individuals may feel a breast lump or change while examining their own breasts, or a physician may note it on a CBE. Additionally, it may be seen on a screening mammogram. It is important to note that not all breast lumps or breast changes signify cancer—they may be benign growths or cysts that need to be removed or drained.
Signs of a possible BRCA1 or BRCA2 alteration in a family, signifying hereditary breast or ovarian cancer, include:
•several relatives with cancer
•close genetic relationships between people with cancer, such as parent-child, sibling-sibling
•earlier ages of cancer onset, such as before ages 45-50
•an individual with both breast and ovarian cancer
•an individual with bilateral or multi-focal breast cancer
•the presence of ovarian, prostate, colon, or pancreatic cancers in the same family
•case(s) of breast cancer in men
Suspicion of a BRCA alteration may be raised if someone has the above features in their family and they are of a particular ethnic group, such as an Ashkenazi Jew. This is because specific BRCA1 and BRCA2 alterations are known to be more common in this group of individuals.
Diagnosis
Once a suspicious breast abnormality has been found, the next step is determining if it is breast cancer. A mammogram can identify an area of increased breast
density, which is a common sign of a malignant tumor. Women in their 20s to 30s naturally have denser breasts, so mammograms may not be as effective in this age group because the increased breast density associated with a tumor is difficult to see. Breast ultrasound, a way of visualizing the breast tissue using sound waves, can be helpful in younger women because breast density is not a large factor in its effectiveness. A breast biopsy can determine specifically whether the breast tissue has undergone a benign or malignant change because the breast tissue is studied directly under a microscope. Sometimes biopsies are performed with a very thin needle (known as fine needle aspiration), or with x ray guidance using a thicker needle (known as a core needle biopsy).
Newer techniques have improved breast cancer screening and diagnosis. Direct digital imaging in mammograms ends the need for film, and the digital images provide finer detail and allow the images to be rotated in order to get several different views of the breasts. Magnetic resonance imaging (MRI) uses magnetic energy to create an image. Its effectiveness is currently the subject of research studies, but MRI often provides very detailed imaging of tumors. MRI is expensive and this is another reason it is not widely used.
As of 2001, there is DNA-based genetic testing to identify a BRCA1 or BRCA2 alteration in an individual. In the United States, Myriad Laboratories in Utah is the only place to offer this costly testing (as of 2001, it is about $2,700 for initial analysis). A blood sample is used and both BRCA genes are studied for alterations. There is also targeted testing for people in high-risk ethnic groups (such as the Ashkenazi Jews) in which only the common BRCA alterations can be tested; this testing is much less costly. Even with current technology (as of 2001), only certain regions of the BRCA genes can be studied, which leaves some alterations unlocated.
With either method of testing, it is best to begin the testing process with an individual who has survived breast and/or ovarian cancer. This is because tests are more likely to find an alteration in a cancer survivor than someone who has not had cancer. A result is abnormal (or “positive”) if a known cancer-causing BRCA alteration is found. If an alteration is found, it is assumed to have caused the cancer(s) in the tested, affected individual. That individual may also identify new cancer risks from the positive result. For example, if a woman survived breast cancer and was found to have a BRCA alteration through testing, she would now be at an increased risk to develop ovarian cancer, as well as a second breast cancer.
For people who go through testing and are not found to have a BRCA alteration (a “negative” result), this result is not informative. There are several possibilities
cancer Breast
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
175 |
Breast cancer
for a negative result. First, there could be a BRCA alteration in the family and the person did not inherit it. In this case, the cancer would be due to reasons unrelated to BRCA1 and BRCA2. Additionally, they could have an alteration in an unknown gene (such as BRCA3), for which there is no testing available (as of 2001). Lastly, they could have a BRCA1 or BRCA2 alteration that is undetectable by available testing methods.
There is a possibility that individuals may have an “unknown alteration” in one of their BRCA genes. In this scenario, a change in the DNA is identified, but its significance is unclear. Therefore, it is unknown whether the gene change causes cancer. In these situations, the results are most often considered uninformative, until more information about the alteration becomes available in the future.
Once an alteration is identified, other at-risk relatives, both affected and unaffected, can pursue targeted analysis for the confirmed familial alteration. This is much quicker and far less expensive than the initial analysis.
Unaffected individuals who test positive for a known alteration in the family are at a significantly increased risk to develop the associated cancers. A woman’s risks associated with a BRCA1 alteration are: 3-85% for breast cancer by age 70, 40–60% for ovarian cancer by age 70. A man’s risk with a BRCA1 alteration is about 8% for prostate cancer by age 70. A woman’s risks with a BRCA2 alteration are: 4–86% for breast cancer by age 70, and 16–27% for ovarian cancer by age 70. Less than 1% of men with a BRCA2 alteration develop breast cancer but they are at a slight or moderate increased risk for prostate cancer. For BRCA2 in men and women, there is an increased risk for colon and pancreatic cancers. Cancers of the larynx (structure in neck that helps with breathing), esophagus (tube-like structure that connects mouth to stomach), stomach, gallbladder (structure that makes bile), bile duct (tube that transports bile between liver and intestine), blood, and melanoma (a form of skin cancer) have been seen in families with BRCA2 alterations.
When a person who has not had cancer tests negative for a known, familial BRCA alteration, they are lowered to the general risk to develop the associated cancers, such as the lifetime risk of 11% for a woman to develop breast cancer. This is because he or she did not inherit the genetic alteration causing cancer in his or her family.
Everyone should receive proper genetic counseling before pursuing any BRCA1 and BRCA2 testing. This should include asking them what they hope to learn from the testing. Many people are not aware of the testing limitations, and may be expecting a clear “yes/no” answer from the results. Asking people what they hope to learn
from testing allows the opportunity to provide them with accurate facts, such as the possibility of a result that is not informative. Common motivations to be tested include the need to make informed medical decisions, financially planning for the future, or just “wanting to know” about cancer risk.
Genetic testing for cancer susceptibility often triggers strong emotional responses. It is important to find out about an individual’s “support system” before they begin testing. Having a close friend, family member, or religious leader to talk with is often helpful for people pursuing testing. Someone who tests positive may be concerned because his or her risks for cancer are now higher than they were before the testing. Additionally, someone may feel “empowered” by the knowledge because they can better plan for medical procedures. Someone with a family history of a BRCA alteration may feel relief if they test negative, because they initially assumed they would develop cancer. Alternatively, someone who tests negative in this situation may feel “survivor guilt” for not having inherited the altered gene. All of these feelings may change the way an individual interacts with his or her family and friends. People may not be aware of the emotional changes that can occur from learning about cancer risk through genetic testing.
It is important to discuss the possibility of insurance coverage for the testing, particularly because it is so expensive. Insurance companies may not routinely cover the testing, unless a physician or genetic counselor describes the need for testing in a letter. Some companies are willing to cover the testing, without wanting to know the results.
Issues of potential “genetic discrimination” should be discussed. Unaffected individuals who test positive for a BRCA1 or BRCA2 mutation may face difficulty when trying to obtain health, life, and/or disability insurance. Fortunately, there are laws in place that can help protect American individuals who have group health insurance, but the exact laws vary by state. As of 2001, there are no laws to protect individuals from life and disability insurance discrimination, nor employer discrimination.
Treatment and management
Breast cancer treatment is determined by the exact size and type of cancer, so it is often unique to an individual. Treatment may include surgeries, such as a lumpectomy (removal of the breast lump) or mastectomy (removal of the entire breast). Breast reconstruction (recreation of the breast) by plastic surgery is an option some individuals may pursue.
Chemotherapy, or using strong chemicals to kill fastgrowing cells, is a common treatment. Side effects from
176 |
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
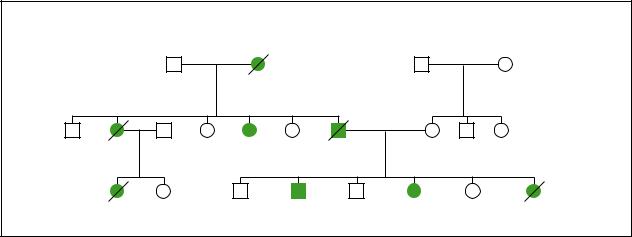
|
Hereditary Breast Cancer |
|
cancerBreast |
|
|
Breast cancer |
|
||
|
|
|
||
|
dx.52y |
|
|
|
Ovarian |
Breast cancer |
Prostate cancer |
|
|
cancer |
dx.42y |
dx.50y |
|
|
dx.34y |
|
|
|
|
Breast cancer |
Prostate cancer |
Ovarian cancer |
Breast cancer |
|
dx.30y |
|
dx.44y |
dx.40y |
dx.35,38y |
(Gale Group)
chemotherapy may include nausea, vomiting, hair loss, exhaustion, and sores in the mouth. Symptoms associated with menopause (such as “hot flashes” and the absence of menstrual periods) may occur, or menopause may actually begin because of chemotherapy. Radiation therapy is another common form of treatment, in which directed radioactive waves are used to kill fast-growing cells. Some side effects of radiation therapy are dry and itchy skin, rashes, exhaustion, nausea, and vomiting.
Sometimes, medications such as Tamoxifen are used to prevent a breast cancer from coming back. Tamoxifen is often used for five years following a breast cancer diagnosis to actively prevent a recurrence. Tamoxifen is only effective in specific types of breast cancer, which again are unique to each individual. Some side effects of Tamoxifen include beginning menopause, as well as an increased risk for uterine cancer. Other drugs, such as Raloxifene, are currently being studied for breast cancer prevention because it may be able to do the same things as Tamoxifen, without the side effects. Research studies are under way to determine whether Tamoxifen or Raloxifene can reduce the risk of breast cancer in women with BRCA alterations.
An example of a screening program for women at high risk to develop breast cancer includes:
•BSEs monthly starting in early adulthood (about 20–25 years of age)
•CBEs every six months or yearly starting at age 25–35
•mammograms yearly starting at age 25–35
Exact screening guidelines may vary between physicians. For men with a BRCA2 alteration, breast cancer screening is recommended, though no formal program is specifically recommended (as of 1997).
In addition to screening, women with BRCA1 or BRCA2 alterations should know about their preventive surgery options. They may consider having their healthy breasts and/or ovaries removed, in order to reduce their risks of developing breast and/or ovarian cancer. Women may be more agreeable to an oophorectomy because ovarian cancer is difficult to detect. Surgeries may greatly reduce a woman’s cancer risk, but they can never eliminate the risk entirely.
For people with cancer or at high risk, there are support and discussion groups available. These may be invaluable to those who feel alone in their situation.
Prognosis
The type and size of breast cancer developed largely determines the overall prognosis for an individual. Those with larger tumors and those with a type of breast tumor that does not usually respond to treatment may have a poorer outcome. Additionally, once cancer has spread to other areas of the body, the prognosis worsens because the cancer is more difficult to treat. The cancer may also be more likely to continue spreading to other areas of the body.
As of 2001, those with BRCA alterations who develop breast cancer have a similar prognosis to those without BRCA alterations that have equivalent cancers. In addition, people with BRCA alterations are treated for their cancers using the same methods as those without alterations.
For cancer-free individuals identified to have BRCA alterations, it is important to remember that they are at an increased risk to develop the associated cancers, but that the risk is not 100%. Though people with BRCA alterations may feel “destined” to develop cancer, it is by no
GALE ENCYCLOPEDIA OF GENETIC DISORDERS |
177 |