

Friesner R.A. (ed.) - Advances in chemical physics, computational methods for protein folding (2002)(en)
.pdf416 |
john l. klepeis et al. |
2.Approach
A systematic approach is presented below for the structure prediction of an antigen binding site based on the crystallographic data of the HLA-DR1 molecule [171]. The approach examines each of the binding sites separately and involves the following steps:
1. The binding sites of HLA-DR1 molecule are evaluated. All amino acids
¼ ˚
within a radius of R 5:0 A of the atoms of the binding amino acid in the crystallographic studies [171] are identified as shown in Table XXXVI. The Program for Pocket Definition, as described in Ref. 234 and Section V.C.3, constructs these pockets through the selection of all residues that are within a radius R of the atoms of the crystallographic binder.
2.The amino acid substitutions between HLA-DR1 and the HLA-II molecule (e.g., HLA-DR3, I-Ek) are identified and are shown in Table XXXVII. Note that pocket 1 of HLA-DR1 requires only one substitution (Gly ! Val in position b86) to give pocket 1 of HLA-DR3. Pockets 4, 6, and 7 involve three substitutions, whereas pocket 9 features only one substitution, in the representation of the corresponding pockets of HLADR3. Note also that all pockets of HLA-DR1 require three or four substitutions in order to give the corresponding pockets of I-Ek.
3.For each one of the substituted residues, the intraand intermolecular energy interactions are modeled. Specifically, the electrostatic, nonbonded, torsional, and hydrogen bonding contributions [38] are considered for each
TABLE XXXVI
¼ ˚
HLA-DR1 Pocket Compositions for R 5:0 A
————————————————————————————————————————
1 |
4 |
6 |
7 |
9 |
|
|
|
|
|
phea24 |
glna09 |
glua11 |
vala65 |
asna69 |
ilea31 |
glua11 |
asna62 |
asna69 |
leua70 |
phea32 |
asna62 |
vala65 |
glub28 |
ilea72 |
trpa43 |
pheb13 |
aspa66 |
tyrb47 |
meta73 |
alaa52 |
leub26 |
leub11 |
trpb61 |
arga76 |
sera53 |
glnb70 |
pheb13 |
leub67 |
trpb09 |
phea54 |
argb71 |
argb71 |
argb71 |
aspb57 |
glua55 |
alab74 |
|
|
tyrb60 |
asnb82 |
tyrb78 |
|
|
trpb61 |
valb85 |
|
|
|
|
glyb86 |
|
|
|
|
pheb89 |
|
|
|
|
thrb90 |
|
|
|
|
|
|
|
|
|

deterministic global optimization and ab initio approaches 417
TABLE XXXVII
Substitutions for HLA-DR3 and I-Ek Binding Sites
Substitutions for HLA-DR3 |
Substitutions for I-Ek |
|
|
|
|
|
b86: Gly !Val |
b85: Val ! Ile |
1 |
b86: Gly ! Phe |
|
|
|
b90: Thr ! Leu |
|
b13: Phe ! Ser |
b13: Phe ! Ser |
4 |
b26: Leu ! Tyr |
b74: Ala ! Glu |
|
b74: Ala ! Arg |
b78: Tyr ! Val |
|
|
b71: Arg ! Lys |
|
b11: Leu ! Ser |
b11: Leu ! Ser |
6 |
b13: Phe ! Ser |
b13: Phe ! Cys |
|
b71: Arg ! Lys |
b71: Arg ! Lys |
|
b28: Glu ! Asp |
b28: Glu ! Val |
7 |
b47: Tyr ! Phe |
b47: Tyr ! Phe |
|
b71: Arg ! Lys |
b67: Leu ! Phe |
|
|
b71: Arg ! Lys |
|
|
a72: Ile ! Val |
9 |
b9: Trp ! Glu |
b9: Trp ! Glu |
|
|
b60: Tyr ! Asn |
substituted residue, as well as the interactions of the substituted residues with the rest of the amino acids that constitute the examined binding site. The solvation energy also is considered through solvent-accessible areas [52,238] as explained in Section III.A.2. The dihedral angles that define the three-dimensional structure of the substituted residues are considered explicitly as variables. The relative position of each amino acid also must be determined, and this is done through the determination of each amino acid’s translation vector and Euler angles. Lower and upper bounds are considered for the N and C0 coordinates of the substituted amino acids, based on the available crystallographic data [171,236,237].
4.Having the mathematical model that includes the intraand intermolecular energetic interactions and the solvation energy, and which has as variables the dihedral angles of the substituted amino acids as well as their translation vectors and Euler angles, we minimize the total potential energy by employing the aBB deterministic global optimization approach [14–18] as described in later sections below.
5.The resulting global minimum energy conformer provides information on the predicted (x; y; z) coordinates of the atoms of the substituted residues. Structure verification is made by superposition of all atoms of the predicted structure and the ones derived from crystallographic data. The superposition is based on the global minimization of the root mean square
deterministic global optimization and ab initio approaches 419
The additional constraints (105–110) represent the bounds on the N and C0 coordinates and express the binding of the specific residue with the rest of the pocket [234], because the substituted residue is part of a longer polypeptide and consequently is not allowed to rotate freely. Because the C0 coordinates can be evaluated as functions of the independent variables, the restrictions on the position of C0 are implemented by the incorporation of a penalty function, P, in the objective function:
P¼ bfhCx0l Cx0 i þ hCx0 Cx0ui
þhCy0l Cy0 i þ hCy0 Cy0ui
þhCz0l Cz0 i þ hCz0 Cz0uig
The h i function is defined as follows: hAi equals A if A is greater than zero; otherwise hAi equals zero. Thus, any coordinate value beyond the specified bounds is multiplied by the penalty parameter b and added to the potential energy. Consequently, the minimization of the objective function eliminates solutions in which the C0 position falls outside the specified bounds.
The global optimization formulation is then as follows:
L |
¼ |
ETotal |
þ |
|
X |
fmL |
|
fm |
|
fmU |
|
fm |
þ |
|
|
cm |
|
|
cm |
|
||||||||||
|
|
|
a |
M |
|
|
|
|
|
|
cmL |
|
|
|
cmU |
|
|
|||||||||||||
|
|
|
|
|
|
|
m¼1 |
|
|
|
|
|
|
K |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
wkmU wkm |
|
|
|
|||||||
|
|
þ omL om omU om þ X wkmL wkm |
|
|
|
|||||||||||||||||||||||||
|
|
þ NxmL Nxm NxmU Nxm k¼1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
þ NymL Nym NymU Nym þ NzmL Nzm NzmU Nzm |
|
|
|
|||||||||||||||||||||||||
|
|
þ e1mL e1m e1mU e1m |
þ |
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
þ 2 |
|
|
2 |
2 |
|
|
2 |
|
3 |
|
3 |
3 |
|
|
|
|
|
|||||||||||
|
|
emL |
|
|
em |
emU |
|
em |
|
|
emL |
|
|
em |
|
emU |
|
em |
|
|
|
|
|
|||||||
where a is a nonnegative parameter that must be greater or equal to the negative one-half of the minimum eigenvalue of the Hessian of ETotal in the considered domain defined by the lower and upper bounds (i.e., xL ¼ p; xU ¼ p) of the dihedral angles, translation variables, and Euler angles. This parameter can be rigorously calculated based on the techniques introduced by Adjiman and Floudas [14] and Adjiman et al. [16,17].
For the problem of determining the binding sites of the unknown HLA molecules, the global variable set includes the f, c, and wk variables. All of the dihedral angles of the substituted residues, as well as their translation vectors
420 |
john l. klepeis et al. |
and Euler angles, are continuous variables in the problem and are treated as local variables.
4.Deterministic Global Optimization
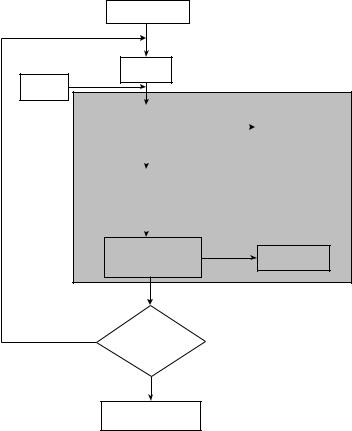
The implementation of the approach involves the connection of the conformational energy program PACK [74], which allows the evaluation of all energy interactions when more than one protein chain is involved in the system, to the deterministic global optimization framework aBB. PACK evaluates all energy components through repeated calls to the ECEPP/3 potential energy function program. The local optimization solver NPSOL is used for the minimization of the overall potential energy provided by PACK and for the minimization of the convexified potential function (L) provided by aBB. MSEED [52], the program for the determination of solvation energy, is interfaced to aBB to allow the consideration of the solvation energy at the local minima. The algorithmic procedure is represented graphically in Fig. 53.
The implementation of the proposed approach is illustrated in Fig. 54 and involves the following steps:
1.The Program for Pocket Definition (PPD) uses the input files residue.pdb and pocket.pdb to generate the coordinates of the residues involved in the considered pocket.
2.The program ARAS is used to determine the translation vectors, Euler angles and dihedral angles of the residues in the pocket given their (x; y; z) coordinates. This information and the initial values for the translation vector, Euler angles, and dihedral angles of the substituted residues are incorporated within the input file name.input.
3.The program prePACK utilizes the residue.data file (a set of initial atomic
coordinates that are based on fixed bond lengths, fixed bond angles, and each variable dihedral angle initially set to 180 ), the mol.in file for each
one of the amino acids involved in the pocket, and the prep.name.abb file (which specifies the fixed and substituted residues) to create a name.date file. The name.date file is the standard input for the potential function program, PACK.
4.The global optimization program aBB requires a name.abb file that
defines the optimization problem, including the variable bounds. aBB also uses the name.input file and the name.bounds file, which contains the C0 bounds used to evaluate the coordinates of C0 as a function of the
independent variables.
5.The program PACK uses the name.date file and is connected with ECEPP/3 in order to evaluate the potential function, which is minimized by the local optimization solver NPSOL.




 residue.pdb
residue.pdb 
deterministic global optimization and ab initio approaches 425
all the atoms involved in the pocket. The relative cRMSD for the whole binding site is 0.0425, which corresponds to 4.25% difference of the predicted Cartesian coordinates of the binding site and the crystallographic data. The cRMSD
a ˚
difference based on the carbons is 0.55 A. The cRMSD for the substituted
˚
residue (Val) is 1.584 A and the relative-cRMSD is 0.04601, which indicates a 4.6% difference between the predicted valine and the valine determined based on the crystallographic data of the HLA-DR3 molecule [236].
To generate pocket 4 of HLA-DR3, three substitutions are made on the composition of the pockets of HLA-DR1 at the positions b13: Phe ! Ser; b26: Leu ! Tyr; and b74: Ala ! Arg. The cRMSD difference for all the
˚
residues in the pocket is 1.11 A, and the overall relative difference of the predicted pocket compared to the crystallographic data is 2.08%. The cRMSD
|
˚ |
|
difference based on the a carbons is 0.49 A. The cRMSD for each one of the |
||
˚ |
˚ |
˚ |
substituted residues is 1.67 A for Ser, 0.83 A for Tyr, and 1.46 A for Arg and correspond to relative differences of 3.2%, 1.2% and 2.3%, respectively.
For pocket 6 of HLA-DR3, the substitutions are at positions b11: Leu to Ser; b13: Phe to Ser; and b71: Arg to Lys. The cRMSD difference for this pocket is
˚ |
|
1.22 A based on all atom deviations, which corresponds to a relative cRMSD of |
|
|
˚ |
4.9%. The cRMSD difference based on the a carbons is 0.61 A. The individual |
|
˚ |
˚ |
cRMSD for Serb11 is 1.26 A, for Serb13 it is 1.62 A, and for Lys b71 it is
˚
1.82 A, which correspond to relative predictive errors of 7.4%, 3.7% and 3.2%, respectively.
For pocket 7 of HLA-DR3, three substitutions are made at the positions b28: Glu to Asp; b47: Tyr to Phe, and b71: Arg to Lys. The cRMSD difference for
˚ |
|
this pocket is 1.94 A based on all atom deviations, which corresponds to a |
|
|
˚ |
4.69% deviation. The cRMSD difference based on the a carbons is 0.71 A. The |
|
˚ |
˚ |
cRMSD for each one of the substituted residues are 1.08 A for Phe, 3.08 A for
˚
Asp, and 3.4 A for Arg and correspond to relative differences of 1.4%, 5.1%, and 4.7%, respectively.
Finally, for pocket 9 only one substitution is needed, namely Trp to Glu in position b9 to obtain pocket 9 of HLA-DR3 from pocket 9 of HLA-DR1. The
|
|
˚ |
resulting pocket is found to have a cRMSD difference of 1.03 A based on all |
||
˚ |
a |
atoms. The relative cRMSD based on all |
atoms and 0.56 A based on the C |
|
|
atom deviations is 37.2%. Considering only the substituted residue, the cRMSD
˚
is 1.67 A. The large predictive deviation in this pocket is due to the large inherent deviation between the HLA-DR1 and the HLA-DR3 crystallographic
˚
data. This cRMSD difference for pocket 9 is 1.05 A, which corresponds to an inherent relative cRMSD of 20.7%.
The results of our prediction approach for all the pockets are summarized in Table XXXVIII. Note that the percentage predictive error is less than 5%, except for pocket 9 where the large inherent deviation between the two crystals prohibits a more accurate prediction.