


227
9
Nanotechnologies for Cellular and Molecular Imaging by MRI
Patrick M. Winter, Shelton D. Caruthers, Samuel A. Wickline, and Gregory M. Lanza
9.1
Introduction
Developments in cellular and molecular biology are extending the horizons of medical imaging from gross anatomic description towards delineation of cellular and biochemical signaling processes. The emerging fields of cellular and molecular imaging aim to diagnose disease noninvasively on the basis of in vivo detection and characterization of complex pathological processes, such as induction of inflammation or angiogenesis. Techniques have been developed recently to achieve molecular and cellular imaging with most imaging modalities, including nuclear [1, 2], optical [2, 3], ultrasound [4], and MRI [5, 6]. This chapter focuses on two techniques developed for detection of atherosclerosis by MRI: cellular imaging of macrophages associated with inflammatory lesions and molecular imaging of angiogenesis that is induced in developing vascular plaques. A selection of the contrast agent formulation and imaging methods will be discussed, as well as the optimization of these techniques for successful cellular and molecular imaging in vivo.
Because individual cells and biochemical molecules are too small to be imaged directly with noninvasive techniques, specific and sensitive site-targeted contrast agents are needed to visualize the epitopes of interest. Historically, nuclear imaging has dominated the fields of cellular and molecular imaging due to the extremely high sensitivity and the relative simplicity of conjugating radioactive tags onto biochemical molecules. For instance, fluorodeoxyglucose (FDG) activity can be imaged with PET scanners to characterize such diverse disease states as tumor metabolism [7] and mental disorders [8]. Radiolabeled somatostatin analogs have also been developed to allow receptor imaging for detection of neuroendocrine tumors [9]. In addition, cellular apoptosis can be detected with technetium-labeled annexin-V, which binds to phosphatidyl serine expressed on the surface of apoptotic cells [10]. Nuclear imaging agents have also been designed to detect gene transfection by imaging the resultant protein products [11].
While nuclear imaging predominated in the early development of cellular and molecular imaging, other modalities, such as optical, magnetic resonance imaging (MRI), and ultrasound, have been pursued to take advantage of the increased resolution, signal-to-noise ratio, and contrast associated with these techniques. The inher-
Nanofabrication Towards Biomedical Applications. C. S. S. R. Kumar, J. Hormes, C. Leuschner (Eds.) Copyright 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
ISBN 3-527-31115-7

228 9 Nanotechnologies for Cellular and Molecular Imaging by MRI
ently lower sensitivity of these methods, however, often requires the use of complex targeted imaging agents in order to incorporate sufficient signal generation motifs. For instance, targeted perfluorocarbon nanoparticles, the first reported molecular imaging agent for ultrasound applications, can significantly increase the reflectivity of thrombi [12–14]. Many other acoustic contrast agents have been developed for similar targeting applications, such as endothelial integrins, tissue factor and thrombi [15–21].
MRI is emerging as a particularly advantageous modality for molecular and cellular imaging given its high spatial resolution and the opportunity to extract both anatomical and physiological information simultaneously [22, 23]. The majority of targeted MRI agents consist of nanoparticles in order to carry sufficient amounts of paramagnetic or superparamagnetic material for in vivo visualization [24]. In cardiovascular disease, novel targeted contrast agents are being developed to detect unstable lesions through identification of fibrin deposited within plaque microfissures [25–28], adhesion or thrombogenic molecules expressed on endothelium of vulnerable plaques [15, 16, 29–31], matrix metalloproteinases in the cores of progressing lesions [32], or angiogenic activity (i.e., expanding vasa vasorum) supporting plaque development [33].
9.2
Cardiovascular Disease
Atherosclerosis, like many chronic human diseases including cancer and diabetes, develops slowly over many years. Unlike most other diseases, however, atherosclerosis is often diagnosed only after an acute, fatal event. Of the 720,000 cardiac deaths per year in America, approximately 60% are “sudden deaths,” occurring without any advanced warning of pathology [34]. Atherosclerosis starts as “fatty streak” lesions in utero [35], and can produce plaques prone to rupture by the early teens [36]. Rupture of unstable atherosclerotic plaques can lead to thrombosis, vascular occlusion, and subsequent myocardial infarction or stroke [37–39].
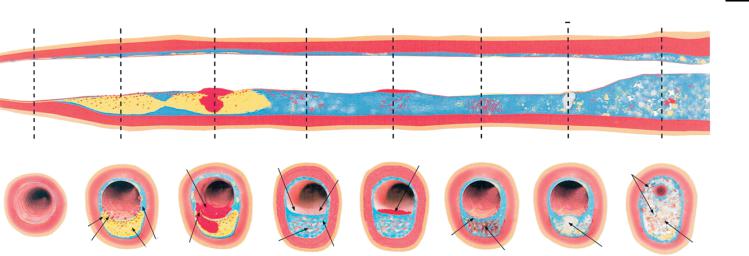
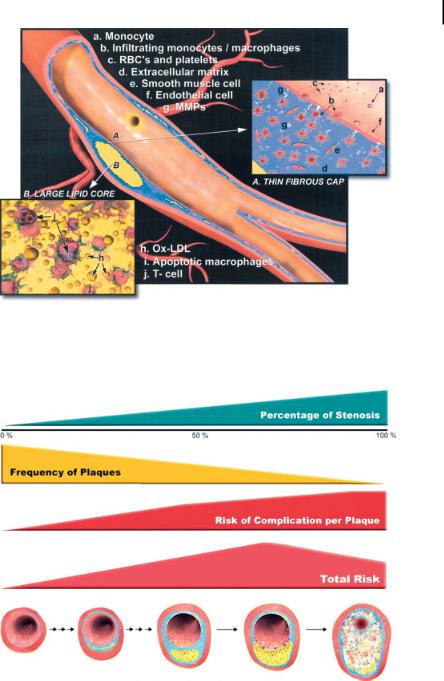
Atherosclerotic plaques grow in discrete stages consisting of repeated episodes of rupture, thrombosis, and healing (Fig. 9.1) [40], leading inevitably to a final event causing complete vascular obstruction [41]. A “vulnerable plaque” is defined as a lesion exhibiting physical and biochemical properties that predispose it to rupture and thrombosis [40]. Typically, these plaques consist of a large lipid core covered by a thin fibrous cap harboring relatively few smooth muscle cells, a population of activated macrophages, and abundant angiogenesis (Fig. 9.2) [40]. The large lipid core is known to destabilize lesions by directing mechanical stress to the fragile shoulderregions of the plaque [42]. Exposure of the lipid core, even through a small localized rupture, can induce the clotting cascade initiating with tissue factor [43]. The lack of smooth muscle cells also weakens the cap [44], facilitating plaque rupture. The accumulation of macrophages as well as other inflammatory cells, which usually secrete high levels of metalloproteinases (MMP), can undermine the fibrous cap, potentially exposing the thrombotic lipid core [45, 46]. Up-regulation of angiogenesis can lead

9.3 Cellular and Molecular Imaging 229
to erosion of the extracellular matrix and replacement with physically fragile neovascular beds, weakening the fibrous cap and promoting plaque rupture [47–49].
In current clinical practice, the diagnosis and characterization of most atherosclerotic plaques is achieved with invasive x-ray catheterization. Highly stenotic lesions, typically with 50% or greater narrowing of the luminal diameter, are identified for immediate therapeutic intervention, while less stenotic lesions are generally deemed clinically insignificant. Ironically, these plaques are often the very lesions prone to rupture leading to heart attack and stroke [50, 51]. Stress tests employing nuclear or ultrasound imaging are also extensively used for detection of flow-limit- ing vascular obstructions during a range of metabolic challenges. Similar to invasive x-ray angiography, however, these techniques are insensitive to plaques with lowgrade stenosis, which are those prone to rupture. Because rupturing atherosclerotic plaques are frequently manifest in arteries with only modest (40–60%) stenosis [52], they remain diagnostically elusive with routine clinical imaging techniques. If recognized and localized, a window of opportunity extending from days to months exists to intervene and/or stabilize plaques medically before more serious clinical sequelae ensue [41]. The principal difficulty revolves around the fact that atherosclerosis produces numerous plaques throughout the vascular system and it is not feasible to treat each lesion individually. Of all the <50% stenotic lesions, only a small fraction may rupture and lead to clinical events (Fig. 9.3) [40]. A significant new opportunity exists for delineating which lesions are prone to rupture and applying therapeutic treatments to only the areas of pathology that pose an immediate danger.
Molecular imaging techniques are being pursued with the goals of both primary and secondary prevention of atherosclerosis. Primary prevention focuses on the identification of patients at the earliest stages of atherosclerotic development, leading to the application of lifestyle and/or pharmacological therapies to prevent further plaque development and perhaps even significant regression of existing unstable or vulnerable lesions. Secondary prevention aims to detect plaques that are prone to impending (e.g., days to weeks) rupture and thromboembolism in patients with advanced atherosclerotic disease, allowing aggressive intervention concentrated on only the most dangerous areas of pathology.
9.3
Cellular and Molecular Imaging
To achieve effective cellular and molecular imaging with MRI, targeted contrast agents must be designed to accomplish long circulating half-life, sensitive, and selective binding to the epitope of interest, prominent contrast-to-noise enhancement, acceptable toxicity, ease of clinical use, and applicability with standard commercially available imaging systems.
Discoveries in molecular biology have elucidated cellular and molecular markers for a wide variety of diseases. These signatures may serve as targets for contrast agents to provide sensitive and specific imaging of the earliest manifestations of
Different Types of Vulnerable Plaque
|
A |
|
B |
Dysfunctional C |
|
D Non-Occlusive |
E |
F |
|
|
G |
||
|
Non-Occlusive |
Endothelium |
|
Mural Thrombus/ |
|
Extensive |
|
||||||
|
|
|
Fibrin |
|
|
|
|
|
|||||
|
|
|
Clot |
|
|
|
|
|
|
Calcification |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Platelets |
|
|
|
|
|
|
|
|
Macro- |
|
|
|
|
|
|
|
|
|
|
|
|
|
phage |
|
|
|
|
|
|
Intact |
|
|
|
|
|
|
|
Collagen |
|
|
|
|
|
|
|
|
|
||
|
Thin Cap |
Smooth |
|
|
|
Cap |
|
|
|
|
Old |
||
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
Calcium |
|
|||
|
|
|
Ruptured |
Muscle Cells |
Proteoglycans |
|
|
|
Thrombus |
||||
|
Large Lipid |
|
|
|
|
Leaking Vasa Vasorum/ |
Nodule |
|
|||||
|
Cap |
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|||
|
Core |
|
|
|
|
|
|
|
Angiogenesis |
|
|
|
|
Normal |
Rupture-Prone |
|
Ruptured/Healing |
Erosion-Prone |
Eroded |
Vulnerable Plaque |
Vulnerable Plaque |
Critically Stenotic |
|||||
|
Vulnerable |
|
Vulnerable |
Vulnerable |
Vulnerable |
with Intra-Plaque |
with Calcified |
Vulnerable Plaque |
|||||
|
Plaque |
|
Plaque |
|
Plaque |
|
Plaque |
|
Hemorrhage |
Nodule |
|
|
|
Figure 9.1. Atherosclerotic plaques develop in discrete steps, leading to a wide range of lesion types in a single patient. Although all these plaque types may lead to thromboembolism, only the most advanced plaques produce luminal stenosis. From left to right: (A) rupture-prone plaque with large lipid core and thin fibrous cap infiltrated by macrophages; (B) ruptured plaque with subocclusive thrombus and early organization; (C) erosion-prone plaque with
proteoglycan matrix in a smooth muscle cellrich plaque; (D) eroded plaque with subocclusive thrombus; (E) intraplaque hemorrhage secondary to leaking vasa vasorum; (F) calcific nodule protruding into the vessel lumen; (G) chronically stenotic plaque with severe calcification, old thrombus, and eccentric lumen. (Reprinted with permission from Ref. [40].)
MRI by Imaging Molecular and Cellular for Nanotechnologies 9 230
9.3 Cellular and Molecular Imaging 231
Figure 9.2. The most common type of nonstenotic vulnerable plaque. The thin fibrous cap, extensive macrophage infiltration, depletion of smooth muscle cells, and large lipid core all contribute to the plaque’s propensity for rupture. (Reprinted with permission from Ref. [40].)
Figure 9.3. Correlation between frequency of plaques, degree of stenosis, and risk of complication per plaque as a function of plaque progression. Although the average absolute risk of severely stenotic plaques may be higher than
the average absolute risk of mildly stenotic plaques, there are more plaques with mild stenoses than plaques with severe stenoses. (Reprinted with permission from Ref. [40].)

232 9 Nanotechnologies for Cellular and Molecular Imaging by MRI
pathology. A variety of different types of ligands can be utilized for targeting, including antibodies, peptides, polysaccharides, aptamers, etc. The emerging fields of cellular and molecular imaging utilize nanotechnologies to develop targeted contrast agents for noninvasive detection of the molecular signatures of disease. Unlike blood pool agents, a targeted contrast agent produces signal enhancement from pathological tissue that would otherwise be difficult to distinguish from surrounding normal tissue. Cellular and molecular imaging techniques have been developed for most clinical imaging modalities, including nuclear, MRI, ultrasound, computed tomographic, and optical. MRI, however, may enjoy several advantages over the other modalities, such as high resolution, high anatomical contrast, high signal-to- noise ratio, widespread clinical availability, and lack of ionizing radiation.
These imaging techniques do not depend upon the native tissue contrast or resolution associated with the underlying imaging modality. Instead, they enhance the conspicuity of very small areas of disease by directly targeting the cellular or molecular processes responsible for the pathological transformation of tissues. Moreover, by targeting the underlying molecular processes, these imaging techniques can provide unique information to characterize the complex metabolic state of the disease. This information could provide crucial insight for various therapeutic aspects, including evaluating potential treatment options, and predicting and monitoring response to therapy.
Image contrast in MRI often depends upon the relaxation characteristics of the tissues [53]. There are three primary relaxation times: longitudinal relaxation (T1), intrinsic transverse relaxation (T2), and apparent transverse relaxation (T2*), each with an associated relaxation rate: 1/T1, 1/T2, and 1/T2* [54,55]. MRI contrast agents are designed to shorten the relaxation times of the tissue of interest and therefore increase the relaxation rates [56]. T1 relaxation describes the regrowth of MRI signal during the pulse sequence, and so shortening T1 allows the signal to recover more quickly. Therefore, contrast agents designed to affect T1 tend to provide increased MRI signal. On the other hand, both T2 and T2* relaxation describes the decay of MRI signal and shortening these relaxation times decreases the MRI signal. Typically, contrast agents increase the relaxation rate in a linear fashion. Therefore, plotting 1/T1 vs. contrast agent concentration yields a straight line. The slope of this line corresponds to the relaxivity of the contrast agent and is given in terms of s–1 mM–1. The relaxivity is a measure of the potency of the contrast agent. A compound with higher relaxivity can provide higher contrast at a given concentration or identical contrast at a lower concentration. Relaxivity is typically measured relative to the paramagnetic or superparamagnetic ion concentration, such as gadolinium or iron. For molecular and cellular imaging applications, however, the relaxivity per nanoparticle, i.e., particle or molecular relaxivity, is more useful for comparing contrast agent effect per binding site.

9.4 Cellular Imaging with Iron Oxides 233
9.4
Cellular Imaging with Iron Oxides
Ultrasmall superparamagnetic iron oxide (USPIO) nanoparticles are potent MRI contrast agents. The iron produces strong local disruptions in the magnetic field of MRI scanners, which lead to increased T2* relaxation. This increased relaxation causes decreased image intensity in areas with iron oxide accumulation, termed “susceptibility artifacts”. Because of the extremely large change in MRI signal induced by superparamagnetic particles, they have been developed for a wide variety of contrast agent applications, including imaging vasculature, bowel, liver, spleen, lymphatics, bone marrow, and tumors, and stem cell therapies [57–59]. In particular, particles with a 15to 25-nm diameter have a very long circulating half-life and are preferentially taken up by macrophages in the body when coated with dextran. These properties have allowed dextran-coated USPIO nanoparticles to be employed for passive targeted imaging of pathological inflammatory processes, such as unstable atherosclerotic plaques, by MRI (Fig. 9.4) [60]. USPIO-labeled macrophages have been shown to preferentially accumulate in unstable and ruptured plaques (75% demonstrating uptake), but not in stable lesions (only 7% showing USPIO uptake) [61].
Several aspects of the USPIO nanoparticle must be optimized for successful visualization of atherosclerotic plaques with MRI. The USPIO formulation must adhere to certain physical and chemical requirements. For instance, particles lacking the dextran coating are not effectively phagocytosed by inflammatory cells. Also, particles with a diameter similar to low-density lipoprotein (LDL), 15–25 nm, have a circulating half-life on the order of hours, providing sufficient time for cellular accu-
A
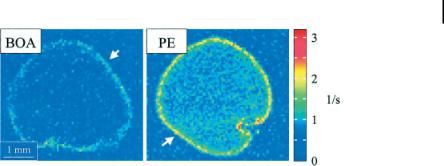
Figure 9.4. Axial 2D gradient-echo MRI at the level of the common iliac arteries, before (A) and 24 hours after (B) USPIO administration. Precontrast image shows small foci of low signal intensity within the wall of the left common iliac artery, possibly representing calcification (A, arrow). On the postcontrast image a
B
pronounced signal loss of the entire crosssection of the artery is seen (B, arrow). The right common iliac artery is not displayed at an acceptable image quality, as it takes an oblique course through the image plane
(B, arrowhead). (Reprinted with permission from Ref. [60].)

234 9 Nanotechnologies for Cellular and Molecular Imaging by MRI
mulation and effective delivery to atherosclerotic plaques [62]. In addition, the dose of USPIOs has a dramatic influence on the appearance by MRI. Increasing the dose by a factor of four produces signal loss in ten times more slices [62].
The imaging protocols are also critically important for USPIO detection by MRI. The susceptibility artifacts created by accumulation of USPIOs are most sensitively imaged with T2*-weighted imaging sequences. T1-weighted and proton density imaging sequences are far less sensitive to the susceptibility artifacts induced by USPIO uptake in tissues [61]. The T2*-weighted sequences typically utilize gradient echo techniques with long echo times. The long echo time accentuates signal loss due to the presence of USPIOs, but these sequences also tend to suffer from a low signal-to-noise ratio. Often, highly specialized coils, such as phased array coils or ap- plication-specific surface coils, are employed in order to maximize the available signal to noise [60,61].
Blood vessels are typically surrounded by fatty tissue, which can interfere with imaging structures within the arterial wall. On MRI, fat appears as a bright signal, which often displays a spatial misregistration artifact relative to other tissues in the body, called a “chemical shift” artifact. This artifact can often overlap signals from the vessel wall and obscure the atherosclerotic plaques under investigation. In order to avoid this problem, imaging sequences employing fat suppression or selective excitation techniques are used for imaging USPIO uptake [60–62]. With these sequences, fat appears dark because its signal is either suppressed or not excited. In a similar manner, bright blood imaging sequences, such as 2D fast low-angle shot (FLASH) sequences, are often used for detection of USPIOs [60, 61]. The bright blood signal allows clearer definition of the vessel lumen, and signal voids in the arterial wall caused by USPIO uptake are much easier to distinguish.
In addition to the imaging protocol itself, the choice of imaging time after USPIO injection is critically important. The long circulating half-life of dextran-coated USPIO nanoparticles is necessary to achieve adequate loading into inflammatory cells, but it can also interfere with obtaining high-quality images. Up to approx. 24 hours after USPIO administration, the blood concentration is high enough to create image artifacts [61, 62], which can obscure visualization of the vessel wall. On the other hand, too long a delay (~72 hours) after USPIO injection can result in no detectable susceptibility artifacts [61].
9.5
Molecular Imaging with Paramagnetic Nanoparticles
As an alternative approach, we proposed and evaluated a ligand-targeted, lipidencapsulated, nongaseous perfluorocarbon nanoparticle emulsion for molecular imaging applications (Fig. 9.5) [12, 15, 26, 28]. The nanoparticles can be formulated to carry paramagnetic gadolinium ions and targeted to a number of important biochemical epitopes, such as fibrin, tissue factor and amb3-integrin. Fibrin deposition is one of the earliest signs of plaque rupture, allowing detection of the “culprit” lesion before a high-grade stenosis has been formed [63]. Tissue factor is another

9.5 Molecular Imaging with Paramagnetic Nanoparticles 235
Payloads
Gd-Chelates
Drugs
Radionuclides
Targeting System “Molecular zip codes ”
Including Antibodies, Peptides, Mimetics, or Aptamers
Figure 9.5. Generalized molecular imaging paradigm for ligand-targeted paramagnetic nanoparticles.
prothrombotic agent and is expressed on the surface of vascular smooth muscle cells, which contribute to restenosis following vascular injury or stent placement [64].
While fibrin and tissue factor can be utilized to delineate unstable cardiovascular diseases, the amb3-integrin is a general marker of angiogenesis and plays an important role in a wide variety of disease states [65]. The amb3-integrin is a well-character- ized heterodimeric adhesion molecule that is widely expressed by endothelial cells, monocytes, fibroblasts, and vascular smooth muscle cells. In particular, amb3-integ- rin plays a critical part in smooth muscle cell migration and cellular adhesion [66,67], both of which are required for the formation of new blood vessels. The amb3- integrin is expressed on the luminal surface of activated endothelial cells but not on mature quiescent cells [68]. We have demonstrated the utility of amb3-integrin-tar- geted nanoparticles for the detection and characterization of angiogenesis associated with growth factor expression [69], tumor growth [70], and atherosclerosis [33].
Angiogenesis plays a critical role in plaque growth and rupture [71]. Normal large-caliber arteries in humans receive oxygen and nutrients from blood that is delivered by the adventitial vasa vasorum, not necessarily from the vessel lumen itself. The normal vasa vasorum carries blood in the same direction as arterial flow and extends perpendicular branches around the vessel wall to supply deep tissues. In regions of atherosclerotic lesions, angiogenic vessels proliferate from the vasa vasorum to meet the high metabolic demands of plaque growth [72]. Atherosclerosis promotes both inflammation and angiogenesis in the arterial wall, probably via a positive feed-back type of system. Inflammatory cells within the lesion stimulate angiogenesis through local molecular signaling, which in turn promotes neovascular growth, thereby providing an avenue for more inflammatory cells to enter the plaque. This process yields a strong correlation between the extent of plaque angiogenesis and local accumulation of inflammatory cells [73].
2369 Nanotechnologies for Cellular and Molecular Imaging by MRI
9.5.1
Optimization of Formulation Chemistry
Successful development of a targeted nanoparticle contrast agent requires optimization of numerous formulation procedures, including the number and chemical structure of the paramagnetic chelates and the targeting ligands. Even the choice of target should be optimized. The target molecule must be expressed at relatively high concentrations in diseased tissue and at very low levels in normal tissue. Also, the target must be accessible to the contrast agent construct. For instance, a 200-nm- diameter nanoparticle could encounter and bind to receptors on the luminal side of an endothelial cell, but perhaps not directly to DNA fragments inside the nucleus of a neuronal cell. The target ligand should have high affinity for the target of interest and minimal binding to similar receptors that may be expressed on normal tissues. In addition, the ligand must retain stability and potency throughout the process of nanoparticle formulation. For example, antibodies or their fragments could denature and lose their targeting abilities during extreme chemical processing steps, such as sterilization of the final nanoparticle product. Other targeting ligands, such as carbohydrates, aptamers, or peptidomimetics, may be less sensitive to these formulation procedures and retain their bioactivity.
Molecular imaging with liquid perfluorocarbon nanoparticles was originally pursued to target fibrin deposits on ruptured atherosclerotic plaques [12,25–28]. By incorporating an anti-fibrin antibody into the particles, a dense layer of nanoparticles can be selectively targeted to the surface of a fibrin clot (Fig. 9.6) [25]. This model provides a robust platform for exploring the consequences of changes to the formulation chemistry of the nanoparticle.

9.5 Molecular Imaging with Paramagnetic Nanoparticles 237
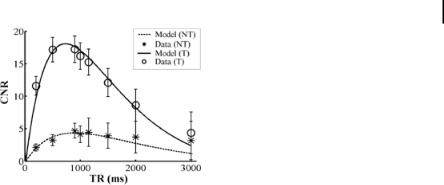
Unlike the USPIO agents discussed previously, the image enhancement characteristics of commercially available paramagnetic contrast agents are too small to visualize the very sparse concentrations, picomolar to nanomolar, of epitopes relevant to molecular imaging. By incorporating vast numbers of paramagnetic complexes (>50,000) onto each particle, the signal enhancement possible for each binding site is magnified dramatically, by a factor of >106. The increased paramagnetic influence arises from two mechanisms: the relaxivity per particle increases linearly with respect to the number of gadolinium complexes, and the relaxivity of each gadolinium complex increases due to the slower tumbling of the molecule when attached to the much larger particle. By studying dilutions of nanoparticles in water, we have observed increased T1 relaxivity with increasing gadolinium payloads (Fig. 9.7) [25]. We also verified that the increased relaxivity seen in solution corresponded to increased signal intensity on T1-weighted images of particles bound to fibrin clots (Fig. 9.8) [25]. Thus, improvements in the formulation chemistry can be assessed in simple phantoms of nanoparticle dilutions and verified in models of physiological systems.
Figure 9.7. T1 relaxation rates as a function of nanoparticle number expressed in picomoles for formulations ranging from 0 to 50 mol% Gd-DTPA-BOA in the outer 2% lipid monolayer. (Reprinted with permission from Ref. [25].)
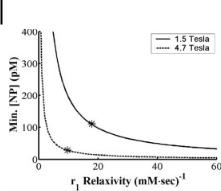
The fibrin clot model also provided a means to evaluate the paramagnetic influence of different gadolinium chelate structures [27]. The T1 relaxivity (r1) of nanoparticles formulated with gadolinium diethylene-triamine-pentaacetic acid bis-oleate [74] (Gd-DTPA-BOA) or gadolinium diethylene-triamine-pentaacetic acid-phosphati- dylethanolamine (Gd-DTPA-PE [75]) has been measured at selected magnetic field strengths: 0.47 T, 1.5 T, and 4.7 T. At all magnetic field strengths, r1 of the Gd-DTPA- PE formulation was approximately two times greater than for the Gd-DTPA-BOA agent [27], indicating that small adjustments to the molecular configuration of a paramagnetic chelate can substantially improve the fundamental relaxation properties.
Variable temperature relaxometry measurements have shown that r1 of the Gd- DTPA-BOA emulsion was largely independent of temperature [27]. In contrast, r1 increased at higher temperatures for the Gd-DTPA-PE emulsion. These tempera- ture-dependence data suggest that the water exchange rate with the paramagnetic ion is higher for the Gd-DTPA-PE chelate than for Gd-DTPA-BOA. At the higher

244 9 Nanotechnologies for Cellular and Molecular Imaging by MRI
Nominal Diameter = 250nm
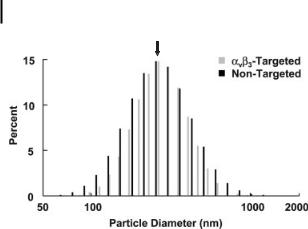
Figure 9.15. Particle size distribution of amb3-targeted (gray) and nontargeted nanoparticles (black) demonstrating identical physical characteristics of the two formulations. (Reprinted with permission from Ref. [70].)
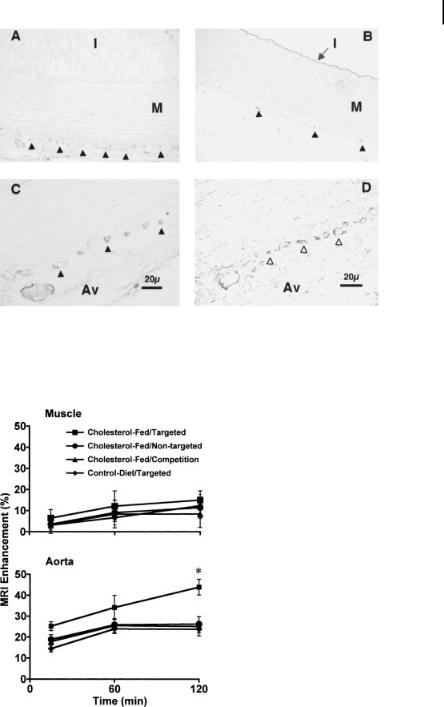
particle accumulation through passive entrapment in the hyperpermeable angiogenic vasculature vs. active targeting to the biochemical epitope of interest (Fig. 9.14) [33]. Differences in particle size, however, may lead to differences in MRI signal enhancement between targeted and nontargeted nanoparticles. Therefore, the particle size distributions for the two formulations must be identical (Fig. 9.15) [33].
Further confirmation of active particle targeting can be achieved with in vivo competition procedures (Fig. 9.14) [33]. Targeted, high-avidity nanoparticles lacking the paramagnetic chelate are injected in order to occupy all available binding sites, but they do not provide MRI signal enhancement. Subsequent injection of targeted paramagnetic nanoparticles will produce image enhancement only through passive particle accumulation. Analysis of MRI signal enhancement in experimental control animals, e.g., control diet rabbits, as well as control tissues, e.g., skeletal muscle, provides even greater discrimination between the ability of the particle to actively target the epitope of interest and passive accumulation of the agent (Fig. 9.14) [33].
In a similar manner, we have also demonstrated molecular imaging of angiogenesis in nascent Vx-2 tumors implanted in the hindlimb of rabbits [70]. Corroboration between MRI signal enhancement and histological staining of angiogenic amb3- integrin expression reaffirms the sensitivity of amb3-targeted nanoparticles. Comparing the amount of MRI signal enhancement achieved with targeted nanoparticles vs. nontargeted or competition formulations provides more supporting evidence for the specificity of our molecular imaging contrast agent. In addition, we have shown that some masses which appeared to be viable tumor on T2-weighted MRI were revealed to be only tumor remnants by histology and consisted mostly of inflammatory cells. These masses did not display MRI enhancement with amb3-targeted nanoparticles, further demonstrating the specificity of these methods for molecular imaging of active angiogenesis.

|
References |
247 |
|
|
|
29 H. W. Kang, L. Josephson, A. Petrovsky, |
38 M. J. Davies, D. M. Krikler, D. Katz, Athero- |
|
R. Weissleder, A. Bogdanov Jr., Magnetic reso- |
sclerosis: inhibition of regression as thera- |
|
nance imaging of inducible E-selectin expres- |
peutic possibilities. Br. Heart J. 1991, 65, |
|
sion in human endothelial cell culture. |
302–310. |
|
Bioconjug. Chem. 2002, 13, 122–127. |
39 P. D. Richardson, M. J. Davies, G. V. Born, |
|
30 S. D. Tiukinhoy, M. E. Mahowald, |
Influence of plaque configuration and stress |
|
V. P. Shively, A. Nagaraj, B. J. Kane, |
distribution on fissuring of coronary athero- |
|
M. E. Klegerman, R. C. MacDonald, |
sclerotic plaques. Lancet 1989, 2, 941–944. |
|
D. D. McPherson, J. S. Matsumura, Develop- |
40 M. Naghavi, P. Libby, E. Falk, S. W. Casscells, |
|
ment of echogenic, plasmid-incorporated, tis- |
S. Litovsky, J. Rumberger, J. J. Badimon, |
|
sue-targeted cationic liposomes that can be |
C. Stefanadis, P. Moreno, G. Pasterkamp, |
|
used for directed gene delivery. Invest. Radiol. |
Z. Fayad, P. H. Stone, S. Waxman, P. Raggi, |
|
2000, 35, 732–738. |
M. Madjid, A. Zarrabi, A. Burke, C. Yuan, |
|
31 J. R. Lindner, J. Song, J. Christiansen, |
P. J. Fitzgerald, D. S. Siscovick, C. L. de Korte, |
|
A. L. Klibanov, F. Xu, K. Ley, Ultrasound |
M. Aikawa, K. E. Juhani Airaksinen, |
|
assessment of inflammation and renal tissue |
G. Assmann, C. R. Becker, J. H. Chesebro, |
|
injury with microbubbles targeted to P-selec- |
A. Farb, Z. S. Galis, C. Jackson, I. K. Jang, |
|
tin. Circulation 2001, 104, 2107–2112. |
W. Koenig, R. A. Lodder, K. March, |
|
32 J. Chen, C. H. Tung, U. Mahmood, |
J. Demirovic, M. Navab, S. G. Priori, |
|
V. Ntziachristos, R. Gyurko, M. C. Fishman, |
M. D. Rekhter, R. Bahr, S. M. Grundy, |
|
P. L. Huang, R. Weissleder, In vivo imaging |
R. Mehran, A. Colombo, E. Boerwinkle, |
|
of proteolytic activity in atherosclerosis. |
C. Ballantyne, W. Insull Jr., R. S. Schwartz, |
|
Circulation 2002, 105, 2766–2771. |
R. Vogel, P. W. Serruys, G. K. Hansson, |
|
33 P. M. Winter, A. M. Morawski, |
D. P. Faxon, S. Kaul, H. Drexler, |
|
S. D. Caruthers, R. W. Fuhrhop, H. Zhang, |
P. Greenland, J. E. Muller, R. Virmani, |
|
T. A. Williams, J. S. Allen, E. K. Lacy, |
P. M. Ridker, D. P. Zipes, P. K. Shah, |
|
J. D. Robertson, G. M. Lanza, S. A. Wickline, |
J. T. Willerson, From vulnerable plaque to vul- |
|
Molecular imaging of angiogenesis in early- |
nerable patient: a call for new definitions and |
|
stage atherosclerosis with amb3-integrin-tar- |
risk assessment strategies: Part I. Circulation |
|
geted nanoparticles. Circulation 2003, 108, |
2003, 108, 1664–1672. |
|
2270–2274. |
41 S. Ojio, H. Takatsu, T. Tanaka, K. Ueno, |
|
34 Z. J. Zheng, J. B. Croft, W. H. Giles, |
K. Yokoya, T. Matsubara, T. Suzuki, |
|
G. A. Mensah, Sudden cardiac death in the |
S. Watanabe, N. Morita, M. Kawasaki, |
|
United States, 1989 to 1998. Circulation 2001, |
T. Nagano, I. Nishio, K. Sakai, K. Nishigaki, |
|
104, 2158–2163. |
G. Takemura, T. Noda, S. Minatoguchi, |
|
35 C. Napoli, C. K. Glass, J. L. Witztum, |
H. Fujiwara, Considerable time from the |
|
R. Deutsch, F. P. D’Armiento, W. Palinski, |
onset of plaque rupture and/or thrombi until |
|
Influence of maternal hypercholesterolaemia |
the onset of acute myocardial infarction in |
|
during pregnancy on progression of early |
humans: coronary angiographic findings |
|
atherosclerotic lesions in childhood: Fate of |
within 1 week before the onset of infarction. |
|
Early Lesions in Children (FELIC) study. |
Circulation 2000, 102, 2063–2069. |
|
Lancet 1999, 354, 1234–1241. |
42 L. H. Arroyo, R. T. Lee. Mechanisms of pla- |
|
36 A. W. Zieske, G. T. Malcom, J. P. Strong, |
que rupture: mechanical and biologic interac- |
|
Natural history and risk factors of athero- |
tions. Cardiovasc. Res. 1999, 41, 369–375. |
|
sclerosis in children and youth: the PDAY |
43 L. Petit, P. Lesnik, C. Dachet, M. Moreau, |
|
study. Pediatr. Pathol. Mol. Med. 2002, 21, |
M. J. Chapman, Tissue factor pathway inhibi- |
|
213–237. |
tor is expressed by human monocyte-derived |
|
37 M. J. Davies, A. C. Thomas, Plaque fissuring |
macrophages: relationship to tissue factor |
|
– the cause of acute myocardial infarction, |
induction by cholesterol and oxidized LDL. |
|
sudden ischaemic death, and crescendo |
Arterioscler. Thromb. Vasc. Biol. 1999, 19, |
|
angina. Br. Heart J. 1985, 53, 363–373. |
309–315. |
|

|
References |
249 |
|
|
|
63 P. Constantinides, Plaque fissuring in |
71 A. N. Tenaglia, K. G. Peters, M. H. Sketch Jr., |
|
human coronary thrombosis. J. Atheroscler. |
B. H. Annex, Neovascularization in atherect- |
|
Res. 1966, 6, 1–17. |
omy specimens from patients with unstable |
|
64 L. Oltrona, C. M. Speidel, D. Recchia, |
angina: implications for pathogenesis of |
|
S. A. Wickline, P. R. Eisenberg, |
unstable angina. Am. Heart J. 1998, 135, |
|
D. R. Abendschein, Inhibition of tissue fac- |
10–14. |
|
tor-mediated coagulation markedly attenuates |
72 Y. Zhang, W. J. Cliff, G. I. Schoefl, G. Higgins, |
|
stenosis after balloon-induced arterial injury |
Immunohistochemical study of intimal |
|
in minipigs. Circulation 1997, 96, 646–652. |
microvessels in coronary atherosclerosis. |
|
65 J. S. Kerr, S. A. Mousa, A. M. Slee, amb3-Integ- |
Am. J. Pathol. 1993, 143, 164–172. |
|
rin in angiogenesis and restenosis. Drug News |
73 K. S. Moulton, K. Vakili, D. Zurakowski, |
|
Perspect. 2001, 14, 143–150. |
M. Soliman, C. Butterfield, E. Sylvin, K. M. Lo, |
|
66 G. G. Bishop, J. A. McPherson, J. M. Sanders, |
S. Gillies, K. Javaherian, J. Folkman, Inhibi- |
|
S. E. Hesselbacher, M. J. Feldman, |
tion of plaque neovascularization reduces |
|
C. A. McNamara, L. W. Gimple, E. R. Powers, |
macrophage accumulation and progression |
|
S. A. Mousa, I. J. Sarembock, Selective |
of advanced atherosclerosis. Proc. Natl. Acad. |
|
amb3-receptor blockade reduces macrophage |
Sci. U. S. A. 2003, 100, 4736–4741. |
|
infiltration and restenosis after balloon angio- |
74 W. Cacheris, T. Richard, R. Grabiak, A. Lee, |
|
plasty in the atherosclerotic rabbit. Circulation |
Paramagnetic complexes of N-alkyl-N-hydro- |
|
2001, 103, 1906–1911. |
xylamides of organic acids and emulsions |
|
67 M. H. Corjay, S. M. Diamond, |
containing same for magnetic resonance im- |
|
K. L. Schlingmann, S. K. Gibbs, |
aging. United States: HemaGen/PFC; 1997. |
|
J. K. Stoltenborg, A. L. Racanelli, amb3, amb5, |
75 C. W. Grant, S. Karlik, E. Florio, A liposomal |
|
and osteopontin are coordinately upregulated |
MRI contrast agent: phosphatidylethanol- |
|
at early time points in a rabbit model of |
amine-DTPA. Magn. Reson. Med. 1989, 11, |
|
neointima formation. J. Cell Biochem. 1999, |
236–243. |
|
75, 492–504. |
76 A. M. Morawski, P. M. Winter, K. C. Crowder, |
|
68 P. C. Brooks, S. Stromblad, R. Klemke, |
S. D. Caruthers, R. W. Fuhrhop, M. J. Scott, |
|
D. Visscher, F. H. Sarkar, D. A. Cheresh, |
J. D. Robertson, D. R. Abendschein, |
|
Antiintegrin amb3 blocks human breast can- |
G. M. Lanza, S. A. Wickline, Targeted nano- |
|
cer growth and angiogenesis in human skin. |
particles for quantitative imaging of sparse |
|
J. Clin. Invest. 1995, 96, 1815–1822. |
molecular epitopes with MRI. Magn. Reson. |
|
69 S. A. Anderson, R. K. Rader, W. F. Westlin, |
Med. 2004, 51, 480–486. |
|
C. Null, D. Jackson, G. M. Lanza, |
77 T. M. Button, R. J. Fiel, Isointense model for |
|
S. A. Wickline, J. J. Kotyk, Magnetic reso- |
the evaluation of tumor-specific MRI contrast |
|
nance contrast enhancement of neovascula- |
agents. Magn. Reson. Imaging 1988, 6, |
|
ture with amb3–targeted nanoparticles. Magn. |
275–280. |
|
Reson. Med. 2000, 44, 433–539. |
78 E. T. Ahrens, U. Rothbacher, R. E. Jacobs, |
|
70 P. M. Winter, S. D. Caruthers, A. Kassner, |
S. E. Fraser, A model for MRI contrast |
|
T. D. Harris, L. K. Chinen, J. S. Allen, |
enhancement using T1 agents. Proc. Natl. |
|
E. K. Lacy, H. Zhang, J. D. Robertson, |
Acad. Sci. U. S. A. 1998, 95, 8443–8448. |
|
S. A. Wickline, G. M. Lanza, Molecular imag- |
79 D. A. Sipkins, D. A. Cheresh, M. R. Kazemi, |
|
ing of angiogenesis in nascent Vx-2 rabbit |
L. M. Nevin, M. D. Bednarski, K. C. Li, |
|
tumors using a novel amb3–targeted nanopar- |
Detection of tumor angiogenesis in vivo by |
|
ticle and 1.5 tesla magnetic resonance imag- |
amb3–targeted magnetic resonance imaging. |
|
ing. Cancer Res. 2003, 63, 5838–5843. |
Nat. Med. 1998, 4, 623–626. |
|
|
80 D. Artemov, N. Mori, R. Ravi, Z. M. Bhujwalla, |
|
|
Magnetic resonance molecular imaging of |
|
|
the HER-2/neu receptor. Cancer Res. 2003, 63, |
|
|
2723–2727. |
|