

книги из ГПНТБ / Новые методы анализа аминокислот, пептидов и белков
..pdf
Г е л ь -ф и л ь т р а ц и я в т о н к о м с л о в |
2 7 5 |
Вильямсон и 'Ал л и с о н [13] сделали попытку усовершенство вать стадию иммунопреципитации с помощью метода Лорелла [63], заключающегося в проведении электрофореза антигена в гелях агарозы, включающих антитела. Согласно предложенной методике, белки после разделения переносят на полоску филь тровальной бумаги, которую затем помещают на слой агарозы с антителами, и проводят электрофорез. Таким путем в сыво ротке мыши удалось обнаружить до 20 компонентов [13, 64]. В другом варианте удаляют лишний сефадекс по Хансону [14], а затем осторожно переносят весь гель агарозы с антителами на полоску сефадекса. По такой методике выполнен анализ мо лекулярновесового распределения а-липопротеина нормальной сыворотки [65], приведенный на рис. 9.
Приведенные примеры ясно показывают, что тонкослойная гель-фильтрация в сочетании с иммунной преципитацией яв ляется простым и удобным методом, который может дополнить иммуноэлектрофорез при идентификации белков в биологиче ских жидкостях или при выделении белков. В случае необходи мости чувствительность и разрешение метода можно повысить, выполняя электрофорез после тонкослойной гель-фильтрации в геле агарозы, содержащем антитела.
СПИСОК ЛИТЕРАТУРЫ
1.Porath J., Flodin Р., Nature, 183, 1657 (1959).
2.Hjertin S., Mosbach R., Anal. Biochem., 3, 109 (1962).
3.Hjertin S., Biochim. Biophys. Acta, 79, 393 (1964).
4.Детерман /'., Гель хроматография, изд-во «Мир», М. 1970.
5.Determann Н., Experientia, 18, 430 (1962).
6. Johansson В. G., Rymo L., Acta Chem. Scand., 16, 2067 (1962).
7.Andrews P., Biochem. J., 91, 222 (1964).
8.Johansson В G., Rymo L., Acta Chem. Scand., 18, 217 (1964).
9.Morris C. J. O. R., J. Chromatog., 16, 167 (1964).
10.Determann H., Michel W., Z. Anal. Chem., 212, 211 (1965).
11.Baron D. N., Economidis J., J. Clin. Pathol., 16, 484 (1963).
12.Maier C. L., Jr., J. Chromatog., 32, 577 (1968).
13.Williamson /., Allison A. C., Lancet, 2, 123 (1967).
14.Hanson L. A., Johansson B. G., Rymo L., Clin. Chim. Acta, 14, 391 (1966).
15.Hobbs J. R., In the Scientific Basis of Medicine: Annual Reviews, Athlone Press, London, 1966, p. 106.
16. Vergani G., Stabilini R., Agosioni A., J. Chromatog., 28, 135 (1967).
17.Kohn J., Immunology, 15, 863 (1968).
18.Fazekas de St. Groth S., Webster R. G., Datyner A., Biochim. Biophys. Acta, 71, 377 (1963).
19.Roberts G. P., J. Chromatog., 22, 90 (1966).
20.Rydon H. M„ Smith P. W. G., Nature, 169, 922 (1952).
21.Wollheim F., Snigurowicsz J., Scand. J. Haematol., 4, 111 (1967).
22.Maier C. L., J. Chromatog., 32, 579 (1968).
23.Grossman H., Wagner H., J. Chromatog., 35, 301 (1968).
24.Stickland R. G., Anal. Biochem., 10, 108 (1965).
25.Agosioni A., Vergani C., Cirla £., Protides Biol. Fluids, Proc. 14th Colloq., Bruges, 1966, Ed. by H. Peeters, Elsevier, Amsterdam, London, New York, 1967,
276 |
ГЛАВА 6 |
26.Hobbs J. R., Jacobs A., Clin. Exptl. Immunol., 5, 199 (1969).
27.Purves L. R., Lancet, 2, 186 (1965).
28. Davis J. S., Flynn F. W„ Plait H. 5., Clin. Chirn. Acta, 21, 357 (1968).
29.Goddard P. F., Hobbs J. R., Proc. Roy. Soc. Med., 61, 335 (1968).
30.Stransky Z., Srcli M., J. Chromatog., 28, 146 (1967).
31.Hanson L. <4., Johansson B. G., Nature, 187, 599 (1960).
32.Cederblad G., Johansson B. G., Ryrno L., Acta Chem. Scand., 20, 2349 (1966).
33.Johansson B. G., Studies on Human Immunoglobulins with special reference to Immunoglobulin A in colostrum, Thesis, University of Lund, 1967.
34.Axelsson H., Johansson B. G., Rymo L., Acta Chem. Scand., 20, 2339 (1966).
35. Hanson L. A., Johansson B. G., Gamma Globulins: Structure and Control of Biosynthesis, Nobel Symposium 3, Ed. by J. Killander, Almqvist and Wicksell, Stockholm, 1967, p. 141.
36.Zink H., Acta Neurol. Scand. 43, Suppl. 28 (1967).
37.Rymo L., personal communication.
38.Lagerkvist U., Waldenstrom J., J. Biol. Chem., 242, 3021 (1967).
39.Whitacker J. R., Anal. Chem., 35, 1950 (1963).
40. Wietand T., Daesberg P., Determann //., Biochem. Z., 337, 303 (1963).
41.Hygstedt O., Jagenburg R., Scand. J. Clin. Lab. Invest., 17, 565 (1965).
42.Yakulis V., Costea N., Heller P., J. Immunol., 102, 488 (1969).
43.Determann H., in «Protides Biol. Fluids, Proc. 14th Colloq.», Ed. by H. Peeters, Elsevier, Amsterdam, London, New York, 1967, p. 563.
44.Johansson B. G., Acta Chem. Scand., 23, 683 (1969).
45.Hjerten S., Johansson B. G., unpublished results.
46.Gelotte B., Flodin P., Killander J., Arch. Biochem. Biophys., Suppl. No. 1, 319 (1962).
47.Dose K., Krause G., Naturwiss., 49, 349 (1962).
48.Chudzik J.. Klein A., J. Chromatog., 36, 262 (1968).
49.Johansson B. G., Rymo L., Biochem. J., 92, 5P (1964).
50.Hanson L A., Roos P., Rymo L., Nature, 212, 948 (1966).
51.Bergmann L., Dencker S. J., Johansson B. G., Svennerholm L., J. Neurochem., 15, 781 (1968).
52.Hanson L., Johansson B. G„ Intern. Arch. Allergy, 31, 380 (1967).
53. |
Carnegie P. |
R., Pacheco G., Proc. |
Soc. Exptl. Biol. Med., |
117, |
137 |
(1964). |
||
54. |
Grant G. H., |
Everall P. H„ J. Clin. |
Pathol., 18, 654 (1965). |
69, |
522 |
(1967). |
||
55. |
Agostoni A., |
Vergani C., Lomanto |
B., J. Lab. Clin. Med., |
|||||
56. |
Grant |
G.H., |
Everall |
P. H., Proc. |
Soc. Anal. Chem., 4, 143(1967). |
|
||
57. |
Grant |
G. H., |
Everall |
P. H., Lancet, |
2, 368 (1967). |
|
|
|
58.Mancini G., Carbonara A. O., Heremans J. F., Immunochemistry, 2, 235 (1965).
59.Hanson L. A., Holmgren J., Johansson B. G., Wadsworth C., unpublished results.
60.Vergani C., Agostoni A., Clin. Chim. Acta, 16, 326 (1967).
61.Solomon A., J. Lab. Clin. Med., 70, 876 (1967).
62.Solomon A., Federation Proc., 26, 529 (1967).
63.Laurell C.-В., Anal. Biochem., 10, 358 (1965).
64.Williamson J., personal communication
65.Johansson B. G., unpublished results.
66.Radola B. J., J. Chromatog., 38, 61 (1968).
67.Radola B. J., J. Chromatog., 38, 78 (1968).
68.Zuidweg M. H., Oostendorp J. G., Bos C. J. K., J. Chromatog., 42, 552 (1969).
69.Ackers G. K-, Advan. Protein Chem., 24, 343 (1970).
70. Porter P„ Noakes D. E., Allen W. D., Immunology, 18, 245 (1970).
71.Radola B. J., Biochim. Biophys. Acta, 194, 335 (1969).
72.Delincee H., Radola B. /., Biochim. Biophys. Acta, 200, 404 (1970).
73 Porter P., Porter M. C., Shanberge J. N., Biochemistry, 6, 1854 (1967).
Г Л А В А 7
Макроэлектрофорез как метод определения а контроля за биосинтезом белка на уровне 10~7— 10~9 г
X. Хидён, Р. В. Ланге
Большинство органов состоит из нескольких видов клеток, причем специфические функции этого органа осуществляются только некоторыми из них. Гетерогенность состава тканей за ставляет прибегать к необычным методам выделения образца и его анализа.
Для этого применимы любые методы, позволяющие опреде лять вещества в количестве 10- 7—10-9 г в объемах 10-5 мкм3 при условии, что экспериментатор имеет чистые образцы разных видов клеток.
За последние пять лет преимущество разделения белков при помощи микрометодов получило общее признание. В 1965 г. Гроссбах [1] описал модификацию диск-электрофореза на поли акриламидном геле для определения 10-9 г количеств белка с помощью стеклянных капилляров. В 1966 г. авторы этой главы предложили микрометод разделения белка в количествах 10-7— 10~9г при использовании полиакриламидного геля в стеклянных капиллярах, имеющих диаметр 200 мк [2]. В 1967 г. Фельгенхауер [3] описал подобный метод для разделения 10—30 мкг белка с разрешающей способностью 1—3 мкг для одного белка. Гофман [4] для микроэлектрофореза белков вместо стеклянных капилляров применил плексигласовую камеру.
Нейхофф [5] увеличил разрешающую способность электрофо ретического метода разделения белка, применяя более высоко концентрированный акриламидный гель с добавкой 0,5% гидантоина. Нейхофф и Шилл [6] элюировали микрофракции белка, полученные этим способом, и показали, что их можно применять для иммунопреципитации. Фельгенгауер [7] добился высокой чув ствительности метода, используя после микроэлектрофореза белка на полиакриламидном геле свою модификацию методики иммунопреципитации. Таким путем он мог обнаружить 10-9 г одного белка. Недавно Маурером [8] был опубликован обзор по диск-электрофорезу, охватывающий также и микрометоды.
278 |
ГЛАВА 7 |
Настоящая работа описывает применение диск-микроэлектро фореза меченных белков из клеток мозга, нервных клеток или глии для определения типа растворимых белков и их обмена в сопоставимых участках мозга или в идентичных структурах, выделенных из мозга различных особей. В полученные величины специфических активностей внесены поправки на локальные ко лебания концентрации предшественников. Эта новая методика является дальнейшим этапом развития ранее опубликованного авторами [2] метода диск-микроэлектрофореза.
7.1.ПРИГОТОВЛЕНИЕ ОБРАЗЦОВ КЛЕТОЧНОГО МАТЕРИАЛА
7.1.1.Выделение нервных клеток и глий
Животных быстро забивают, дают вытечь крови и немед ленно извлекают мозг. Затем приготовляют срез из интересую щего исследователя участка мозга. Этот срез толщиной 2—4 мм помещают в 0,25 М раствор сахарозы или раствор Кребса — Рингера и рассматривают на охлаждаемом столике стереомикро скопа. Стереомикроскоп снабжен окулярами с 20-кратным уве личением и вспомогательной передней линзой, дающей 20— 160-кратное увеличение. Источники света закрывают инфра красными фильтрами, поглощающими тепло.
Срез мозговой ткани, тщательно сохраняемый под слоем раствора, заливают 0,01 М раствором метиленового синего и оставляют на 20 с, затем промывают его и в дальнейшем сохра няют под слоем раствора сахарозы или соли. В результате та кой обработки тела нервных клеток слегка окрашиваются в си ний цвет, который сохраняется не менее 5 мин. По истечении этого времени окрашивание можно повторить. Однако даже без повторного окрашивания тренированный глаз различает нервные клетки на ослепительно белом фоне глий, так как они имеют желтоватый оттенок, а синаптические узлы на краю клетки еще сохраняют синее окрашивание. Это было описано ранее [9].
Клетки вместе с частью глии осторожно отделяют от среза, вводя под клетку заостренную проволочку из нержавеющей стали («Nicrothal L., АВ Kanthal», Халлстахаммер, Швеция) диаметром 18—20 мкм. Каждую клетку извлекают из тканевого среза и помещают в раствор Кребса — Рингера. Эти манипуля ции производят вручную, поэтому стереомикроскоп должен быть снабжен упорами для рук.
Свободно плавающие в растворе нервные клетки со значи тельной частью дендритов освобождают от глии осторожными манипуляциями снизу. На рис. 1 изображены шесть изолирован ных нервных клеток сфотографированные в проходящем свете.
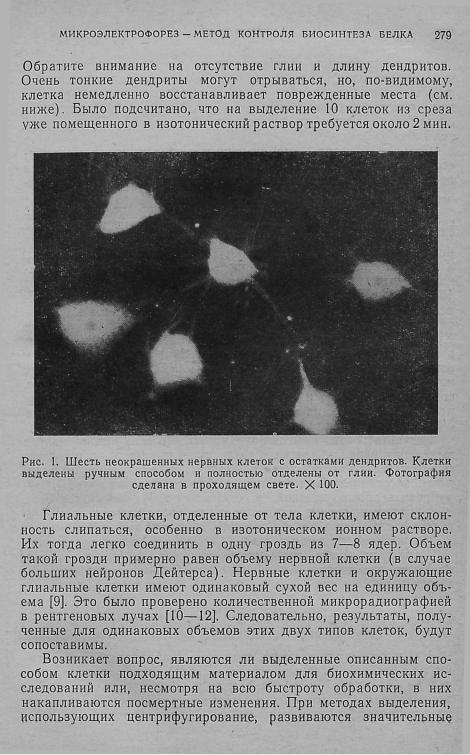
280 ГЛАВА 7
усилия, разрывающие глиальные клетки; они могут повредить и нервные клетки. Малые силы, используемые при микроманипу ляциях, не возбуждают таких разрывающих усилий. Как пока зали опыты авторов, возможные механические воздействия в приведенном методе приготовления образцов на много поряд ков ниже.
Следует отметить, что опытному экспериментатору требу ется 5 мин на то, чтобы забить животное, поместить срез мозга на термостатированный столик микроскопа и отделить 10 нерв ных клеток.
Такие клетки помещают в ионный раствор, не содержащий субстрата, в течение 2 ч в них микроманометрически измеряют поглощение кислорода [3]. В это время скорость поглощения кислорода, выражаемая в мкл 0 2-Ю-4 на клетку, была близка к нулю. После добавления глюкозы или p-оксибутирата, дыха ние немедленно восстанавливается и в течение 10 мин достигает 10 мкл 0 2-Ю-4 на клетку, сухой вес клеток равен 2-10-8 г. Вы деленные нервные клетки сохраняют также способность к фосфо рилированию [14]. Далее выделенные клетки помещали в раствор Кребса—Рингера с добавками бикарбоната и глюкозы. Затем в них под визуальным контролем вводили микроэлектроды и измерялись мембранные потенциалы [15]. Средний мембранный потенциал 120 клеток при 23 °С достигал 40 мВ. В атмосфере, содержащей 95% N2 и 5% С 02, мембранные потенциалы падали до 20 мВ. При замене азота атмосферы на кислород (95% 0 2 и 5% С02) мембранный потенциал вновь возрастал.
Результаты этих опытов показывают, что если при процедуре выделения нервная клетка и была сколько-нибудь повреждена, то потом мембрана клеточных выростов и перикарион, по-види мому, восстанавливают все повреждения настолько, чтобы пре дупредить потерю клеткой ее содержимого. Из нервных клеток мозга млекопитающих вышеописанным способом можно в при сутствии глиальных клеток получить культуру ткани [16].
Из имеющихся данных ясно, что нервные клетки определен ных участков мозга, выделенные вручную из свежего среза, являются прекрасным материалом для биохимических исследо ваний.
7.1.2.Образцы, выделенные из областей мозга, содержащих гомогенные клетки
Некоторые участки мозга, например гранулярный слой рети ны или слой пирамидных клеток в гиппокампе, имеют сравни тельно однородный клеточный состав. Образцы из этих участ ков мозга можно получать по методике, описанной в предыду щем параграфе. Для приготовления срезов, состоящих из 100—
МИКРОЭЛЕКТРОФОРЕЗ - МЕТОД КОНТРОЛЯ БИОСИНТЕЗА БЕЛКА |
281 |
1000 клеток, применяют инструменты из нержавеющей стали, сделанные под стереомикроскопом. Клеточный материал гомогенизуют, как описано ниже, и определяют концентрацию белка по Лоури [17]. Ниже приведен анализ слоя пирамидных клеток из гиппокампа (схема 1).
7.1.3.Гомогенизация
Клеточный материал гомогенизуют в стеклянном капилляре диаметром 400 мкм. Чистая вода растворяет 39% суммарного белка из образца нервных клеток (гиппокамп крысы). При ис пользовании раствора тушс-буфера, имеющего pH 7,4, содержа щего 0,1% Тритона X-100, экстрагируется 72% суммарного белка. Как показали радиометрические измерения, в результате электрофореза 92% этого белка переходит в полиакриламидный гель. Если кроме Тритона Х-100 к экстрагирующему раствору добавлять 0,1% натрийдодецилсульфат, то в раствор переходит 92% суммарного белка. Более 90% этого материала при элек трофорезе переходит в гель. Образец гомогенизуют, вводя в
раствор с клетками прикрепленную к отрезку тефлоновой трубки петлю из стальной проволоки толщиной 28—70 мкм. Эту ме шалку прикрепляют к зубоврачебной бор-машине, вращающейся со скоростью 12 000 об/мин [18]. Гомогенизацию ведут 1—3 ми нуты, наблюдая за ее ходом в микроскоп. Раствор центрифуги руют 5—10 мин в гематокритной центрифуге при скорости вра щения 11 000 об/мин. При такой обработке получается прозрач ная надосадочная жидкость.
7.2.ПРИГОТОВЛЕНИЕ КАПИЛЛЯРОВ И ПОЛИМЕРИЗАЦИЯ ГЕЛЕЙ
Обычно в продаже имеются капилляры различной толщины. Авторы этой главы применяли капилляры длиной 33 мм и диа метром 215 мкм или 440 мкм. Капилляры кипятили в растворе детергента RBS 25 («Kistner», Гетеборг, Швеция), ополаскивали горячей, затем дистиллированной водой, 95%-ным этанолом и ацетоном. В капилляры засасывали раствор силиклада (Siliclad, фирмы «Clay-Adams, Inc», Нью-Йорк) разбавленный в от ношении 1:100. После этого капилляры промывали дистиллиро ванной водой и сушили.
Для приготовления геля использовали мономеры, перекристаллизованные по Ленингу [19]. Ранее применявшиеся мето дики для приготовления растворов были немного изменены ав торами согласно рекомендациям Нейхоффа [5] (см. разд. 7.2.1). Основное отличие заключалось в том, что при приготовлении 25%-ного полиакриламидного геля авторы добавляли 0,5%-ный
282 ГЛАВА ?
гидантоин. Гидантоин в 10 раз увеличивает время полимериза ции, а также размеры пор и стабилизует гель. Под действием капиллярных сил капилляры заполняются акриламидным раст вором наполовину своей высоты. Полимеризация идет в течение ночи в атмосфере азота. Чтобы устранить возможные арте факты, вызванные действием персульфата аммония [20] при электрофорезе, на поверхность геля наливают немного трис- буфера (20 мкм) и тиогликолята, pH 8,4.
Перед каждым циклом электрофореза в капилляр осторожно вносят микропипеткой 5%-ный верхний гель высотой 2 мм. Этот гель полимеризуют под действием света в течение 5—10 мин. Перед тем как внести образец, излишнюю воду, скопившуюся на поверхности геля, отсасывают. Затем на несколько минут вносят небольшое количество раствора Б (см. разд. 7.2.1), разбавлен ного в отношении 1 : 8, и тоже отсасывают. Микропипеткой вно сят 0,5—1,5 мкл образца. Таким образом, капиллярная трубка малого объема и диаметром 200 мкм содержит 0,5 мкл образца, 0,07 мкл верхнего геля и 0,5 мкл разделяющего геля. Капилляр
ная |
трубка диаметром |
400 мкм содержит 1,7 мкл образца, |
0,3 |
мкм верхнего геля и |
2,5 мкл разделяющего геля. |
7.2.1.Полимеризация нижнего геля
Раствор А. pH 8,8. К 860 мг специально очищенного трис-бу-
фера «The British Drug Houses» добавляют 0,063 мл N,N,N',N'-
тетраметилэтилендиамина, 3,6 н. H2SO4 до pH 8,8 и воду до об щего объема 10 мл.
Раствор Б. pH 6,7. К 2,85 г трыс-буфера добавляют 1 М раст вор Н3РО4 до значения pH 6,7 и воду до общего объема 50 мл. Раствор В. К 20 г перекристаллизованного акриламида Eastman Organic Chemicals» добавляют 200 мг N, N'-метиленбисакрила- мида, 3,75 мг K3Fe(CN)6 и воду до общего объема 37,5 мл.
Раствор Г. К 70 мг (NH4)2S208 добавляют 25 мл 2%-ного раст вора Тритон Х-100 и разбавляют водой до объема 50 мл.
раствор Д. К 5,98 г триса добавляют 0,46 |
мл N.N.N'.N'-TeTpa- |
||
метилэтилендиамина, |
1 М Н3РО4 до pH 6,7 |
и разбавляют водой |
|
до объема 100 мл. |
(NH4)2S20 8 добавляют 5 мл |
2%-ного Три |
|
Раствор Е. К 200 мг |
|||
тона Х-100 и разбавляют водой до общего объема |
10 мл. |
||
Приготовление. К одному мл смеси (0,5 мл раствора А и 1,5 мл раствора В) добавляют 1,0 мл раствора Г, получая 20%-ный раствор. К нему добавляют 100 мг акриламида,. 10 мг гидантоина, в результате чего получают 25%-ный полиакриламидный гель.
Гомогенизация. К раствору образца в 0,25 М сахарозе, содер
жащей 0,5% Тритона |
Х-100 |
и забуференной раствором Б до |
pH 6,7, добавляют 20 |
мкмоль |
натрийтиогликолята. |