

Gale Encyclopedia of Genetic Disorder / Gale Encyclopedia of Genetic Disorders, Two Volume Set - Volume 1 - A-L - I
.pdfFemale carriers usually have no symptoms and have normal visual fields, normal electroretinograms (a measurement of electrical activity of the retina), and normal visual acuity. However, female carriers sometimes show abnormalities of the interior lining of the eye in the form of pigment spotting with tiny patches of RPE depigmentation. Brownish granular pigmentation and changes in the RPE and choroid may occur later. There is also some evidence to suggest that mild progression of symptoms— and even the full disease—may occur in a small number of female carriers.
Diagnosis
Although there is no treatment for choroideremia because the disorder is so rare and has received relatively little research attention, a diagnostic blood test developed by Canadian researchers allows early diagnosis of the disorder. Patients with the abnormal choroideremia gene lack a protein called Rab Escort Protein-1 (REP-1), which is involved in the lipid (any one of a group of fats or fat-like substances) modification of protein—a process called prenylation. The test uses a monoclonal antibody (an antibody of exceptional purity and specificity, derived from a single cell) to determine the presence or absence of the REP-1 protein in blood samples. The REP-1 test is unable to determine carrier status, however; the REP-1 protein is present in female carriers.
Because no biochemical abnormality has been found in choroideremia, no single laboratory test is available for diagnosis. Rather, the diagnosis is based on the typical retinal abnormalities, abnormal electroretinogram findings, the progressive course of the disorder, and the combination of typical symptoms. Family history is also helpful in diagnosing the disorder. When the diagnosis is in doubt, examination of the mother usually reveals the pigmentary changes and other retinal abnormalities typically found in carriers.
Choroideremia is one of the few retinal degenerative disorders that may be detected before birth in some cases (in women who have been found to be carriers due to family history or abnormal ophthalmologic findings). All family members with a history of choroideremia are encouraged to consult an ophthalmologist and to seek genetic counseling. These professionals can explain the disease and the inheritance risk for all family members and for future offspring.
Treatment and management
There is no treatment for choroideremia because further research is needed to understand the exact mechanism causing this progressive loss of vision. It is not known whether any external environmental factors, such
K E Y T E R M S
Choriocapillaris—Capillary layer of the choroid.
Choroid—A vascular membrane that covers the back of the eye between the retina and the sclera and serves to nourish the retina and absorb scattered light.
Electroretinogram (ERG)—A measurement of electrical activity of the retina.
Retina—The light-sensitive layer of tissue in the back of the eye that receives and transmits visual signals to the brain through the optic nerve.
Retinal pigment epithelium (RPE)—The pigmented cell layer that nourishes the retinal cells; located just outside the retina and attached to the choroid.
Retinitis pigmentosa—Progressive deterioration of the retina, often leading to vision loss and blindness.
as light, contribute to the progression of the disease, or if genetic factors alone are responsible for the great variability observed. However, patients diagnosed with the disorder early are better able to make decisions regarding family planning and the onset of blindness.
Assistance for individuals with choroideremia is available through low-vision aids, including optical, electronic, and computer-based devices. Personal, educational, and vocational counseling, as well as adaptive training skills are also available through community resources.
Prognosis
Progression of the disease continues throughout the individual’s life, although both the rate and degree of visual loss are variable among those affected, even within the same family.
Resources
BOOKS
Cremers, F.P.M., and F.F. Ropers. “Choroideremia.” In The Metabolic and Molecular Basis of Disease. Ed. C.R. Scriver, A.L. Beaudet, W.S. Sly, and D. Valled, 4311–23, vol. 3. New York: McGraw Hill, 1995.
PERIODICALS
MacDonald, I. M., et al. “A Practical Diagnostic Test for Choroideremia.” Opthalmology 105 (1998): 1637–40.
Majid, M. A., et al. “Unusual Macular Findings in a Known Choroideremia Carrier.” Eye 12 (1998): 740–41.
Choroideremia
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
231 |

Chromosomal abnormalities
Syed N., et al. “Evaluation of Retinal Photoreceptors and pigment epithelium in a female carrier of choroideremia.” Opthalmology 108, no. 4 (April 2001): 711–20.
ORGANIZATIONS
American Foundation for the Blind. 11 Penn Plaza, Suite 300, New York, NY 10001. (800) 232-5463.
Choroideremia Research Foundation. 23 E. Brundreth St., Springfield, MA 01109. http://www.choroideremia
.org .
National Association for Parents of the Visually Impaired. PO Box 317, Watertown, MA 02472. (617) 972-7441 or (800) 562-6265. http://www.spedex.com/napvi .
National Eye Institute. 31 Center Dr., Bldg. 31, Room6A32, MSC 2510, Bethesda, MD 20892-2510. http://www.nei
.nih.gov .
National Federation for the Blind. 1800 Johnson St., Baltimore, MD 21230. (410) 659-9314. epc@roundley.com.http://www.nfb.org .
National Organization for Rare Disorders (NORD). PO Box 8923, New Fairfield, CT 06812-8923. (203) 746-6518 or (800) 999-6673. Fax: (203) 746-6481. http://www
.rarediseases.org .
WEBSITES
The Choroideremia Group.http://www.onelist.com/subscribe.cgi.choroideremia .
Genevieve T. Slomski, PhD
I Chromosomal abnormalities
Chromosomal abnormalities describe changes in the normal number of chromosomes or structural problems within the chromosomes themselves. These abnormalities occur when an egg or sperm with an incorrect number of chromosomes, or a structurally faulty chromosome, unites with a normal egg or sperm during conception. Some chromosome abnormalities occur shortly after conception. In this case, the zygote, the cell formed during conception that eventually develops into an embryo, divides incorrectly.
Chromosomal abnormalities can cause serious mental or physical disabilities. Down syndrome, for instance, is caused by an extra chromosome 21. People with Down syndrome are mentally retarded and may have a host of physical abnormalities, including heart disorders. Other individuals, called Down syndrome mosaics, have a mixture of normal cells and cells with three copies of chromosome 21, resulting in a milder form of the disorder. Most abnormalities in chromosome number lead to the death of the embryo. Zygotes that receive a full extra set of chromosomes, a condition
called polyploidy, usually do not survive inside the uterus and are spontaneously aborted (a process sometimes called a miscarriage).
Normal number and structure of human chromosomes
A chromosome consists of the body’s genetic material, the deoxyribonucleic acid, or DNA, along with many kinds of proteins. Within the chromosomes, the DNA is tightly coiled around these proteins (called histones) allowing approximately 6 ft (2 m) strands of DNA to occupy a microscopic space within the nucleus of the cell. When a cell is not dividing, the chromosomes are invisible within the cell’s nucleus. Just prior to cell division, the chromosomes begin to replicate and condense. As the replicated DNA condenses, each chromosome looks somewhat like a fuzzy “X” under the microscope. Chromosomes contain the genes, or segments of DNA that code for proteins, of an individual. When a chromosome is structurally faulty, or if a cell contains an abnormal number of chromosomes, the types and amounts of the proteins encoded by the genes is changed. When proteins are altered in the human body, the result can be serious mental and physical changes and disease.
Humans have 46 chromosomes—22 pairs of autosomal chromosomes and one pair of sex chromosomes. These chromosomes may be examined by constructing a karyotype, or organized depiction, of the chromosomes. To construct a karyotype, a technician stops cell division just after the chromosomes have replicated and condensed using a chemical, such as colchicine. The chromosomes are visible within the nucleus at this point. The image of the chromosomes seen through the microscope is photographed. Each chromosome is cut out of the picture, and arranged on another sheet in the correct sequence and orientation. The chromosome pairs are identified according to size, shape, and characteristic stripe patterns (called banding).
Normal cell division
In most animals, two types of cell division take place: mitosis and meiosis. In mitosis, each cell division produces two cells that are identical to the parent cell, i.e. one parent cell produces two daughter cells. Compared to its parent chromosome, each daughter cell has exactly the same number of chromosomes and identical genes. This preservation of chromosome number and structure is accomplished through the replication of the entire set of chromosomes just before mitosis.
Sex cells, such as eggs and sperm, undergo a different type of cell division called meiosis. Because sex cells
232 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

each contribute half of a zygote’s genetic material, sex cells must carry only half the full number of chromosomes. This reduction in the number of chromosomes within sex cells is accomplished during two rounds of cell division, called meiosis I and meiosis II. Before meiosis I, the chromosomes replicate. During meiosis I, a cell with 46 replicated chromosomes divides to form two cells that each contain 23 replicated chromosomes. Normally, the meiosis I division separates the 23 pairs of chromosomes evenly, so that each daughter cell contains one chromosome from each chromosome pair. No replication occurs between meiosis I and meiosis II. During meiosis II, the two daughter cells containing 23 replicated chromosomes divide to form four daughter cells, each containing 23 non-replicated chromosomes. Mistakes can occur during either meiosis I or meiosis II. Chromosome pairs may fail to separate during meiosis I, or a replicated chromosome may fail to separate during meiosis II.
Meiosis produces four daughter cells, each with half the normal number of chromosomes. These sex cells are called haploid cells (haploid means “half the number”). Non-sex cells in humans are called diploid (meaning “double the number”) since they contain the full number of normal chromosomes. Human diploid cells normally each have 46 chromosomes, and haploid cells normally each have 23 chromosomes.
Alterations in chromosome number
Two kinds of chromosome number alterations can occur in humans: aneuploidy, an abnormal number of chromosomes, and polyploidy, more than two complete sets of chromosomes.
Aneuploidy
Most alterations in chromosome number occur during meiosis. During normal meiosis, chromosomes are distributed evenly among the four daughter cells. Sometimes, however, an uneven number of chromosomes are distributed to the daughter cells. As noted in the previous section, chromosome pairs may not move apart in meiosis I, or the chromosomes may not separate in meiosis II. The result of both kinds of mistakes (called nondisjunction of the chromosomes) is that one daughter cell receives an extra chromosome, and another daughter cell does not receive any chromosome.
When an egg or sperm that has undergone faulty meiosis and has an abnormal number of chromosomes unites with a normal egg or sperm during conception, the zygote formed will have an abnormal number of chromosomes. This condition is called aneuploidy. There are several types of aneuploidy. If the zygote has an extra chromosome, the condition is called trisomy. If the
|
Nondisjunction |
|
Chromosomal |
|
|
in meiosis I |
|
Haploid # of this |
|
|
|
|
||
|
|
|
cell is 2. |
|
|
|
|
|
|
Normal |
|
|
abnormalities |
|
meiosis II |
|
|
||
|
|
|
|
|
|
|
Gametes |
|
|
(a) |
Extra |
|
Missing |
|
chromosome |
chromosome |
|
||
Normal |
|
|
|
|
meiosis I |
|
|
|
|
|
Nondisjunction |
|
|
|
|
in meiosis II |
|
|
|
|
|
Gametes |
|
|
(b) |
Extra |
Missing |
Normal number |
|
chromosome |
chromosome |
of chromosomes |
|
|
Figure 1. (Gale Group)
zygote is missing a chromosome, the condition is called monosomy.
If the zygote survives and develops into a fetus, the chromosomal abnormality is transmitted to all of its cells. The child that is born will have symptoms related to the presence of an extra chromosome or absence of a chromosome.
Examples of aneuploidy include trisomy 21, also known as Down syndrome, and trisomy 13, also called
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
233 |

Chromosomal abnormalities
K E Y T E R M S
Amniocentesis—A procedure performed at 16-18 weeks of pregnancy in which a needle is inserted through a woman’s abdomen into her uterus to draw out a small sample of the amniotic fluid from around the baby. Either the fluid itself or cells from the fluid can be used for a variety of tests to obtain information about genetic disorders and other medical conditions in the fetus.
Aneuploidy—An abnormal number of chromosomes in a cell. Trisomy 18 and trisomy 13 are examples of aneuploid conditions.
Angelman syndrome—A syndrome caused by a deletion in the maternally inherited chromosome 15 or uniparental disomy of the paternal chromsome 15.
Chorionic villus sampling (CVS)—A procedure used for prenatal diagnosis at 10-12 weeks gestation. Under ultrasound guidance a needle is inserted either through the mother’s vagina or abdominal wall and a sample of cells is collected from around the fetus. These cells are then tested for chromosome abnormalities or other genetic diseases.
Chromosome—A microscopic thread-like structure found within each cell of the body and consists of a complex of proteins and DNA. Humans have 46 chromosomes arranged into 23 pairs. Changes in either the total number of chromosomes or their shape and size (structure) may lead to physical or mental abnormalities.
Cri du chat syndrome—A syndrome caused by a deletion in chromosome 5; characterized by a strange cry that sounds like the mewing of a cat.
Deletion—The absence of genetic material that is normally found in a chromosome. Often, the genetic material is missing due to an error in replication of an egg or sperm cell.
Deoxyribonucleic acid (DNA)—The genetic material in cells that holds the inherited instructions for growth, development, and cellular functioning.
Diploid—Means “double number.” The normal number of chromosomes (two) for all cells of the human body, except for the sex cells.
Down syndrome—A genetic condition characterized by moderate to severe mental retardation, a characteristic facial appearance, and, in some individuals, abnormalities of some internal organs. Down syndrome is always caused by an extra copy of chromosome 21, or three rather than the normal two. For this reason, Down syndrome is also known as trisomy 21.
Duplication—A chromosomal abnormality in which a broken segment of a chromosome attaches to the chromosome pair resulting in extra chromosomal material.
Edwards syndrome—A syndrome caused by trisomy 18; characterized by multi-system disorders; and usually lethal by age 1.
(continued)
Patau syndrome. Trisomy 13 occurs in one out of every 5,000 births, and its symptoms are more severe than those of Down syndrome. Children with trisomy 13 often have cleft palate and eye defects, and always have severe physical and brain malformations. Trisomy 18, known as Edwards syndrome, results in severe mutliple defects. Children with trisomy 13 and trisomy 18 usually survive less than a year after birth (Figure 1).
Aneuploidy of sex chromosomes
Sometimes, nondisjunction occurs in the sex chromosomes. Humans have one set of sex chromosomes. These sex chromosomes are called “X” and “Y” after their approximate shapes in a karyotype. Males have both an X and a Y chromosome, while females have two X chromosomes. Disorders associated with abnormal numbers of sex chromosomes are less severe than those asso-
ciated with abnormal numbers of autosomes. This is thought to be because the Y chromosome carries few genes, and extra X chromosomes are inactivated shortly after conception. Nevertheless, aneuploidy in sex chromosomes causes changes in physical appearance and in fertility (Figure 2).
Individuals with Klinefelter syndrome, for instance, are men with two X chromosomes (XXY). This condition occurs in one out of every 600 male births. Men with Klinefelter syndrome have small testes and are usually sterile. Some men with Klinefelter develop enlarged breasts. Males who are XXY are of normal intelligence. However, mental retardation is not unusual in males with more than two X chromosomes, such as XXXY, XXXXY, or XXXXXY.
Males with an extra Y chromosome (XYY) have no physical defects, although they may be taller than aver-
234 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

KEY TERMS ( C O N T I N U E D )
Fragile X syndrome—A condition caused by an abormality of a region on the X chromosome which may be expressed in males or females, and may increase in severity when inherited from the mother.
Gene—A building block of inheritance, which contains the instructions for the production of a particular protein, and is made up of a molecular sequence found on a section of DNA. Each gene is found on a precise location on a chromosome.
Haploid—Means “half the number;” the number of chromosomes in a sex cell.
Inversion—A type of chromosomal defect in which a broken segment of a chromosome attaches to the same chromosome, but in reverse position.
Klinefelter syndrome—A syndrome that occurs in XXY males; characterized by sterility and small testes; normal intelligence.
Meiosis—The process in which a cell in the testes or ovaries undergoes chromosome separation and cell division to produce sperms or eggs.
Metafemale—An out of date term for XXX females, also called triple X syndrome.
Mitosis—The process by which a somatic cell—a cell not destined to become a sperm or egg—dupli- cates its chromosomes and divides to produce two new cells.
Monosomy—Missing an entire copy of a chromosome or a piece of one copy of a chromosome.
Nucleus—The central part of a cell that contains most of its genetic material, including chromosomes and DNA.
Patau syndrome—A syndrome caused by trisomy 13; characterized by cleft palate, severe mental retardation, and many other physical defects; usually lethal by age 1.
Polyploidy—A condition in which a cell receives more than two complete sets of chromosomes.
Prader-Willi syndrome—A syndrome caused by a deletion in the paternally inherited chromosome 15 or by uniparental disomy of the maternal chromosome 15.
Tetraploidy—A form of polyploidy; four sets of chromosomes.
Translocation—The transfer of one part of a chromosome to another chromosome during cell division. A balanced translocation occurs when pieces from two different chromosomes exchange places without loss or gain of any chromosome material. An unbalanced translocation involves the unequal loss or gain of genetic information between two chromosomes.
Triploidy—A form of polyploidy; three sets of chromosomes.
Trisomy—The condition of having three identical chromosomes, instead of the normal two, in a cell.
Turner syndrome—Chromosome abnormality characterized by short stature and ovarian failure, caused by an absent X chromosome. Occurs only in females.
Zygote—The cell formed by the uniting of egg and sperm.
age. XYY males occur in one out of every 1,000 male births.
Females with an extra X chromosome (XXX) are sometimes said to have “triple X syndrome” and were sometimes called metafemales. This defect occurs in one out of every 1,000 female births. Females with XXX do not usually have mental retardation; pubertal development and fertility are normal.
Females with only one X chromosome (XO) have Turner syndrome. Turner syndrome is also called monosomy X and occurs in one out of every 2,000-5,000 female births. The sex organs of females with Turner syndrome do not mature at puberty; therefore these women are usually sterile. They are of short stature and have no
mental deficiencies. Heart defects are more common in girls with Turner syndrome.
Polyploidy
Polyploidy is lethal in humans. Normally, humans have two complete sets of chromosomes. Normal human cells, other than sex cells, are thus described as diploid. In polyploidy, a zygote receives more than two complete chromosome sets. Examples of polyploidy include triploidy, in which a zygote has three sets of chromosomes, and tetraploidy, in which a zygote has four sets of chromosomes. Triploidy could result from the fertilization of an abnormal diploid sex cell with a normal sex cell or from the fertilization of one egg by two sperm.
abnormalities Chromosomal
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
235 |

Chromosomal abnormalities
Figure 2. (Gale Group)
Tetraploidy could result from the failure of the zygote to divide after it replicates its chromosomes. Human zygotes with either of these conditions usually die before birth, or soon after. Interestingly, polyploidy is common in plants and is essential for the proper development of certain stages of the plant life cycle. Also, some kinds of cancerous cells have been shown to exhibit polyploidy.
Alterations in chromosome structure
Another kind of chromosomal abnormality is changes of chromosome structure. Structural defects arise during replication of the chromosomes just before a meiotic cell division. Meiosis is a complex process that often involves the chromosomes exchanging segments with each other in a process called crossing-over. If the process is faulty, the structure of the chromosomes changes. Sometimes these structural changes are harmless to the zygote; other structural changes, however, can be lethal.
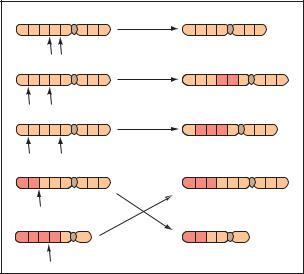
Four types of general structural alterations occur during replication of chromosomes (Figure 3). All four types begin with the breakage of a chromosome during replication. In a deletion, the broken segment of the chromosome is “lost”. Thus, all the genes that are present on this segment are also lost. In a duplication, the segment is inserted into the homologous chromosome as extra (duplicated) DNA. In an inversion, the segment attaches to the original chromosome, but in a reverse position. In a translocation, the segment attaches to an entirely different chromosome.
Because chromosomal structural changes cause the loss or misplacement of genes, the results can be quite severe. Deletions and duplications lead to missing and extra chromosomal material, meaning that there are too many or too few genes in that region. Translocations may or may not be harmful. If the translocation is balanced, meaning that all of the DNA is present and none is missing, the only effect may be a higher risk for abnormal
sperm or eggs. If the translocation is not balanced, the chance of associated physical and cognitive abnormalities increases. Inversions of DNA may also be harmless except for a risk of abnormal sperm or eggs. However, both inversions and balanced translocations may have clinical consequences, depending on where the breakage and rejoining of DNA occurred.
A structural abnormality in chromosome 21 occurs in about 4% of people with Down syndrome. In this abnormality, a translocation, a piece of chromosome 21 breaks off during meiosis of the egg or sperm cell and attaches to chromosome 13, 14, or 22. The parents of a child with Down syndrome due to this type of translocation could be balanced carriers for the translocation, and if so, are at increased risk to have another child with Down syndrome.
Some structural chromosomal abnormalities have been implicated in certain cancers. For instance, myelogenous leukemia is a cancer of the white blood cells. Researchers have found that the cancerous cells contain a translocation of chromosome 22, in which a broken segment switches places with the tip of chromosome 9.
Syndromes associated with chromosomal deletions
Many syndromes are associated with chromosomal deletions. These include Cri du chat syndrome, velocardiofacial syndrome, Prader-Willi syndrome, Angelman syndrome, Wolf-Hirschhorn syndrome, SmithMagenis syndrome, Miller-Dieker syndrome, LangerGiedion syndrome, and the trichorhinophalangeal syndromes.
Cri du chat means “cat cry” in French. Children with this syndrome have an abnormally developed larynx that makes their cry sound like the meowing of a cat in distress. They also have a small head, misshapen ears, and a rounded face, as well as other systemic abnormalities and mental retardation. Cri du chat is caused by a deletion of a segment of DNA in chromosome 5.
Velocardiofacial syndrome is also called DiGeorge syndrome or Shprintzen syndrome. More recently, it has been called deletion 22q11 syndrome because it is caused by a deletion of part of chromosome 22. Individuals with velocardiofacial syndrome may have congenital heart disease, cleft palate, learning difficulties, and subtle characteristic facial features.
Two syndromes caused by a chromosome abnormality illustrate an interesting concept: the severity or type of symptoms associated with a chromosomal defect may depend upon whether the child receives the changed gene from the mother or the father. Both Prader-Willi syn-
236 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
drome and Angelman syndrome are usually caused by a deletion in chromosome 15. Prader-Willi syndrome is characterized by mental retardation, obesity, short stature, and small hands and feet. Angelman syndrome is characterized by jerky movements and neurological symptoms. People with this syndrome also have an inability to control laughter, and may laugh inappropriately at odd moments. If a child inherits the changed chromosome from its father, the result is Prader-Willi syndrome. But if the child inherits the changed chromosome from its mother, the child will have Angelman syndrome.
A person may have Prader-Willi or Angelman syndrome, but not have the chromosomal deletion usually associated with these conditions. This may be due to a chromosomal error called uniparental disomy. Usually, one of each chromosome pair is inherited from each parent, and every section of DNA has two copies—one maternally inherited and the other paternally inherited. Uniparental disomy refers to the mistake of both copies of a section of DNA being inherited from one parent. Two copies of a maternally inherited chromosome 15 (no paternal gene present) causes Prader-Willi syndrome, and two copies of a paternally inherited chromosome 15 causes Angelman syndrome.
The sequence of events leading to Prader-Willi and Angelman syndrome is unknown. Researchers have determined that the genes in this region on chromosome 15 may be “turned off,” depending on which parent contributed the chromosome. This process of gene inactivation is called imprinting. Some people have Prader-Willi and Angelman syndrome because the mechanism controlling the imprinting malfunctions.
Expansion of chromosomal material
Not only can the sex of the parent from whom a gene is inherited determine whether it is turned “on” or turned “off,” but the sex of the parent may also influence whether certain abnormal sections of chromosomes become more abnormal. For example, the sex of the parent contributing the X chromosome may increase or decrease the chance that a child will be affected with fragile X syndrome.
Fragile X syndrome occurs in one out of 1,000 male births and one out of 2,000 female births. Males are affected more severely than females and the syndrome may be more pronounced if the child inherits the disorder from his/her mother. Part of this is explained by the fact that fragile X syndrome is caused by an abnormality of the X chromosome. Remember that a male is XY and a female is XX. A male child receives a Y chromosome from the father and an X chromosome from the mother. A female child, however, can receive an X from either the
A B C D E F G H |
Deletion |
A B C E F G H |
|
Chromosomal |
|||
|
|
|
|
|
|||
A B C D E F G H |
Duplication |
A B C B C E F G H |
|||||
|
|
|
|
|
|||
|
|
|
|
|
|
||
A B C D E F G H |
Inversion |
A D C B E F G H |
|
abnormalities |
|||
|
|
|
|
|
|||
A B C D E |
F G H |
Reciprocal |
M N O C D E |
F G |
H |
||
|
|||||||
|
|
translocation |
|
|
|
|
|
M N O P Q R |
|
A B P Q R |
|
|
|
||
Figure 3. (Gale Group)
mother or the father. Girls with fragile X syndrome are less severely affected than boys because they have a normal X chromosome that helps to protect them from the abnormal X chromosome. However, it was somewhat perplexing that girls were affected at all.
This mystery was solved when researchers learned that there is a range of abnormality in the fragile X chromosome. If the abnormality of the fragile X region of the chromosome is severe, the influence can be strong enough to affect females. If the abnormality is mild, females will not have symptoms of fragile X syndrome. Furthermore, the fragile X region of the X chromosome may become more severe when it is maternally inherited. The sex of the parent that the region is inherited from affects whether the chromosome abnormality remains stable or becomes greater.
Many other conditions are associated with similar chromosome abnormalities and may remain stable or become more severe depending upon whether the chromosome region is inherited from the mother or the father. In some of these conditions, the region becomes more abnormal when it is paternally inherited. Huntington disease, an adult onset neurological disease, is one such condition.
Maternal age and prenatal diagnosis
Currently, no cures exist for any of the syndromes caused by chromosomal abnormalities. For most of the conditions caused by aneuploidy, the risk to give birth to a child with a chromosomal abnormality increases with the mother’s age. The risk for Down syndrome, for instance, jumps from one in 1,000 when the mother is age
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
237 |
Chromosomal abnormalities
15-30 to one in 350 at age 35. This is most likely because the risk for nondisjunction as the eggs finish forming increases as maternal age increases. A man’s age does not increase the nondisjunction risk because of differences in the way eggs and sperms develop. Sperm are maturing and reproducing throughout a man’s adult life. Women, on the other hand, are born with all of the eggs they will ever have. At birth these eggs are part way through meiosis I, and each month as a woman ovulates, one egg finishes meiosis I and begins meiosis II.
People at high risk for chromosomal abnormalities may opt to know whether the fetus they have conceived has one of these abnormalities. Amniocentesis is a procedure in which some of the amniotic fluid that surrounds and cushions the fetus in the uterus is sampled with a needle placed in the uterus. Real-time ultrasound is used to guide the procedure. The amniotic fluid contains fetal cells that can be tested for chromosomal, DNA, and biochemical abnormalities. Another test, chorionic villi sampling (CVS), involves taking a piece of tissue from the devloping placenta. Undergoing either amniocentesis or CVS increases the risk of miscarriage slightly. Women and couples considering the procedure should be fully informed of the risks, benefits, and limitations of each procedure. If an abnormality is detected, the prenatal care provider discusses the options available with the woman or couple. Chromosomal abnormalities cannot be corrected. Some parents may terminate the pregnancy. Other parents choose to continue the pregnancy and use the time to prepare for the birth of a child with special needs.
Many resources are available to parents learning of abnormalities before or after birth. In the case of a sex chromosome abnormality, it is common for people to learn of the abnormality as a teenager or even as an adult. A primary care physician, obstetrician, or support group can recommend a specialist from whom more information may be obtained. This specialist is often a medical geneticist, perinatologist, or genetic counselor. Many organizations also provide resources and information to individuals and families.
In conclusion, the division of chromosomes during developmental and during sperm and egg formation is a complex process. Most of the time, however, the process occurs normally. Mistakes that are made can result in changes in chromosome number as well as abnormal chromosomes. Extra or missing chromosomal material usually leads to physical and congnitive defects. Changes in sex chromosome compliment are often associated with milder problems. Some problems with chromosomes are relatively common and are associated with well defined syndromes. Other problems with chromosomes occur rarely and problems associated with the change are only seen in a few individuals.
Resources
BOOKS
Baker, Diane, et al. Chromosome Abnormalities and Genetic Counseling. New York: Wiley-Liss, 1998.
Gardner, R. J. M., et al. A Guide to Genetic Counseling. New York: Oxford University Press, 1996.
PERIODICALS
Bos, A. P., et. al. “Avoidance of emergency surgery in newborn infants with trisomy 18.” The Lancet 339 no. 8798, (April 11, 1992): 913-6.
Kubas, C. “Noninvasive means of identifying fetuses with possible Down syndrome: a review.” The Journal of Perinatal and Neonatal Nursing 13 no. 2, (September 1999): 27-46.
Newberger, D. S. “Down syndrome: prenatal risk assessment and diagnosis.” American Family Physician 62 no.4, (August 2000): 837-8.
Sanders, Roger C. Structural Fetal Abnormalities: The Total Picture. St. Louis: Mosby, 1996.
ORGANIZATIONS
American Association for Klinefelter Syndrome Information and Support (AAKSIS) 2945 W. Farwell Ave., Chicago, IL 60645-2925. (773) 761-5298 or (888) 466-5747. Fax: (773) 761-5298. aaksis@aaksis.org http://www.aaksis
.org .
Angelman Syndrome Foundation. 414 Plaza Dr., Suite 209, Westmont, IL 60559-1265. (630) 734-9267 or (800) 432-6435. Fax: (630) 655-0391. info@angelman.org.http://www.angelman.org .
Chromosome Deletion Outreach, Inc. PO Box 724, Boca Raton, FL 33429-0724. (561) 391-5098 or (888) 2366880. Fax: (561) 395-4252. cdo@worldnet.att.net.http://members.aol.com/cdousa/cdo.htm .
Genetic Alliance. 4301 Connecticut Ave. NW, #404, Washington, DC 20008-2304. (800) 336-GENE (Helpline) or (202) 966-5557. Fax: (888) 394-3937 info @geneticalliance. http://www.geneticalliance.org .
Klinefelter Syndrome and Associates, Inc. PO Box 119, Roseville, CA 95678-0119. (916) 773-2999 or (888) 9999428. Fax: (916) 773-1449. ksinfo@genetic.org.http://www.genetic.org/ks .
National Down Syndrome Congress. 7000 PeachtreeDunwoody Rd., Bldg 5, Suite 100, Atlanta, GA 303281662. (770) 604-9500 or (800) 232-6372. Fax: (770) 604-9898. ndsccenter@aol.com. http://www.ndsccenter
.org .
National Down Syndrome Society. 666 Broadway, New York, NY 10012-2317. (212) 460-9330 or (800) 221-4602. Fax: (212) 979-2873. http://www.ndss.org info@ndss.org .
National Fragile X Foundation. PO Box 190488, San Francisco, CA 94119-0988. (800) 688-8765 or (510) 763-6030. Fax: (510) 763-6223. natlfx@sprintmail.com. http://nfxf
.org .
Prader-Willi Syndrome Association. 5700 Midnight Pass Rd., Suite 6, Sarasota, FL 34242-3000. (941) 312-0400 or (800) 926-4797. Fax: (941) 312-0142. http://www
.pwsausa.org PWSAUSA@aol.com .
238 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

Triple X syndrome support. 231 W. Park Ave., Sellersville, PA 18960. (215) 453-2117. edr@starbyte.com http://www
.voicenet.com/~markr/triple.html .
Velo-Cardio-Facial Syndrome Research Institute. Albert Einstein College of Medicine, 3311 Bainbridge Ave., Bronx, NY 10467. (718) 430-2568. Fax: (718) 430-8778. rgoldber@aecom.yu.edu. http://www.kumc.edu/gec/ vcfhome.html .
WEBSITES
“Angelman Syndrome” NCI Genes and Disease.
http://www.ncbi.nlm.nih.gov/disease/angelman.html . “Fragile X Syndrome” NCI Genes and Disease.
http://www.ncbi.nlm.nih.gov/disease/FMR1.html . “Velocardiofacial Syndrome” NCI Genes and Disease.
http://www.ncbi.nlm.nih.gov/disease/DGS.html .
Michelle Bosworth, MS, CGC
I Chromosome
Chromosomes are microscopic units containing organized genetic information, located in the nuclei of diploid and haploid cells (e.g. human somatic and sex cells), and are also present in one-cell non-nucleated organisms (unicellular microorganisms), like bacteria, which do not have an organized nucleus. The sum-total of genetic information contained in different chromosomes of a given individual or species are generically referred to as the genome.
In humans, chromosomes are structurally made of roughly equal amounts of proteins and DNA. Each chromosome contains a double-strand DNA molecule, arranged as a double helix, and tightly coiled and neatly packed by a family of proteins called histones. DNA strands are comprised of linked nucleotides. Each nucleotide has a sugar (deoxyribose), a nitrogenous base, plus one to three phosphate groups. Each nucleotide is linked to adjacent nucleotides in the same DNA strand by phosphodiester bonds. Phosphodiester is another sugar, made of sugar-phosphate. Nucleotides of one DNA strand link to their complementary nucleotide on the opposite DNA strand by hydrogen bonds, thus forming a pair of nucleotides, known as a base pair, or nucleotide base. Genes contain up to thousands of sequences of these base pairs. What distinguishes one gene from another is the sequence of nucleotides that code for the synthesis of a specific protein or portion of a protein. Some proteins are necessary for the structure of cells and tissues. Others, like enzymes, a class of active (catalyst) proteins, promote essential biochemical reactions, such as digestion, energy generation for cellular activity, or
metabolism of toxic compounds. Some genes produce several slightly different versions of a given protein through a process of alternate transcription of base pair segments known as codons.
Amounts of autosomal chromosomes differ in cells of different species; but are usually the same in every cell of a given species. Sex determination cells (mature ovum and sperm) are an exception, where the number of chromosomes is halved. Chromosomes also differ in size. For instance, the smallest human chromosome, the sex chromosome Y, contains 50 million base pairs (bp), whereas the largest one, chromosome 1, contains 250 million base pairs. All three billion base pairs in the human genome are stored in 48 chromosomes. Human genetic information is therefore stored in 24 pairs of chromosomes (totaling 48), 24 inherited from the mother, and 24 from the father. Two of these chromosomes are sex chromosomes (chromosomes X and Y). The remaining 46 are autosomes, meaning that they are not sex chromosomes and are present in all somatic cells (i.e., any other body cell that is not a germinal cell for spermatozoa in males or an ovum in females). Sex chromosomes specify the offspring gender: normal females have two X chromosomes and normal males have one X and one Y chromosome.
Each set of 24 chromosomes constitutes one allele, containing gene copies inherited from one of the parents. The other allele is complementary or homologous, meaning that it contains copies of the same genes and on the same positions, but originated from the other parent. As an example, every normal child inherits one set of copies of gene BRCA1, located on chromosome 13, from the mother and another set of BRCA1 from the father, located on the other allelic chromosome 13. Allele is a Greek-derived word that means “one of a pair,” or any one of a series of genes having the same locus (position) on homologous chromosomes.
The first chromosome observations were made under light microscopes, revealing rod-shaped structures in varied sizes and conformations; commonly J-, or V-shaped in eukaryotic cells and ring-shaped chromosome in bacteria. Staining reveals a pattern of light and dark bands. Today those bands are known to correspond to regional variations in the amounts of the two nucleotide base pairs: adenine-thymine (A-T or T-A) in contrast with amounts of guanine-cytosine (G-C or C-G).
Genetic abnormalities and diseases occur when one of the following events happens: a) one chromosome copy is missing, b) extra copies of a chromosome are present, c) a chromosome breaks and its fragment is fused into another chromosome (insertion), d) a fragment is deleted, e) a gene is transferred from one chromosome to another (translocation), f) duplication of a chromosomal segment occurs, g) inversion of a chromosomal seg-
Chromosome
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
239 |

Cleft lip and palate
False-colour light micrograph of normal human chromosomes, obtained by amniocentesis. (Photo Researchers, Inc.)
ment occurs. Down syndrome, for instance, is caused by the presence of a third copy of chromosome 21.
In non-dividing cells, it is not possible to distinguish morphological details of individual chromosomes because they remain elongated and entangled to each other. However, when a cell is dividing, i.e., undergoing mitosis, chromosomes become highly condensed and each individual chromosome occupies a well-defined spatial location.
Mitotic chromosomes present a constricted region, to which the spindle fibers attach during cellular division. Such a constricted region, known as a centromere or primary constriction, may be located in three different positions in chromosomes. Centromeric position allows the classification of chromosomes in three groups: a) acrocentric: centromere lies very near one end; b) metacentric: centromere at the middle, dividing the chromosome in two equal parts or arms; and c) submetacentric: centromere near middle, but dividing chromosome in two unequal arms.
When a chromosome loses its centromere, it is known as acentric. As the centromere is essential for both division and retention of chromosome copies in the new cells, acentric chromosomes will not pass to the daughter cells during the parental cell division. Therefore, daughter cells will miss one chromosome in their karyotype. A karyotype map shows mitotic chromosomes in the mitotic phase, known as metaphase. In metaphase, chro-
mosomes align in pairs. In a normal human karyotype, there are 22 pairs of autosomal chromosomes and two sex chromosomes (X and Y). Each pair of autosomal chromosomes contains two complementary or homologous chromosomes, a maternal and a paternal copy.
Some chromosomes also present a secondary constriction that always appears at the same site. They are also useful, along with centromere position and chromosome size, for identifying and characterizing individual chromosomes, in a karyotype.
Karyotype analysis was the first genetic screening utilized by geneticists to assess inherited abnormalities, like additional copies of a chromosome or a missing copy, as well as DNA content and gender of the individual. With the development of new molecular screening techniques and the growing number of identified individual genes, detection of other more subtle chromosomal mutations is now possible (e.g., determinations of gene mutations, levels of gene expression, etc). Such data allow scientists to better understand disease causation and to develop new therapies and medicines for those diseases.
Sandra Galeotti, MS
Chromosome mapping see Gene mapping
Chronic pancreatitis see Hereditary pancreatitis
Cleft lip see Cleft lip and palate
I Cleft lip and palate
Definition
A cleft is a birth defect that occurs when the tissues of the lip and or palate of the fetus do not fuse very early in pregnancy. A cleft lip, sometimes referred to as a harelip, is an opening in the upper lip that can extend into the base of the nostril. A cleft palate is an opening in the roof of the mouth.
Description
Infants born with cleft lips will have an opening involving the upper lip. The length of the opening ranges from a small notch to a cleft that extends into the base of the nostril. Cleft lips may involve one or both sides of the lip.
240 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |