

Нарушение метаболизма железа
Клинические проявления дефицита железа многообразны и зависят от степени его выраженности и длительности существования.
Причины дефицита железа:
● потеря крови (желудочно – кишечные кровотечения, менструации и т. д.); ● увеличение физиологического дефицита в железе возникает при росте, беременности, родах, лактации, голодании;
● нарушение всасывания железа в кишечнике, нарушение кислото– и ферменто образующей функции желудка;
● хронические инфекции, интоксикации, гиповитаминозы (особенно С - гиповитаминоз), опухолевые процесы;
● элементарный дефицит железа;
● прием некоторых лекарственных препаратов. Длительное применение нестеридных противовоспалительных препаратов аспирин, вольтарен связан с риском хронической кровопотери, через изъязвление слизистую желудка. Одним из самых распространенных заболеваний на земном шаре является железодефицитная анемия (ЖДА). При дефиците Fe страдает весь организм, как правило железодефицитные состояния встречаются у детей, молодых женщин, они сопровождают различные хронические заболевания. Клинические появления зависят от этиологии, выраженности и скорости развития анемии. Болезни сердца и легких усугубляют тяжесть анемии, как правило при уровне гемоглобина ниже 70 г/л появляются признаки тканевой гипоксии.
● ЖДА развивается при недостатке поступления железа с пищей нарушение всасывания Fe из-за заболеваний ЖКТ, беременности в период роста организма. Хроническая кровопотеря – частая причина железодефицитной анемии у взрослых (70% всех анемий). Ранних клинических симптомов дефицита Fe не существует, а симптомы дефицита Fe появляются только после истощения запасов Fe в организме. Недостаточное поступление Fe с пищей – распространенная причина дефицита Fe у нас в стране, которая зависит от низкой доли продуктов животного происхождения в рационах. Возникает отрицательный баланс Fe, т. е. потери превышают поступление с пищей. Это часто бывает у беременных, при кормлении грудью, занятиях спортом. При ЖДА уменьшается размер эритроцитов и их пигментация (гипохромные эритроциты малых размеров). В эритроцитах уменьшается содержание гемоглобина (гипохромная анемия), понижается степень насыщения железом, повышается ОЖСС, в тканях и плазме крови снижается концентрация ферритина.
Повышенное содержание железа в организме, превышающее объем ферритинового депо ведет к развитию гемохроматоза. Образующийся гемосидерин вследствие низкой растворимости в воде откладывается в виде гранул в печени, поджелудочной железе, селезенке. Ионы железа оказывают токсичное действие на ферменты окислительно - восстановительных систем клеток, индуцируют образование активных форм кислорода (регуляция фентона) и инициируют перекисное окисление липидов, избыток ферритина и гемосидерина лизосомы, и илзосомальные ферменты вызывают повреждение клеточных структур. В сыворотке выявляют: повышенный уровень Fe, снижение ОЖСС, повышение уровня ферритина.
Гемостаз, определение, компоненты, стадии.
Гемостаз - остановка кровотечения при повреждении кровеносных сосудов. Механизмы, обеспечивающие гемостаз, реализуются не только при кровотечении, но и при любом повреждении интимы сосудистой стенки, вызванном физическими, гемодинамическими, химическими факторами, воспалением, действием иммунных комплексов, нарушением метаболизма (атеросклероз, коллагеновые болезни) и т.д.
Система гемостаза является функциональной системой организма, обеспечивающей постоянство его внутренней среды, предупреждение и остановку кровотечений, сохранение жидкого состояния циркулирующей крови.
Система гемостаза состоит из:
Свертывающая система (Тромбоцитарно-сосудистый – первичный гемостаз и коагуляционный (плазменный) – вторичный гемостаз)
Противосвертывающая система (Система антикоагулянтов и система фибринолиза)
В настоящее время установлено, что свертывание крови очень сложный процесс, так, коагуляционный гемостаз протекает в 3 стадии и включает:
1 стадия: образование протромбиназ /тканевой и кровяной/;
2 стадия: образование тромбина;
3 стадия: образование фибрина.
Функции сосудистого эндотелия, субэндотелия и тромбоци- тов. Сосудисто-тромбоцитарный гемостаз (первичный). Участие тромбоксана и простациклина в регуляции первичного гемоста- за.
Сосудистый эндотелий вырабатывает факторы, влияющие на развитие и течение воспаления.
Они делятся на провоспалительные и противовоспалительные.
Провоспалительные факторы:
Фактор некроза опухоли-α (ФНО-α, кахектин) - это пироген, во многом дублирует действие ИЛ-1, но кроме того, играет важную роль в патогенезе септического шока, вызванного грамотрицательными бактериями. Под влиянием ФНО-α резко увеличивается образование макрофагами и нейтрофилами Н2О2 и других свободных радикалов. При хроническом воспалении ФНО-α активирует катаболические процессы и тем самым способствует развитию кахексии.
Большая часть компонентов субэндотелия синтезируется и секретируется эндотелиальными клетками.
Клетки, находящиеся под эндотелием (макрофаги, фибробласты), имеют на поверхности тканевой фактор (TF, тканевой тромбопластин). При связывании тканевого фактора с фактором VIIa и при наличии ионов Ca2+ формируется комплекс, активирующий фактор Х. Эта реакция является пусковой для процесса свертывания крови.
Сосудисто-тромбоцитарный гемостаз:
1) обеспечивает остановку кровотечения из сосудов микроциркулярного русла и в сосудах с низким АД;
2) является предфазой коагулляционного гемостаза.
Фазы:
Рефлекторный спазм поврежденных сосудов. Обеспечивается БАВ, которые выделяются из разрушенных тромбоцитов (серотонин, НА, Адр.) – временно прекращают кровотечение. Эта реакция увеличивается при охлаждении поврежденного участка.
Спазм сосудов дополняется: адгезией тромбоцитов. В силу электростатического взаимодействия (тромбоцит “- „), обнажаются волокна коллагена стенки «+», происходит прилипание тромбоцитов к стенке (3 – 10с).
Обратимая агрегация (скучивание) тромбоцитов. Начинается почти одновременно с адгезией. Катализатор этого процесса АДФ, выделяемая из поврежденных тканей сосуда – внешняя АДФ, из тромбоцитов и эритроцитов – «внутренняя». Образуется рыхлая тромбоцитарная пробка, пропускающая плазму – белый тромб.
Необратимая агрегация – тромбоцитарная пробка становится непроницаемой для плазмы. Происходит это под влиянием тромбина, который меняет структуру мембраны тромбоцитов, и они сливаются в гомогенную массу.
Ретракция белого тромба. Это сокращение и уплотнение белого тромба, за счет сокращения нитей фибрина. Этим путем (сосудисто-тромбоцитарным) останавливается кровотечение из сосудов МЦР за 3 – 4 минуты при бытовых травмах.
Сосудисто-тромбоцитарный (первичный) гемостаз:
Включается сразу после повреждения сосуда
Реализуется эндотелием при участии гладкой мускулатуры сосудов и тромбоцитами
3 этапа: местная вазоконстрикция, адгезия, агрегация
Образуется первичный белый тромб
Нормальный эндотелий обладает высоким антитромботическим потенциалом, что определяется следующими его свойствами:
Рефлекторный (кратковременный) спазм сосудов, который возникает при травме. Он значительно уменьшает объем кровотока через поврежденный сосуд или даже прекращает в нем движение крови. Затем спазм сосудов поддерживается действием серотонина, адреналина, тромбоксана, эндотелинов, которые выделяются из тромбоцитов или клеток поврежденных сосудов
Продукцией под влиянием мембранной фосфолипазы и фермента тромбоксан-синтетазы мощного вазодилататора и ингибитора агрегации тромбоцитов – простагландина (простациклина)
Простациклин образуется из арахидоновой кислоты в эндотелиии сосудов и поступает в кровь. Синтез и секрецию простациклина эндотелиальными клетками стимулируют тромбин, гистамин, ангиотензин II и калликреин. Он реализует своё действие через аденилатциклазную систему передачи сигнала. Взаимодействие простациклина с рецептором вызывает активацию протеинкиназы А. Активная протеинкиназа А фосфорилирует и таким образом активирует Са2+-АТФ-азу и Са2+-транслоказа. Это приводит к снижению уровня содержания Са2* в цитоплазме тромбоцитов, сохранению ими дисковидной формы и снижению способности к агрегации. Активация тромбоцитов сопровождается появлением на поверхности плазматической мембраны отрицательно заряженных участков, образованных фосфатидилсерином. Основные индукторы активации и агрегации тромбоцитов - фактор фон Виллебранда, коллаген, тромбин, АДФ.
Характеристика плазменных факторов свёртывая крови. Роль витамина К и ионов Са2+ в гемокоагуляции. Внешний и внутренний пути гемокоагуляции.
Коагуляционный гемостаз – ферментативный процесс, в результате которого происходит образование фибринового сгустка. Этот фибриновый сгусток («красный тромб») плотно закупоривает поврежденный сосуд.
Красный тромб состоит из сети переплетенных волокн фибрина-полимера и захваченных в эту сеть г.о. эритроцитов, тромбоцитов и плазмы. Волокна фибрина-полимера также прикрепляются к пораженным поверхностям кровеносных сосудов. Таким образом красный тромб крепится к любому отверстию сосуда, предупреждая дальнейшую потерю крови.
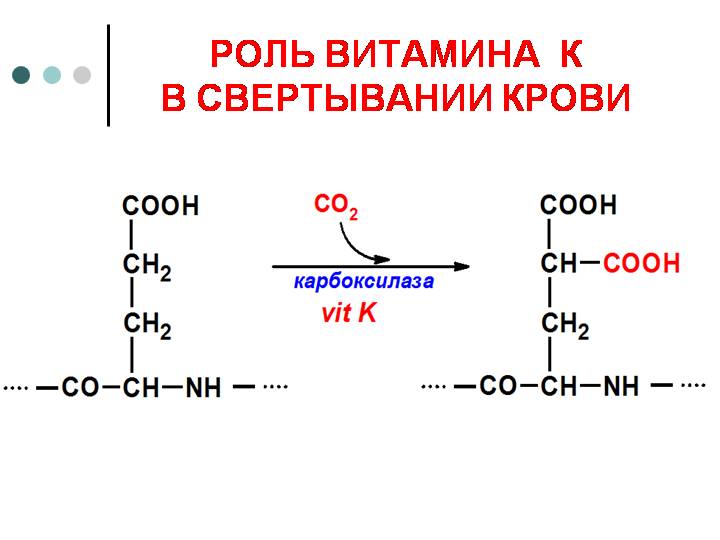
Роль витамина «К» в гемокоагуляции. Витамин К (К1, К2, К3, викасол и другие) является антигеморрагическим фактором. Он принимает участие в посттрансляционном созревании факторов II, VII, IX и Х свѐртывающей системы крови (а также в созревании витамин К-зависимых антикоагулянтов — протеинов С и S). Гамма-карбоксилирование остатков глутаминовой кислоты в молекуле этих белков протекает после трансляции, в эндоплазматическом ретикулуме гепатоцитов с участием глутамилкарбоксилазы. Роль кофактора в составе этого фермента выполняет восстановленная форма витамина К.
Наличие дополнительной -карбоксильной группы в остатках глутаминовой кислоты придает этим белкам способность при посредстве ионов кальция связываться на фосфолипидной поверхности и участвовать в реакциях гемокоагуляции. При авитаминозе К содержание витамин К-зависимых факторов системы свѐртывания в плазме крови не изменяется, но нарушается их способность связываться на поверхности тромбопластинов.
Длительная и выраженная гиперкоагуляция создает благоприятные условия для тромбообразования. Аномалии или дефицит факторов гемокоагуляции (коагулопатии) ведут к нарушению коагуляционного гемостаза, что сопровождается кровотечениями.
Роль кальция в гемостазе огромна. Большинство белков гемостаза имеют сайты связывания кальция. При удалении кальция из плазмы (например, при смешивании крови с цитратом натрия) активировать гемостатические реакции практически невозможно. Наиболее важные из известных функций кальция в гемостазе:
• Участие в образовании связей витамин-К-за-висимых факторов (II, VII, IX, X, протеин С,протеин S) с фосфолипидной поверхностью.
• Участие в активации фактора XIII.
• Участие в образовании связи ф.VII и тканевого фактора.
• Ускорение процесса роста фибринового сгустка, участие в стабилизации фибриновогосгустка, ограничение протеолиза фибрина и фибриногена плазмином, защита фибриногена и фибрина от температурной и щелочной денатурации.
• Стабилизация структуры многих белков гемостаза и опосредование взаимодействия между ними.
• Участие в процессах активации тромбоцитов и других клеток.
• Кальций необходим для формирования цитоскелета и возбуждения клетки. Он участвует в полимеризации актина и миозина и формировании актин-миозиновых волокон. Без него невозможны процессы изменения формы активированных клеток, их движение, секреция.
• Кальций участвует в регуляции большинства внутриклеточных процессов как внутриклеточный мессенджер (посредник) перемещения молекул.
Противосвёртывающая и фибринолитическая система
Противосвертывающая система.
К ним относят физиологические ингибиторы свёртывания, которые сохраняют кровь в жидком состоянии.
К ним относят три группы веществ:
а) Тромбомодулин
б) Система протеина – С
в) Ингибиторы активированных факторов свёртывания крови
а) Тромбомодулин (ТМ) – гликопротеид, интегральный белок мембран эндотелиальных клеток. Тм содержится в эндотелии вен, артерий капилляров и лимфатической системе большинства органов(по степени убывания сердце > поджелуд железы > легкие > скелетные мышцы > почки > печень > плацента) за исключением головного мозга . Тм не требует протеометической активации. Основная функция : Тм связывает тромбин(IIа )в присутствии Ca 2+ ! тем самым его инактивирует . Образуя Тм – IIа – Ca 2+ . Синтез Тм стимулируется тромбином, ц – АМФ, ретиноевой кислотой, пентоксифеллином и ИЛ – 4. Синтез Тм ингибируется ИЛ – 1, гипоксией, эндотоксином, герпетической инфекции.
б) Система протеина – С
Пр С – синтезируется в печени, в неактивной форме. В N в плазме= 4 г /л, Пр С активируется комплексом Тм – IIа – Ca 2+ путем частичного протеолиза. Активированный Пр С связывается с протеином S и с Ca 2+ с образованием акт Пр С – пр S – Ca 2+. Этот тройной комплекс инактивирует Vа и VIIIа путем гидролиза этих факторов. Сам же Пр S – синтезируется в печени при участии вит К и образует комплекс с Ca 2+ . Однако Пр С – S - Ca 2+ активирует плазминоген превращая его в плазмин.
в) Ингибиторы активированных факторов свёртывания крови:
α2 –малоглобулин Антиконвертин α1-антитрипсин
Антитромбин III - белок плазмы крови. Место синтеза печень и небольшое его количество синтезирует эндотелий. В свободном состоянии находится в неактивной форме. В физ. условиях антитромбин III связывается с гепарином и образует комплекс антитромбин III -геп. Который обладает активностью. 11 Гепарин- мукополисахарид , который синтезируется тучными клетками, имеющимися в различных тканях , и депонируются в них. Комплекс Антитромбин III – Гепарин – сильный ингибитор свёртывания крови. Он ингибирует IIа , IXа , Xа , XIIа, XIVa. Комплекс Анти III - геп не ингибирует VIIа и не влияет на факторы в составе мембранных комплексов, а устраняет ферменты, находящиеся в плазме, препятствуя распространения тромбообразования в кровотоке.
α2 –макроглобулин – синтезируется в печени, ингибирует IIа • Ca 2+.
Антиконвертин (тканевой ингибитор внешнего пути свёртывания)Синтезируется в эндотелии сосудов. Он связывается с комплексом III- VIIа - Ca2+ • ФЛ, после чего улавливается печенью и разрушается в ней.
α1 – антитрипсин- основное количество синтезируется печенью и находится в крови. В легких он синтезируется альвеолярными макрофагами.
α1 АТр неспецефический ингибитор IIа, XIa , XIVa.
α1 АТр ингибирует лейкоцитарную эластазу. Лейкоцитарная эластаза синтезируется в нейтрофиллах и выделяется во внеклеточное пространство. Лейкоцитарная эластаза (Эластаза нейтрофилов) разрушает эластин.
