

Gale Encyclopedia of Genetic Disorder / Gale Encyclopedia of Genetic Disorders, Two Volume Set - Volume 1 - A-L - I
.pdf
|
|
|
|
|
|
Hydrogen bonds |
|
|
DNA |
|||
|
|
|
|
|
|
5' |
|
|
|
3' |
|
|
5' |
3' |
|
5' |
3' |
|
|
|
|
|
(deoxyribonucleic |
||
|
O |
|
|
H |
|
OH |
|
|||||
|
|
|
|
–O |
P |
O |
|
|
|
|
||
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
O |
T |
H |
|
A |
|
|
|
|
|
|
T |
A |
O |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
O |
|
|||
|
|
|
|
|
|
|
|
|
O |
P |
O– |
|
|
|
|
|
|
O |
|
H |
|
|
O |
|
acid) |
|
|
|
|
–O |
P |
O |
|
|
|
|
||
|
|
|
C |
G |
O |
C |
|
H |
G |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
O |
|
|
||
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
P |
O– |
|
|
|
|
|
|
O |
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
–O |
P |
O |
|
H |
|
|
|
|
3' |
|
|
G |
C |
O |
G |
H |
|
C |
|
|
|
5' |
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
P |
O– |
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
5' |
|
O |
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
–O |
P |
O |
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3' |
A |
T |
O |
A |
|
H |
T |
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
O |
P |
O– |
|
|
3' |
5' |
3' |
5' |
OH |
H |
|
|
|
O |
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
3' |
|
|
|
5' |
|
||
The structure of a DNA molecule. (Gale Group)
It was not clear how this relatively simple structure could assume enough different conformations to “code” for hundreds of thousands of genetic traits. In comparison, a single protein molecule contains various arrangements of twenty fundamental units (amino acids) making it a much better candidate as a carrier of genetic information.
Yet, experimental evidence began to point to a possible role for nucleic acids in the transmission of hereditary characteristics. That evidence implicated a specific sub-family of the nucleic acids known as the deoxyribonucleic acids, or DNA. DNA is characterized by the presence of the sugar deoxyribose in the sugar-phosphate backbone of the molecule and by the presence of adenine, cytosine, guanine, and thymine, but not uracil.
As far back as the 1890s, the German geneticist Albrecht Kossel (1853–1927) obtained results that pointed to the role of DNA in heredity. In fact, historian
John Gribbin has suggested that the evidence was so clear that it “ought to have been enough alone to show that the hereditary information... must be carried by the DNA.” Yet, somehow, Kossel himself did not see this point, nor did most of his colleagues for half a century.
As more and more experiments showed the connection between DNA and genetics, a small group of researchers in the 1940s and 1950s began to ask how a DNA molecule could code for genetic information. The two who finally resolved this question were a somewhat unusual pair, James Watson, a 24-year old American trained in genetics, and Francis Crick, a 36-year old Englishman, trained in physics and self-taught in chemistry. The two met at the Cavendish Laboratories of Cambridge University in 1951, and became instant friends. They were united by a common passionate belief that the structure of DNA held the key to understanding how genetic information is stored in a cell and how it is transmitted from one cell to its daughter cells.
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
343 |
DNA (deoxyribonucleic acid)
In one sense, the challenge facing Watson and Crick was a relatively simple one. A great deal was already known about the DNA molecule. Few new discoveries were needed, but those few discoveries were crucial to solving the DNA-heredity puzzle. Primarily the question was one of molecular architecture. How were the various parts of a DNA molecule oriented in space such that the molecule could hold genetic information?
The key to answering that question lay in a technique known as x-ray crystallography. When x rays are directed at a crystal of some material, such as DNA, they are reflected and refracted by atoms that make up the crystal. The refraction pattern thus produced consists of a collection of spots and arcs. A skilled observer can determine from the refraction pattern the arrangement of atoms in the crystal.
The technique is actually more complex than described here. For one thing, obtaining satisfactory x-ray patterns from crystals is often difficult. Also, interpreting x-ray patterns—especially for complex molecules like DNA—can be extremely difficult.
Watson and Crick were fortunate in having access to some of the best x-ray diffraction patterns that then existed. These “photographs” were the result of work being done by Maurice Wilkins and Rosalind Elsie Franklin at King’ s College in London. Although Wilkins and Franklin were also working on the structure of DNA, they did not recognize the information their photographs contained. Indeed, it was only when Watson accidentally saw one of Franklin’s photographs that he suddenly saw the solution to the DNA puzzle.
Racing back to Cambridge after seeing this photograph, Watson convinced Crick to make an all-out attack on the DNA problem. They worked continuously for almost a week. Their approach was to construct tinker- toy-like models of the DNA molecule, shifting atoms around into various positions. They were looking for an arrangement that would give the kind of x-ray photograph that Watson had seen in Franklin’s laboratory.
Finally, on March 7, 1953, the two scientists found the answer. They built a model consisting of two helices (corkscrew-like spirals), wrapped around each other. Each helix consisted of a backbone of alternating sugar and phosphate groups. To each sugar was attached one of the four nitrogen bases, adenine, cytosine, guanine, or thymine. The sugar-phosphate backbone formed the outside of the DNA molecule, with the nitrogen bases tucked inside. Each nitrogen base on one strand of the molecule faced another nitrogen base on the opposite strand of the molecule. The base pairs were not arranged at random, however, but in such a way that each adenine was paired with a thymine, and each cytosine with a guanine.
The Watson-Crick model was a remarkable achievement, for which the two scientists won the 1954 Nobel Prize in Chemistry. The molecule had exactly the shape and dimensions needed to produce an x-ray photograph like that of Franklin’s. Furthermore, Watson and Crick immediately saw how the molecule could “carry” genetic information. The sequence of nitrogen bases along the molecule, they said, could act as a genetic code. A sequence, such as A-T-T-C-G-C-T . . . etc., might tell a cell to make one kind of protein (such as that for red hair), while another sequence, such as G-C-T-C-T-C-G . . . etc., might code for a different kind of protein (such as that for blonde hair). Watson and Crick themselves contributed to the deciphering of this genetic code, although that process was long and difficult and involved the efforts of dozens of researchers over the next decade.
Watson and Crick had also considered, even before their March 7th discovery, what the role of DNA might be in the manufacture of proteins in a cell. The sequence that they outlined was that DNA in the nucleus of a cell might act as a template for the formation of a second type of nucleic acid, RNA (ribonucleic acid). RNA would then leave the nucleus, emigrate to the cytoplasm and then itself act as a template for the production of protein. That theory, now known as the Central Dogma, has since been largely confirmed and has become a critical guiding principal of much research in molecular biology.
Scientists continue to advance their understanding of DNA. Even before the Watson-Crick discovery, they knew that DNA molecules could exist in two configurations, known as the “A” form and the “B” form. After the Watson-Crick discovery, two other forms, known as the “C” and “D” configurations, were also discovered. All four of these forms of DNA are right-handed double helices that differ from each other in relatively modest ways.
In 1979, however, a fifth form of DNA known as the “Z” form was discovered by Alexander Rich and his colleagues at the Massachusetts Institute of Technology. The “Z” form was given its name partly because of its zig-zag shape and partly because it is different from the more common A and B forms. Although Z-DNA was first recognized in synthetic DNA prepared in the laboratory, it has since been found in natural cells whose environment is unusual in some respect or another. The presence of certain types of proteins in the nucleus, for example, can cause DNA to shift from the B to the Z conformation. The significance and role of this most recently discovered form of DNA remains a subject of research among molecular biologists.
Judyth Sassoon, ARCS, PhD
344 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

I Donohue syndrome
Definition
Donohue syndrome, also formerly called leprechaunism, is a genetic disorder caused by mutations in the insulin receptor gene. W. L. Donohue first described this rare syndrome in 1948.
Description
Donohue syndrome is a disorder that causes low birth weight, unusual facial features, and failure to thrive in infants. Donohue syndrome is associated with the over-development of the pancreas, a gland located near the stomach. It is also considered to be the most insulin resistant form of diabetes.
Donohue syndrome results from a mutation of the insulin receptor gene which prevents insulin in the blood from being processed. Therefore, even before birth, the fetus exhibits “insulin resistance” and has high levels of unprocessed insulin in the blood. Insulin is one of two hormones secreted by the pancreas to control blood sugar (glucose) levels. Donohue syndrome is known as a progressive endocrine disorder because it relates to the growth and functions of the endocrine system, the collection of glands and organs that deliver hormones via the bloodstream.
Hormones are chemicals released by the body to control cellular function (metabolism) and maintain equilibrium (homeostasis). These hormones are released either by the endocrine system or by the exocrine system. The endocrine system consists of ductless glands that secrete hormones into the bloodstream. These hormones then travel through the blood to the parts of the body where they are required. The exocrine system consists of ducted glands that release their hormones via ducts directly to the site where they are needed. The pancreas is both an endocrine and an exocrine gland. As part of the endocrine system, the pancreas acts as the original producer of estrogen and other sex hormones in fetuses of both sexes. It also regulates blood sugar through its production of the hormones insulin and glucagon. The pancreas releases insulin in response to high levels of glucose in the blood. Glucagon is released when glucose levels in the blood are low. These two hormones act in direct opposition to each other (antagonistically) to maintain proper blood sugar levels. As an exocrine gland, the pancreas secretes digestive enzymes directly into the small intestine.
In an attempt to compensate for the high blood insulin level, the pancreas overproduces glucagon as well as the female hormone estrogen and other related (estro-
genic) hormones. As excess estrogen and related hormones are produced, they affect the development of the external and internal sex organs (genitalia) of the growing baby.
Insulin mediates the baby’s growth in the womb through the addition of muscle and fat. A genetic link between fetal insulin resistance and low birthweight has been suggested. Without the proper processing of insulin, the fetus will not gain weight as fast as expected. Therefore, the effects of Donohue syndrome tend to become visible during the seventh month of development when the fetus either stops growing entirely or shows a noticeable slowdown in size and weight gain. This lack of growth is further evident at birth in affected infants, who demonstrate extreme thinness (emaciation), difficulty gaining weight, a failure to thrive, and delayed maturation of the skeletal structure.
Genetic profile
Donohue syndrome is a non-sex-linked (autosomal) recessive disorder. In 1988, Donohue syndrome was identified as the first insulin receptor gene mutation directly related to a human disease. The gene responsible for the appearance of Donohue syndrome is the insulin receptor gene located at 19p13.2. Over 40 distinct mutations of this gene have been identified. Besides Donohue syndrome, other types of non-insulin-dependent (Type II) diabetes mellitus (NIDDM) can result from mutations of this gene, including Rabson-Mendenhall syndrome and type A insulin resistance.
Demographics
Donohue syndrome occurs in approximately one out of every four million live births. As in all recessive genetic disorders, both parents must carry the gene mutation in order for their child to have the disorder. Therefore, Donohue syndrome has been observed in cases where the parents are related by blood (consanguineous). Parents with one child affected by Donohue syndrome have a 25% likelihood that their next child will also be affected with the disease.
Signs and symptoms
Infants born with Donohue syndrome have characteristic facial features that have been said to exhibit “elfin” or leprechaun-like qualities, such as: a smallish head with large, poorly developed and low-set ears; a flat nasal ridge with flared nostrils, thick lips, a greatly exaggerated mouth width, and widely spaced eyes. They will be very thin and have low blood sugar (hypoglycemia) due to their inability to gain nutrition through insulin pro-
syndrome Donohue
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
345 |

Donohue syndrome
K E Y T E R M S
Autosomal—Relating to any chromosome besides the X and Y sex chromosomes. Human cells contain 22 pairs of autosomes and one pair of sex chromosomes.
Chorionic villus sampling (CVS)—A procedure used for prenatal diagnosis at 10-12 weeks gestation. Under ultrasound guidance a needle is inserted either through the mother’s vagina or abdominal wall and a sample of cells is collected from around the fetus. These cells are then tested for chromosome abnormalities or other genetic diseases.
Consanguineous—Sharing a common bloodline or ancestor.
Endocrine system—A system of ductless glands that regulate and secrete hormones directly into the bloodstream.
Fibroblast—Cells that form connective tissue fibers like skin.
Hirsutism—The presence of coarse hair on the face, chest, upper back, or abdomen in a female as a result of excessive androgen production.
Histologic—Pertaining to histology, the study of cells and tissues at the microscopic level.
Hypoglycemia—An abnormally low glucose (blood sugar) concentration in the blood.
Insulin—A hormone produced by the pancreas that is secreted into the bloodstream and regulates blood sugar levels.
Insulin receptor gene—The gene responsible for the production of insulin receptor sites on cell surfaces. Without properly functioning insulin receptor sites, cells cannot attach insulin from the blood for cellular use.
Insulin resistance—An inability to respond normally to insulin in the bloodstream.
Insulin-like growth factor I—A hormone released by the liver in response to high levels of growth hormone in the blood. This growth factor is very similar to insulin in chemical composition; and, like insulin, it is able to cause cell growth by causing cells to undergo mitosis (cell division).
Pachyderma—An abnormal skin condition in which excess skin is produced that appears similar to that of an elephant (pachyderm).
Pancreas—An organ located in the abdomen that secretes pancreatic juices for digestion and hormones for maintaining blood sugar levels.
Serological—Pertaining to serology, the science of testing blood to detect the absence or presence of antibodies (an immune response) to a particular antigen (foreign substance).
cessing. They will exhibit delayed bone growth and maturation, and difficulty in gaining weight and developing (failure to thrive).
Donohue syndrome patients are prone to persistent and recurrent infections. Delayed bone growth not only leads to skeletal abnormalities, it also leads to a compromised immune system. Many of the chemicals used by the body to fight infection are produced in the marrow of the bones. When bone maturation is delayed, these chemicals are not produced in sufficient quantities to fight off or prevent infection.
At birth, affected individuals can also have an enlarged chest, with possible breast development, excessive hairiness (hirsutism), as well as overdeveloped external sex organs, because of increased estrogen production caused by an overactive pancreas. As an additional side effect of the increased sex hormones released in Donohue syndrome, these individuals often have extremely large hands and feet relative to their non-affected peer group. As the result of a lack of insulin, the infant is likely to
have a relatively small amount of muscle mass, very little fat, and a distended abdomen (due to malnutrition). Additional symptoms of Donohue syndrome include pachyderma, or elephant skin, in which there is excess skin production causing large, loose folds; and abnormal coloration (pigmentation) of the skin. These individuals are also quite susceptible to both umbilical and inguinal hernias.
In addition to the defect in the insulin receptor gene, Donohue syndrome is associated with problems in the epidermal growth factor receptor, which controls growth of the skin. An abnormal functioning of the epidermal growth factor receptor has been identified in three unrelated individuals affected with Donohue syndrome. This suggests that the probable cause of leprechaunism is more than just the insulin receptor. These observations may help explain the physical symptom of pachyderma in those affected with Donohue syndrome. It has also been suggested that the high concentrations of insulin close to the cell membranes lead to receptor activity at these loca-
346 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
tions. This lowered growth hormone activity, in turn, causes slowed cellular growth which leads to systemic growth failure in affected patients.
Diagnosis
In families with a history of the disease, diagnosis in utero before birth of the fetus is possible through molecular DNA analysis of tissue samples from the chorionic villi, which are cells found in the placenta. After birth, the diagnosis of Donohue syndrome is usually made based on the blood tests that show severe insulin resistance coupled with hypoglycemia. The presence of several of the physical symptoms listed above in addition to positive results in a test for severe insulin resistance, such as an insulin receptor defect test or a fasting hypoglycemia test, is usually sufficient for a diagnosis of Donohue syndrome. The diagnosis of Donohue syndrome may be confirmed by observed cellular (histologic) changes in the ovaries, pancreas, and breast that are not normal for the age of the patient.
Treatment and management
Genetic counseling of parents with a Donohue syndrome affected child may help prevent the conception of additional children affected with this genetic disorder. After birth, affected infants may require treatment for malnutrition as well as insulin resistant diabetes. Patients with a demonstrated residual insulin receptor function may survive past infancy. In these cases, the treatment regimen must certainly include on-going insulin resistant diabetes care and dietetic counseling to assist with weight gain. It may also be necessary to administer growth hormone therapy to certain patients to spur growth, but this is only indicated in those individuals who show signs of functioning growth hormone receptors and no signs of higher than normal resistance to growth hormone.
The revolutionary impact of recombinant DNA technology, whereby scientists can mass produce genetic material for use in medicine, has made possible another treatment method which involves the introduction of recombinant human insulin-like growth factor 1 (rhIGF- 1) into the body. A case study has been reported of a female affected with Donohue syndrome and low levels of insulin-like growth factor 1 (IGF-1), which is indicative of a higher than normal resistance to growth hormone.
Examination of the patient’s fibroblasts showed normal binding of IGF-1 and normal functioning of these fibroblasts in response to IGF-1. Fibroblasts are connec-
tive tissue cells that accomplish growth in humans by differentiating into chondroblasts, collagenoblasts, and osteoblasts, all of which are the precursor cells necessary to produce bone growth in humans. This case report indicates that if enough IGF-1 could get to the fibroblasts in the patient’s body, there is every reason to believe that these fibroblasts would function normally and mature into the precursor cells needed for bone growth. This finding made the patient an ideal candidate for rhIGF-1 treatments.
The longand short-term effects on growth patterns and glucose metabolism in the patient were studied after the treatment with recombinant human insulin-like growth factor 1 (rhIGF-1). The rhIGF-1 that was not immediately utilized by the patient was rapidly destroyed in the cellular conditions produced by Donohue syndrome. Therefore, to maintain the desired levels of rhIGF-1 in the blood, the patient received rhIGF-1 both in injection form prior to every meal and via a continuous subcutaneous infusion method similar to that used to continuously pump insulin for some patients with diabetes. Recombinant human IGF-1 was administered to this patient over a period of six years with an observation of normal blood glucose levels and a return to normal growth patterns. Moreover, the treatment did not cause negative side effects. The results of this case study offer a promising new treatment for certain individuals affected with Donohue syndrome. As of 2001, other clinical studies of treatments with rhIGF-1 are in progress.
Prognosis
Individuals born with Donohue syndrome generally die in infancy from either malnutrition or recurrent and persistent infection. All individuals affected with Donohue syndrome that survive past infancy have severe mental retardation and profound motor skill impairment. Survival into childhood is thought to be due to some remaining insulin receptor function and the ability of extremely high insulin concentrations to transmit signals through alternate pathways.
Resources
PERIODICALS
Desbois-Mouthon, C., et al. “Molecular analysis of the insulin receptor gene for prenatal diagnosis of leprechaunism in two families.” Prenatal Diagnosis (July 1997): 657–63.
Hattersley, A. “The fetal insulin hypothesis: an alternative explanation of the association of low birthweight with diabetes and vascular disease.” Lancet (May 1999): 1789–92.
Nakae, J., et al. “Long-term effect of recombinant human insulin-like growth factor I on metabolic and growth control in a patient with leprechaunism.” Journal of Clinical Endocrinology and Metabolism (February 1998): 542–9.
syndrome Donohue
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
347 |

Down syndrome
Psiachou, H., et al. “Leprechaunism and homozygous nonsense mutation in the insulin receptor gene.” Lancet (October 1993): 924.
Reddy, S., D. Muller-Wieland, K. Kriaciunas, C. Kahn. “Molecular defects in the insulin receptor in patients with leprechaunism and in their parents.” Journal of Laboratory and Clinical Medicine (August 1989): 1359–65.
ORGANIZATIONS
Children Living with Inherited Metabolic Diseases. The Quadrangle, Crewe Hall, Weston Rd., Crewe, Cheshire, CW1-6UR. UK 127 025 0221. Fax: 0870-7700-327.http://www.climb.org.uk .
National Center for Biotechnology Information. National Library of Medicine, Building 38A, Room 8N805, Bethesda, MD 20894. (301) 496-2475. http://www.ncbi
.nlm.nih.gov .
National Organization for Rare Disorders (NORD). PO Box 8923, New Fairfield, CT 06812-8923. (203) 746-6518 or (800) 999-6673. Fax: (203) 746-6481. http://www
.rarediseases.org .
WEBSITES
“Leprechaunism.” National Organization for Rare Disorders,
Inc. http://www.stepstn.com/cgi-win/nord.exe?proc GetDocument&rectype 0&recnum 387 . (05 February 2001).
OMIM— Online Mendelian Inheritance in Man. http://www
.ncbi.nlm.nih.gov/htbin-post/Omim/dispmim?246200 . (05 February 2001).
Paul A. Johnson
I Down syndrome
Definition
Down syndrome is the most common chromosome disorder and genetic cause of mental retardation. It occurs because of the presence of an extra copy of chromosome 21. For this reason, it is also called trisomy 21.
Description
When a baby is conceived, the sperm cell from the father and the egg cell from the mother undergo a reduction of the total number of chromosomes from 46 to 23. Occasionally an error occurs in this reduction process and instead of passing on 23 chromosomes to the baby, a parent will pass on 24 chromosomes. This event is called nondisjunction and it occurs in 95% of Down syndrome cases. The baby therefore receives an extra chromosome at conception. In Down syndrome, that extra chromosome is chromosome 21. Because of this extra chromosome 21, individuals affected with Down syndrome have 47 instead of 46 chromosomes.
Genetic profile
In approximately one to two percent of Down syndrome cases, the original egg and sperm cells contain the correct number of chromosomes, 23 each. The problem occurs sometime shortly after fertilization—during the phase when cells are dividing rapidly. One cell divides abnormally, creating a line of cells with an extra copy of chromosome 21. This form of genetic disorder is called mosaicism. The individual with this type of Down syndrome has two types of cells: those with 46 chromosomes (the normal number), and those with 47 chromosomes (as occurs in Down syndrome). Individuals affected with this mosaic form of Down syndrome generally have less severe signs and symptoms of the disorder.
Another relatively rare genetic accident that causes Down syndrome is called translocation. During cell division, chromosome 21 somehow breaks. The broken off piece of this chromosome then becomes attached to another chromosome. Each cell still has 46 chromosomes, but the extra piece of chromosome 21 results in the signs and symptoms of Down syndrome. Translocations occur in about 3–4% of cases of Down syndrome.
Once a couple has had one baby with Down syndrome, they are often concerned about the likelihood of future offspring also being born with the disorder. Mothers under the age of 35 with one Down syndromeaffected child have a 1% chance that a second child will also be born with Down syndrome. In mothers 35 and older, the chance of a second child being affected with Down syndrome is approximately the same as for any woman at a similar age. However, when the baby with Down syndrome has the type that results from a translocation, it is possible that one of the two parents is a carrier of a balanced translocation. A carrier has rearranged chromosomal information and can pass it on, but he or she does not have an extra chromosome and therefore is not affected with the disorder. When one parent is a carrier of a translocation, the chance of future offspring having Down syndrome is greatly increased. The specific risk will have to be assessed by a genetic counselor.
Demographics
Down syndrome occurs in about one in every 800 live births. It affects an equal number of male and female babies. The majority of cases of Down syndrome occur due to an extra chromosome 21 within the egg cell supplied by the mother (nondisjunction). As a woman’s age (maternal age) increases, the risk of having a Down syndrome baby increases significantly. By the time the woman is age 35, the risk increases to one in 400; by age
348 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

40 the risk increases to one in 110; and, by age 45, the risk becomes one in 35. There is no increased risk of either mosaicism or translocation with increased maternal age.
Down syndrome occurs with equal frequency across all ethnic groups and subpopulations.
Signs and symptoms
While Down syndrome is a chromosomal disorder, a baby is usually identified at birth through observation of a set of common physical characteristics. Not all affected babies will exhibit all of the symptoms discussed. There is a large variability in the number and severity of these characteristics from one affected individual to the next. Babies with Down syndrome tend to be overly quiet, less responsive to stimuli, and have weak, floppy muscles. A number of physical signs may also be present. These include: a flat appearing face; a small head; a flat bridge of the nose; a smaller than normal, low-set nose; small mouth, which causes the tongue to stick out and to appear overly large; upward slanting eyes; bright speckles on the iris of the eye (Brushfield spots); extra folds of skin located at the inside corner of each eye and near the nose (epicanthal folds); rounded cheeks; small, misshapen ears; small, wide hands; an unusual deep crease across the center of the palm (simian crease); an inwardly curved little finger; a wide space between the great and the second toes; unusual creases on the soles of the feet; overly flexible joints (sometimes referred to as being double-jointed); and shorter-than-normal stature.
Other types of defects often accompany Down syndrome. Approximately 30–50% of all children with Down syndrome are found to have heart defects. A number of different heart defects are common in Down syndrome. All of these result in abnormal patterns of blood flow within the heart. Abnormal blood flow within the heart often means that less oxygen is sent into circulation throughout the body, which can cause fatigue, a lack of energy, and poor muscle tone.
Malformations of the gastrointestinal tract are present in about 5–7% of children with Down syndrome. The most common malformation is a narrowed, obstructed duodenum (the part of the intestine into which the stomach empties). This disorder, called duodenal atresia, interferes with the baby’s milk or formula leaving the stomach and entering the intestine for digestion. The baby often vomits forcibly after feeding, and cannot gain weight appropriately until the defect is repaired.
Another malformation of the gastrointestinal tract seen in patients with Down syndrome is an abnormal connection between the windpipe (trachea) and the digestive tube of the throat (esophagus) called a tracheo-
K E Y T E R M S
Chromosome—A microscopic thread-like structure found within each cell of the body and consists of a complex of proteins and DNA. Humans have 46 chromosomes arranged into 23 pairs. Changes in either the total number of chromosomes or their shape and size (structure) may lead to physical or mental abnormalities.
Karyotype—A standard arrangement of photographic or computer-generated images of chromosome pairs from a cell in ascending numerical order, from largest to smallest.
Mental retardation—Significant impairment in intellectual function and adaptation in society. Usually associated an intelligence quotient (IQ) below 70.
Mosaic—A term referring to a genetic situation in which an individual’s cells do not have the exact same composition of chromosomes. In Down syndrome, this may mean that some of the individual’s cells have a normal 46 chromosomes, while other cells have an abnormal 47 chromosomes.
Nondisjunction—Non-separation of a chromosome pair, during either meiosis or mitosis.
Translocation—The transfer of one part of a chromosome to another chromosome during cell division. A balanced translocation occurs when pieces from two different chromosomes exchange places without loss or gain of any chromosome material. An unbalanced translocation involves the unequal loss or gain of genetic information between two chromosomes.
Trisomy—The condition of having three identical chromosomes, instead of the normal two, in a cell.
esophageal fistula (T-E fistula). This connection interferes with eating and/or breathing because it allows air to enter the digestive system and/or food to enter the airway.
Other medical conditions occurring in patients with Down syndrome include an increased chance of developing infections, especially ear infections and pneumonia; certain kidney disorders; thyroid disease (especially low or hypothyroid); hearing loss; vision impairment requiring glasses (corrective lenses); and a 20 times greater chance than the population as a whole of developing leukemia.
Development in a baby and child affected with Down syndrome occurs at a much slower than normal
syndrome Down
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
349 |

Down syndrome
The sibling on the right has Down syndrome. (Photo Researchers, Inc.)
rate. Because of weak, floppy muscles (hypotonia), babies learn to sit up, crawl, and walk much later than their unaffected peers. Talking is also quite delayed. The level of mental retardation is considered to be mild-to- moderate in Down syndrome. The degree of mental retardation varies a great deal from one child to the next. While it is impossible to predict the severity of Down syndrome at birth, with proper education, children who have Down syndrome are capable of learning. Most children affected with Down syndrome can read and write and are placed in special education classes in school. The majority of individuals with Down syndrome become semi-independent adults, meaning that they can take care of their own needs with some assistance.
As people with Down syndrome age, they face an increased chance of developing the brain disease called Alzheimer’s (sometimes referred to as dementia or senility). Most people have a 12% chance of developing Alzheimer disease, but almost all people with Down syndrome will have either Alzheimer disease or a similar type of dementia by the age of 50. Alzheimer disease causes the brain to shrink and to break down. The number of brain cells decreases, and abnormal deposits and structural arrangements occur. This process results in a loss of brain functioning. People with Alzheimer’s have
strikingly faulty memories. Over time, people with Alzheimer disease will lapse into an increasingly unresponsive state.
As people with Down syndrome age, they also have an increased chance of developing a number of other illnesses, including cataracts, thyroid problems, diabetes, and seizure disorders.
Diagnosis
Diagnosis is usually suspected at birth, when the characteristic physical signs of Down syndrome are noted. Once this suspicion has been raised, genetic testing (chromosome analysis) can be undertaken in order to verify the presence of the disorder. This testing is usually done on a blood sample, although chromosome analysis can also be done on other types of tissue, including the skin. The cells to be studied are prepared in a laboratory. Chemical stain is added to make the characteristics of the cells and the chromosomes stand out. Chemicals are added to prompt the cells to go through normal development, up to the point where the chromosomes are most visible, prior to cell division. At this point, they are examined under a microscope and photographed. The photograph is used to sort the different sizes and shapes of
350 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
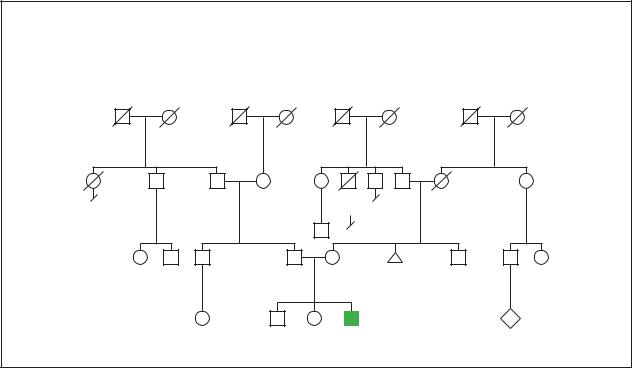
Down Syndrome
Chromosomal
Sporadic trisomy 21
Stomach |
d.63y |
cancer |
Melanoma |
2
50y 42y
3 |
P |
17y 11y 4y
47,XY,+21
(Gale Group)
chromosomes into pairs. In most cases of Down syndrome, one extra chromosome 21 will be revealed. The final result of such testing, with the photographed chromosomes paired and organized by shape and size, is called the individual’s karyotype. An individual with Down syndrome will have a 47 XX 21 karyotype if they are female and a 47 XY 21 karyotype if they are male.
Women who become pregnant after the age of 35 are offered prenatal tests to determine whether or not their developing baby is affected with Down syndrome. A genetic counselor meets with these families to inform them of the risks and to discuss the types of tests available to make a diagnosis prior to delivery. Because there is a slight risk of miscarriage following some prenatal tests, all testing is optional, and couples need to decide whether or not they desire to take this risk in order to learn the status of their unborn baby.
Screening tests are used to estimate the chance that an individual woman will have a baby with Down syndrome. A test called the maternal serum alpha-fetoprotein test (MSAFP) is offered to all pregnant women under the age of 35. If the mother decides to have this test, it is performed between 15 and 22 weeks of pregnancy. The MSAFP screen measures a protein and two hormones that are normally found in maternal blood during pregnancy. A specific pattern of these hormones and protein can indicate an increased risk for having a baby born with
Down syndrome. However, this is only a risk and MSAFP cannot diagnose Down syndrome directly. Women found to have an increased risk of their babies being affected with Down syndrome are offered amniocentesis. The MSAFP test can detect up to 60% of all babies who will be born with Down syndrome.
Ultrasound screening for Down syndrome is also available. This is generally performed in the midtrimester of pregnancy. Abnormal growth patterns characteristic of Down syndrome such as growth retardation, heart defects, duodenal atresia, T-E fistula, shorter than normal long-bone lengths, and extra folds of skin along the back of the neck of the developing fetus may all be observed via ultrasonic imaging.
The only way to definitively establish (with about 99% accuracy) the presence or absence of Down syndrome in a developing baby is to test tissue during the pregnancy itself. This is usually done either by amniocentesis, or chorionic villus sampling (CVS). All women under the age of 35 who show a high risk for having a baby affected with Down syndrome via an MSAFP screen and all mothers over the age of 35 are offered either CVS or amniocentesis. In CVS, a tiny tube is inserted into the opening of the uterus to retrieve a small sample of the placenta (the organ that attaches the growing baby to the mother via the umbilical cord, and provides oxygen and nutrition). In amniocentesis, a small
syndrome Down
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
351 |
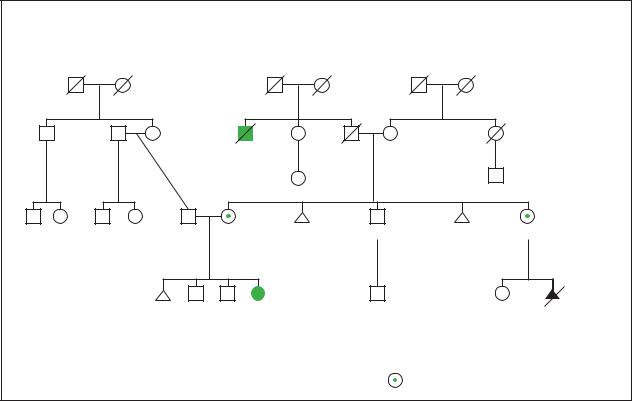
Downsyndrome |
|
Down Syndrome |
|
|
Family Robertsonian Translocation |
|
|||
|
|
|
|
|
Heart |
Down syndrome |
d.62y |
|
|
disease |
d.40y |
|
Heart |
2 |
|
|
3 |
attack |
|
2 |
|
|
|
|
29y |
31y |
|
36y |
34y |
4y |
2y |
1y |
9y |
3y |
|
46, XX, der(14;21)(q10;q10), +21 |
46, XX, der(14;21) |
||
|
(q10;q10), +21 |
|||
|
|
|
|
|
|
|
|
|
= 45, XX, t(14; 21)(q10; q10), –14,+21 |
(Gale Group)
amount of the fluid in which the baby is floating is withdrawn with a long, thin needle. CVS may be performed as early as 10 to 12 weeks into a pregnancy. Amniocentesis is generally not performed until at least the fifteenth week. Both CVS and amniocentesis carry small risks of miscarriage. Approximately 1% of women miscarry after undergoing CVS testing, while approximately one-half of one percent miscarry after undergoing amniocentesis. Both amniocentesis and CVS allow the baby’s own karyotype to be determined.
Approximately 75% of all babies diagnosed prenatally as affected with Down syndrome do not survive to term and spontaneously miscarry. In addition, these prenatal tests can only diagnose Down syndrome, not the severity of the symptoms that the unborn child will experience. For this reason, a couple might use this information to begin to prepare for the arrival of a baby with Down syndrome, to terminate the pregnancy, or in the case of miscarriage or termination, decide whether to consider adoption as an alternative.
Treatment and management
No treatment is available to cure Down syndrome. Treatment is directed at addressing the individual con-
cerns of a particular patient. For example, heart defects may require surgical repair, as will duodenal atresia and T-E fistula. Many Down syndrome patients will need to wear glasses to correct vision. Patients with hearing impairment benefit from hearing aids.
While some decades ago all children with Down syndrome were quickly placed into institutions for lifelong care, research shows very clearly that the best outlook for children with Down syndrome is a normal family life in their own home. This requires careful support and education of the parents and the siblings. It is a life-changing event to learn that a new baby has a permanent condition that will affect essentially all aspects of his or her development. Some community groups help families deal with the emotional effects of raising a child with Down syndrome. Schools are required to provide services to children with Down syndrome, sometimes in separate special education classrooms, and sometimes in regular classrooms (this is called mainstreaming or inclusion).
As of May 2000, the genetic sequence for chromosome 21 was fully determined, which opens the door to new approaches to the treatment of Down syndrome through the development of gene-specific therapies.
352 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |