The Nitro Group in Organic Synthesis
.pdf
|
|
|
|
7.1 R–Nu FROM R–NO2 |
191 |
|
|
NO2 |
SnCl4, CH2Cl2 |
|
SPh |
(7.34) |
|
PhS |
RT, 6 h |
|
|
|||
|
||||||
|
|
|
|
|||
|
Me |
|
||||
|
|
|
|
|
||
Me |
|
|
|
|||
|
|
70% (trans/cis = 2/98) |
|
|||
|
|
|
|
|
||
Ring expansion of cyclic ketones via nitro-aldol reaction of α-nitrosulfides followed by treatment with AlCl3 has been reported (Eq. 7.35).36
O |
PhS NO2 |
|
|
O |
|
|
PhS NO2 |
HO |
AlCl3 |
|
SPh |
|
|
||||
Me |
Me |
|
|||
–80 ºC |
0 ºC, 30 min |
|
|||
|
|
||||
|
|
|
|||
Ph |
Ph |
|
Ph |
Me (7.35) |
|
|
|
74% |
|||
|
|
|
|
||
|
|
|
|
|
|
7.1.3 Intramolecular Nucleophilic Substitution Reaction
Although the base-catalyzed addition of nitroalkanes to electron-deficient olefins has been extensively used in organic synthesis (see Michael addition Chapter 4), it is only recently that the reaction has been extended to the cyclopropanation reaction. In 1978, it was reported that the anion of nitromethane reacts with certain highly electron-deficient olefins to produce cyclopropanes in good yield (Eq. 7.36).36 More recently, this reaction has been extended to more general cyclopropanations, as shown in Eqs. 7.37 and 7.38, in which potassium salts of nitroalkanes are employed in DMSO as alkylidene transfer reagents.37–39
|
|
Me |
|
|
|
|
Me |
|
|
|
|
|
|
CO2Me |
|
|
|
CO2Me |
|
|
|
||
|
|
|
|
NaCH2NO2 |
|
|
|
|
|
|
|
MeOH |
|
|
|
|
|
|
|
100 ºC, 2 h |
|
|
|
MeO |
|
|
|
|
|
MeO |
(7.36) |
|
|
|
|
|
|
85% |
|
|
|
|
|
|
|
|
|
|
CO2Me |
|
NO2 |
DMSO |
|
|
CN |
|
+ |
|
K+ |
25 ºC, 6 h |
|
|
(7.37) |
|
CN |
|
|
|
CO2Me |
||
|
|
|
|
|
|
86% |
|
R |
CO2Me |
K |
+ |
DMSO |
R |
CN |
(7.38) |
|
+ |
|
|
||||
|
|
25 ºC |
H |
CO2Me |
|
||
H |
CN |
NO2 |
|
||||
Alumina-supported KF is an effective reagent for Michael addition of nitroalkanes to electron-deficient olefins. Subsequent cycloalkylations afford cyclopropanes.37 However, the reaction of α,β-unsaturated ketones with nitroalkanes in the presence of KF-Al2O3 in acetonitrile gives 4,5-dihydrofuranes (Eq. 7.39).40
|
O |
|
|
Ph |
CO2Me |
Ph |
Me |
|
KF-Al2O3 |
|
|
|
+ |
NO2 |
|
|
(7.39) |
|
MeCN |
||||
H |
CO2Me |
|
O |
||
|
80 ºC, 13 h |
90% |
|||
|
|
|
|
|
|
192 SUBSTITUTION AND ELIMINATION OF NO2 IN R–NO2
Yosikoshi reported the synthesis of furan derivatives by the reaction of 1,3-diketones with nitroalkenes, in which the Michael addition of the anions of 1,3-diketones and the subsequent
intramolecular displacement of the nitro group by enolate oxygen are involved as key steps (Eq. 7.40).42
O |
|
SPh |
|
O |
|
O |
|
|
|
|
|
||
|
|
KF |
|
|
NaIO4, CCl4 |
|
|
+ |
|
|
SPh |
||
|
NO2 |
xylene |
|
reflux |
||
|
|
O |
|
|||
O |
|
H |
reflux |
|
O |
|
|
81% |
|
91% |
|||
|
|
|
|
|
(7.40) In some cases, no cycloalkylation is observed by the reaction of nitromethane with electrondeficient olefins with cyano and methoxycarbonyl groups. The reaction affords new, highly functionalized cyclohexenes in the presence of catalytic amount of piperidine under solvent-free
conditions with focused microwave irradiation (Eq. 7.41).42
|
|
Ph |
NC CO2Me |
Ph |
CN |
NH2 |
|
NH |
|
|
+ |
MeNO2 |
|
|
(7.41) |
|
microwave |
O2N |
|||
H |
CO2Me |
|
CO2Me |
||
|
irradiation |
|
Ph |
||
|
|
|
|
|
|
|
|
|
|
|
70% |
The tandem Michael and cyclopropanation reaction of lithium enolates with nitroalkenes gives tricyclic ketones in one pot, as shown in Eq. 7.42.43
OLi |
|
|
Me |
Me |
|
|
|
|
|
|
|
|
THF/HMPA |
O |
Me |
|
|
+ |
|
(7.42) |
|||
NO2 –78 ºC |
65 ºC |
|
|
||
|
|
|
|||
Me
63%
7.1.4 Allylic Rearrangement
Allylic nitro compounds undergo [2.3]sigmatropic rearrangement to afford rearranged alcohols, as shown in Eq. 7.4346 and Eq. 7.44.47 Because the allylic nitro compounds used in these reactions are readily prepared either by the Henry reaction or the Michael addition, these reactions may be useful in organic synthesis.
OAc OAc |
OAc OAc |
|
|
|
NO2 |
tetralin |
|
|
|
|
|
(7.43) |
|
|
|
|
|
|
|
|
|
OH |
||
|
|
|
190 ºC |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
AcO |
|
|
|
|
|
AcO |
|
|
|
|
|
|
|
|
|
|
|
85% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
Me |
NO2 |
|
Me |
|
OH |
|
Me |
|
H |
|
|
|
|
|
Me |
+ |
|
|
Me |
(7.44) |
|
|
Me |
214 ºC |
|
|
|
|
||||
Ph |
Ph |
O |
|
Ph |
O |
|
||||
O Me |
Me |
|
Me |
|
||||||
|
|
|
||||||||
|
|
|
|
|
|
|||||
|
|
|
|
15% |
|
|
|
18% |
|
|
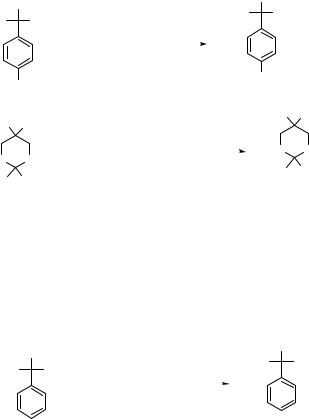
194 SUBSTITUTION AND ELIMINATION OF NO2 IN R–NO2 |
|
|
|||||||
|
|
H H O |
|
|
|
|
CN |
O |
|
CN |
O |
|
C |
NH2 HMPA |
|||||
|
Et C CH2CH2 |
C Me |
|||||||
Et C CH2CH2 C Me |
+ |
|
|||||||
|
|
|
25 ºC |
|
H |
|
|||
NO2 |
|
N |
|
|
|
||||
|
|
|
|
|
60% |
(7.48) |
|||
|
|
CH2Ph |
|
|
|
|
|
||
|
Me |
|
|
|
|
|
|
Me |
|
|
|
|
|
|
Me |
H |
|
||
Me |
NO2 |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
||
|
+ |
NaTeH |
C2H5OH |
|
|
(7.49) |
|||
|
|
25 ºC |
|
|
|||||
|
|
|
|
|
|
||||
SO2Ph
SO2Ph
82%
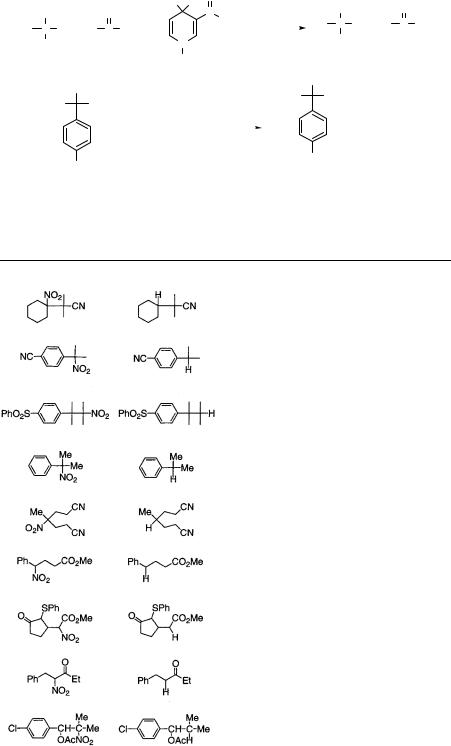
Table 7.1. Denitration of nitro compounds with MeSNa or Bu3SnH
R-NO2 |
Product |
Reagent |
Yield (%) |
Ref. |
|
|
MeSNa |
95 |
49 |
|
|
DMSO, 3 h |
|
|
|
|
Bu3SnH, (PhCO)2O2 |
95 |
52 |
|
|
benzene, 18 h |
|
|
|
|
MeSNa |
82 |
49 |
|
|
HMPA, 16 h |
|
|
|
|
Bu3SnH, (PhCO)2O2 |
90 |
52 |
|
|
benzene, 18 h |
|
|
|
|
MeSNa |
83 |
49 |
|
|
DMF, 8 h |
|
|
|
|
Bu3SnH, (PhCO)2O2 |
85 |
52 |
|
|
benzene, 18 h |
|
|
|
|
Bu3SnH, AIBN |
92 |
51 |
|
|
benzene (80 °C) |
|
|
|
|
1.5 h |
|
|
|
|
Bu3SnH, AIBN |
92 |
51 |
|
|
benzene, 1.5 h |
|
|
|
|
Bu3SnH, AIBN |
64 |
51 |
|
|
benzene, 1.5 h |
|
|
|
|
Bu3SnH, AIBN |
75 |
51 |
|
|
benzene, 1.5 h |
|
|
|
|
Bu3SnH, AIBN |
78 |
51 |
|
|
benzene, 1.5 h |
|
|
|
|
Bu3SnH, AIBN |
83 |
51 |
|
|
benzene, 1.5 h |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7.2 |
R–H FROM R–NO2 |
195 |
|||||||||||
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
H |
|
||||||
|
|
|
|
|
|
|
Na2S2O4 |
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
+ Et3SiH |
Ph |
|
C |
|
C |
|
Me |
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
Ph |
|
C |
|
C |
|
Me |
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
HMPA-H2O |
|
|
O H |
(7.50) |
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
O NO2 |
|||||||||||||||||||||
|
|
|
25 ºC |
70% |
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
Tanner and coworkers investigated the denitrohydrogenation reaction and proposed an electron-transfer mechanism, which is analogous to the denitration mechanism using MeSNa.52. These nitro compounds are all tertiary and rather special. The results are summarized in Table 7.1. On the other hand, Ono and coworkers have selected the more general nitro compounds which are formed by the conventional reactions such as the Henry reaction or the Michael reaction, for denitration with tin radical. They have found that such nitro groups are cleanly denitrated with Bu3SnH. Now, the sequence of the Henry reaction or Michael reaction and denitration provides a very useful strategy for organic synthesis. The regio-controlled carboncarbon bond formation and functional selective denitration are very attractive, as exemplified in Eqs. 7.51–7.53. The nitro group is selectively removed from the compounds, which contain other functional groups such as Cl, OH, C?O, CN, SO, and SO2 groups. The sequence of the Michael addition of nitroalkanes and denitration provides a new and general method for conjugate additions of primary and secondary alkyl groups.56
Ph |
NO2 |
|
|
|
|
|
|
|
|
|
|
|
|
NO2 |
|
Bu SnH, AIBN |
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
Ph |
|
|
|
CO2Me |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ph |
|
|
|
|
|
|
|
|
CO2Me |
|
benzene, 80 ºC |
|
|
|
|||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||
|
|
|
CO2Me |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(7.51) |
|||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
O |
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
O |
|
|
TMG |
|
|
|
|
|
DBU |
|
|
|
|
Bu3SnH |
|
|
|
|
|||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
NO2 |
|
|
|
|
|
|
NO2 |
|
|
|
SOPh |
|||||||||||||||||||||||||||||||
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SOPh |
|
|
AIBN |
|
|
||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SOPh |
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
NO2 |
|
|
|
|
72% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(7.52) |
|||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
72% |
|
|
|
|
|
|
|
|
|
70% |
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|||||||||
CH3NO2 |
|
|
TMG |
|
|
|
|
NO2 |
|
|
|
|
|
|
TMG |
|
|
|
|
|
|
|
NO2 |
|
|
CO2Me |
|||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CO2Me |
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
48% |
|
|
|
|
|
|
|
|
|
|
|
|
|
74% |
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
TMG |
|
O2N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Bu3SnH |
|
|
|
|
|
|
|
|
|
SO2Ph |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CO2Me |
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CO Me |
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
AIBN |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
SO2Ph |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SO2Ph |
|
|
|
|
|
|
O |
|
|
(7.53) |
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
73% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
85% |
|
|||||
Synthetic chemists have long lauded the radical reactivity profile of Bu3SnH, but bemoaned its separation of organic tin compounds from the product and toxicity problems. To overcome these drawbacks of tin reagents, several alternatives to Bu3SnH have been devised: such as polymeric tin hydrides,57 acid soluble tin hydrides,58 water-soluble tin hydrides,59 and tris(2- (perfluorohexyl)ethyl)tin hydride.60 The last reagent is used in C6H5CF3. Tin reagents and the denitrated products are separated by liquid-liquid extraction using perfluoromethylcyclohexane and CH2Cl2. This is a new technique for the purification and separation of organic compounds using organic fluorine compounds (Eq. 7.54).
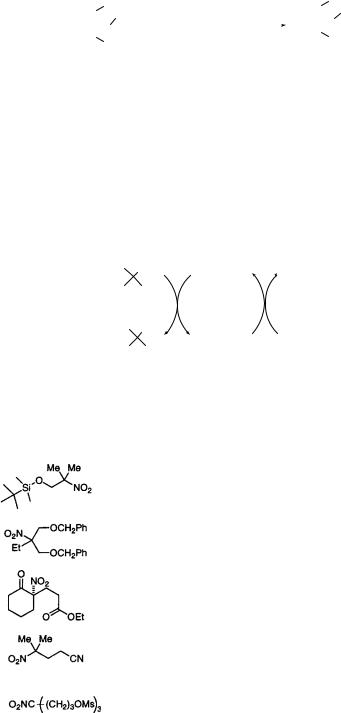
196 SUBSTITUTION AND ELIMINATION OF NO2 IN R–NO2 |
|
|
|
|
||||||
|
|
OSO2Me |
|
|
|
|
OSO2Me |
|||
|
|
|
|
|
|
|
OSO2Me |
|||
|
|
|
OSO2Me (C |
F |
CH CH ) SnH |
|
|
|
||
|
|
|
H |
|
|
|||||
O2N |
|
6 |
|
13 2 2 3 |
(7.54) |
|||||
|
|
|
|
|
|
|
|
|||
|
|
|
AIBN, C6H5CF3 |
|
|
|||||
|
|
|
|
|
|
OSO2Me |
||||
|
|
|
|
|
||||||
|
|
OSO2Me |
|
reflux, 24 h |
|
|
88% |
|||
|
|
|
|
|
|
|
|
|
||
Silicon hydrides, in particular (Me3Si)3SiH, can serve as substituents for Bu3SnH in a number of radical-mediated processes.61 However, silicon hydrides cannot promote the reduction of tertiary nitroalkanes to alkanes.62 In 1998, Fu reported the catalytic cycle for the Bu3SnH-cata- lyzed reduction of nitroalkanes to alkanes, using 10% Bu3SnH as the catalyst and PhSiH3 as the reducing agent (see Eq. 7.55 and Scheme 7.5). The new catalytic reaction proceeds with efficiency, comparable to the stoichiometric Bu3SnH method (Table 7.2).63 Like the stoichiometric method, the catalytic reaction is effective for the reduction of tertiary nitroalkanes and activated secondary nitroalkanes and is compatible with functionality, such as ethers, acetals, ketones, esters, nitriles, and mesylates. The conversion of nitroalkanes to alkanes is currently most often accomplished with stoichiometric Bu3SnH, but the environmentally friendlier Bu3SnH-catalyzed variant may become the method of choice for effecting these important transformations.
|
NO2 |
Bu3SnH |
PhSiONO |
|
|
||
|
H |
Bu3SnONO |
PhSiH3 |
|
|
||
|
Scheme 7.5. Bu3SnH-catalyzed reduction of nitroalkanes to alkanes |
||
Table 7.2. Bu3SnH-catalyzed reduction of nitroalkane to alkanes |
|||
|
|
|
|
|
|
|
Yield (%) |
Substrate |
|
Catalyzeda |
Stoichb |
|
|
76 |
77 |
71 |
74 |
75 |
78 |
70 |
67 |
61 |
58 |
|
|
=10% Bu!SnH, PhSiH! (0.5 equiv), ACHN (0.2 equiv), toluene, 110 °C, 5 h.
>Bu!SnH (1.5 equiv), ACHN (0.2 equiv), toluene, 110 °C, 5 h.

|
|
|
|
7.2 |
R–H FROM R–NO2 197 |
R |
NO2 |
10% Bu3SnH |
|
R |
H |
R |
R |
0.5 equiv PhSiH3 |
|
R |
(7.55) |
∆ |
R |
||||
|
|
initiator, toluene, |
|
|
Although the nitro compounds, which can be denitrated, are mostly tertiary ones, the radial denitration using Bu3SnH can also be applied to secondary nitro compounds. The nitro groups at benzylic and allylic positions are readily denitrated with Bu3SnH by the procedure that is used for the denitration of tertiary nitro compounds (60–80% yield). The secondary nitro groups of α-nitro ketones or α-nitro esters are also cleanly replaced by hydrogen on treatment with Bu3SnH (Eq. 7.56).56
|
SPh |
SPh |
CO2Me |
||||
|
|
|
|||||
|
|
|
CO2Me Bu3SnH, AIBN |
O |
|
|
|
|
|
|
|
||||
O |
|
|
|
(7.56) |
|||
benzene |
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
NO2 |
|
|
|
|
|
|
|
|
|
75% |
|
|
The unactivated secondary nitro groups are rather difficult to be replaced by hydrogen. Rather drastic conditions using a large excess of Bu3SnH in refluxing toluene are required for the denitration of simple secondary nitro groups.64 Although the yield is moderate (40–60%), the direct removal of the secondary nitro groups is synthetically useful. Some examples are presented in Eq. 7.57,64 Eq. 7.58,65 and Eq. 7.59.66 The high yield reported in Eq. 7.59 is exceptional. Any other methods without using Bu3SnH fail in denitrating secondary nitro groups.
O |
Me |
|
O Me |
|
|
O |
Bu3SnH, AIBN |
|
O |
|
(7.57) |
|
toluene |
|
|
|
|
|
|
|
|
|
|
|
NO2 |
|
48% |
|
|
|
O |
|
|
O |
|
|
|
|
|
|
|
|
NO2 |
CO |
Me |
|
CO2Me |
|
|
2 |
|
|
|
|
|
|
Bu3SnH |
|
|
Si O |
|
Si |
O |
|
|
|
|
|
AIBN |
O |
|
|
O Si |
|
toluene |
(7.58) |
|
|
|
|
Si |
||
|
|
|
|
30% |
|
OAc |
|
|
OAc |
|
|
|
CO2Me |
Bu3SnH, AIBN |
CO2Me |
(7.59) |
|
|
NO2 |
benzene |
|
|
|
|
|
|
93% |
|
|
Primary nitro groups are much more difficult to be replaced by hydrogen. Indirect methods are required for the conversion of a nitromethyl group to a methyl group. The Nef reaction and subsequent reduction via the hydroxymethyl group and radical deoxygenation may be the method of choice for this conversion (see Scheme 7.2). However, in 1995, Witczak and Li reported the removal of primary nitro groups by reaction with Bu3SnH in the presence of 1,1′-azobis(cyclohexanecarbonitrile)(ABCN) (Eq. 7.60).67 This is the only report of the radical