

Концентрационная цепь, составленная из электродов II рода: Ag, AgCl | KCl(a1) || KCl(a2) | AgCl, Ag
0 0 |
RT |
|
RT |
|
RT |
|
a1 |
|
|
Уравнение Нернста: E=Eпр −E лев=Eпр−E лев+ |
|
ln a1− |
|
ln a2= |
|
ln |
|
. |
|
F |
F |
F |
a2 |
||||||
|
|
|
|
|
Полярность определяется путем сравнения концентраций. Правым (положительным) будет тот электрод, который опущен в раствор с более высокой концентрацией хлорид-ионов, т. к. только в этом случае E > 0.
Формальная кинетика
Скорость, константа скорости, основной постулат химической кинетики
Формальная кинетика рассматривает скорость химической реакции как функцию только концентрации.
Скорость любой гомогенной реакции по i-ому компоненту определяется как изменение количества этого компонента в единицу времени в единице объема реакционного пространства. Для реакции aA + bB → cC + dD:
ri =− |
1 dnA |
=− |
1 |
|
dnB |
= |
1 |
|
dnC |
= |
1 |
|
dnD |
. |
|||
|
|
|
|
|
|
|
|
|
|
|
|
||||||
V d τ |
V d τ |
V d τ |
V d τ |
||||||||||||||
|
|
|
|
|
|||||||||||||
Количество вещества в единице объема — это концентрация вещества:
r=− |
1 dC A |
=− |
1 dC B |
= |
1 dCC |
= |
1 dC D |
|||||||
|
|
|
|
|
|
|
|
|
|
|||||
a d τ |
b d τ |
c d τ |
d d τ |
|||||||||||
|
|
|
|
|||||||||||
Скорость всегда больше нуля, поэтому при определении скорости через исходное вещество ставим знак «-», т. к. концентрация исходных веществ убывает во времени.
Скорость зависит от концентраций исходных веществ, температуры, давления (в случае газофазной реакции).
Основной постулат химической кинетики — скорость реакции в каждый момент времени пропорциональна произведению концентраций исходных веществ, возведенных в некоторые
степени: r=k [ A]a [ B]b .
Коэффициент пропорциональности k называется константой скорости реакции. Константа скорости численно равна скорости химической реакции при единичной концентрации реагирующих веществ.
На размерность константы скорости влияет порядок реакции. Так для первого порядка k = [1/с], для второго порядка k = [л/моль*с]. Константа не зависит от концентрации реагирующих веществ, она постоянна при данной температуре.
Влияние температуры на константу скорости приближенно определяется правилом ВантГоффа: при повышении температуры на каждые 10 градусов скорость реакции увеличивается
|
k2 |
T 2−T1 |
|
в 2-4 раза. |
=γ 10 , где k2 – константа при новой температуре, k1 – константа при |
||
|
|||
|
k1 |
||
исходной температуре, γ — коэффициент Вант-Гоффа.
Кинетические кривые
Кинетической кривой называется график зависимости концентрации i-го компонента

реакции от времени.
Найдем зависимость концентрации от времени для реакций 0-3 порядков. Скорость реакции нулевого порядка не изменяется с течением времени:
|
−dC A |
=k dC A=−kdt C A=−kt+C0 - линейная убывающая функция для исходного |
|
||||||
|
|
||||||||
|
dt |
|
|
|
|
|
|
|
|
компонента. C0 – концентрация компонента A в начале реакции. |
|
|
|
||||||
Реакция первого порядка: |
−dC A |
=k [ A] |
dC A |
=−kdt lnC A=−kt+lnC0 |
C A=C0 e |
−kt |
- для |
||
dt |
[ A] |
|
|||||||
|
|
|
|
|
|
|
|
||
исходного компонента А график — убывающая экспонента.
Реакция второго порядка, если концентрации исходных веществ равны:
−dC A |
2 |
dC A |
|
1 |
|
|
1 |
|
|
|
|
C0 |
|
||||
|
|
=k [ A] |
|
=−kdt − |
|
=−kt− |
|
|
C A= |
|
|
|
|
|
|
- убывающая гипербола. |
|
|
dt |
[ A]2 |
C A |
C 0 |
C0 kt+1 |
||||||||||||
Реакция третьего порядка, если концентрации исходных веществ равны: |
|||||||||||||||||
−dC A |
|
dC A |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
1 |
|
|
1 |
|
|
1 |
|
|
|
|
|||||
|
|
=k [ A] |
|
=−kdt − |
|
=−kt− |
|
|
|
C A= |
|
|
|
|
|
|
|
|
dt |
3 |
2 |
|
2 |
|
1 |
|
|
|
|||||||
|
|
|
[ A] |
|
C A |
|
|
C0 |
|
√kt+ |
|
|
|
||||
|
|
|
|
|
|
|
C02 |
|
|||||||||
Тангенс угла наклона касательной, проведенной к какой-либо точке кривой, будет равен скорости реакции в данный момент времени по данному компоненту.
Для наглядности кинетические кривые строят в таких координатах, в которых кинетическая кривая становится прямой, т. е. линеаризуется. Для реакции 1 порядка это координаты lnC=f(t), для 2 порядка — 1/C = f(t).
Порядок реакции r=k [ A]a [ B]b
Частным порядком реакции называют порядок реакции по i-му компоненту. a – порядок реакции по компоненту a, b – порядок реакции по компоненту b. Общий порядок реакции — это сумма частных порядков: n = a + b.
Молекулярностью реакции называется количество частиц, принимающих участие в одном элементарном акте химического превращения.
Различают моно-, би- и тримолекулярные реакции. Участие в реакции более трех частиц следует рассматривать как сложную реакцию, протекающую через несколько элементарных стадий, каждая из которых моно-, биили тримолекулярна. Для простых одностадийных гомогенных реакций молекулярность совпадает с порядком реакции.
Временем полупревращения называется такой промежуток времени, за который исходная концентрация реагентов снизится вдвое.
Найдем время полупревращения для реакций 0-3 порядков.
Реакция нулевого порядка: C A=−kt+C 0 C20 =−kt1/ 2+C0 t1 /2= C2k0 Реакция первого порядка: C A=C0 e−kt C20 =C0 e−kt1/2 ln 12 =−kt1 /2 t1/ 2= ln2k

Реакция второго порядка: |
C |
= |
|
|
C0 |
|
C0 |
= |
|
|
|
C0 |
|
|
|
C |
kt |
|
+1=2 t |
|
= |
1 |
|||||||
C0 kt+1 |
2 |
|
C |
0 kt1/ 2+1 |
|
|
kC0 |
||||||||||||||||||||||
|
A |
|
|
|
|
|
|
|
0 |
|
1/ 2 |
|
|
|
1/ 2 |
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
1 |
|
|
|
|
|
|
1 |
1 |
1 |
|
|
|
|
|
3 |
|
|
|
|
|||||||
Реакция третьего порядка: |
C A=√ |
|
|
|
|
k= |
|
( |
|
− |
|
) t1 / 2= |
|
|
|
|
|
||||||||||||
kt+ |
1 |
|
2t |
C 2 |
C02 |
2kC02 |
|
|
|
|
|||||||||||||||||||
|
C02 |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
Степень превращения — это отношение количества молей вещества, вступившего в реакцию,
к исходному числу молей: α |
= |
n |
= |
C0 −C |
=1− |
C |
. |
|
ni |
C0 |
C 0 |
||||||
i |
|
|
|
|
Интегральные методы нахождения порядка реакции.
1)Метод подстановки — подставляем полученные значения времени и концентрации исходного вещества в кинетические уравнения разных порядков, находим константы реакции. Порядок реакции тот, в каком уравнении константа остается постоянной для всех значений.
2)Графический метод — строим графики в координатах C – t, lnC – t, 1/C – t, 1/C2 – t. Какой из этих графиков окажется прямой линией, такой порядок у реакции.
3)Способ определения времени полупревращения — проводим несколько опытов при различных начальных концентрациях и определяем периоды полупревращения. По зависимости периода полупревращения от начальной концентрации определяем порядок реакции. В рамках этого способа можно вычислить порядок реакции по этой
формуле: n= lg t'1'/ 2−lg t'1/ 2 +1 , зная периоды полупревращения при двух различных lg c''0 −lg c'0
начальных концентрациях. Порядок реакции может быть дробным.
4)Метод Эмануэля-Кнорре позволяет вычислить порядок реакции по формуле:
|
ln[ |
t ' |
|
] |
|
|
|
||
n=1+ |
(t ' ' − |
t ') |
, где α '=1− |
c' |
- степень превращения вещества в момент |
||||
lg (1−α ') |
|
c0 |
|||||||
|
|
|
|||||||
времени t', t' – произвольно выбранный момент времени, t'' – такой момент времени, в который c'' = (c')2.
Дифференциальные методы определения порядка реакции.
Реакция aA + bB → продукты.
|
1 dcA |
nA |
nB |
|
|||
Скорость реакции r=− |
|
|
|
=k1 cA |
cB |
, где nA, nB – частные порядки реакций. |
|
a dt |
|||||||
|
|
|
|
||||
Метод изоляции Оствальда. Проводим нужную реакцию сначала в условиях избытка всех исходных веществ, кроме одного. Тогда можно пренебречь изменением концентраций всех реагентов, кроме того, по которому ведется расчет. Тогда концентрацию всех компонентов можно внести в постоянный коэффициент, и скорость реакции можно записать так:
−rA=− |
dc1 |
=k1 c1n1 , где |
k1=akc20n2 |
, n1 – порядок реакции по первому веществу. |
|
||||
|
dt |
|
|
|
Способ логарифмирования |
|
|
||
Прологарифмируем это выражение: |
ln−rA=ln k 1+n1 ln c1 . |
|||
Построим график в координатах ln(-rA) – lnc1, все точки должны лечь на прямую линию.
Тангенс угла наклона этой прямой будет равен порядку реакции по веществу А. Этим методом определяется концентрационный порядок.
Влияние температуры на скорость химической реакции
Влияние температуры на константу скорости приближенно определяется правилом ВантГоффа: при повышении температуры на каждые 10 градусов скорость элементарной
|
k2 |
T 2−T1 |
|
гомогенной реакции увеличивается в 2-4 раза. |
=γ 10 , где k2 – константа при новой |
||
|
|||
|
k1 |
||
температуре, k1 – константа при исходной температуре, γ — коэффициент Вант-Гоффа. Коэффициент Вант-Гоффа является функцией температуры, при очень высоких и очень низких температурах он становится равным единице, т. е. реакция перестает зависеть от изменения Т.
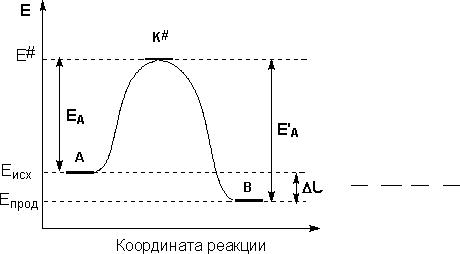
Более полно зависимость скорости реакции от температуры описывает уравнение Аррениуса:
дlnk |
= |
Ea |
, где Ea – энергия активации — минимальная избыточная энергия по |
|
дT |
RT 2 |
|||
|
|
сравнению со средней, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию.
|
Ea |
; k= Ae− |
Ea |
|
|
Проинтегрировав это уравнение, получаем: lnk=lnA− |
RT |
. A – |
|||
RT |
|||||
|
|
|
|
предэкспоненциальный множитель — численно равен константе скорости химической реакции при бесконечно большой температуре.
Построив график в координатах lnk – 1/T, получим прямую линию, которая отсекает на вертикальной оси отрезок длиной lnA, а тангенс угла наклона которой равен Ea/R.
Для расчета энергии активации необходимо знать значений констант скоростей при различных температурах, чтобы построить данный график.
Уравнение Аррениуса позволяет рассчитать константу скорости при определенной температуре, зная константу скорости при другой температуре:
ln kk21 = ERa (T11 − T12 )
Для определения энергии активации в первом приближении достаточно знать две константы скорости при двух различных Т.
Температурный коэффициент Вант-Гоффа можно связать с энергией активации:
|
|
k |
2 |
|
|
|
Ea |
( |
1 |
|
|
|
|
1 |
) |
k |
2 |
=exp[ |
Ea |
( |
1 |
|
1 |
)]=γ |
T 2−T1 |
|
|
||||||||||||
ln |
= |
|
− |
− |
10 |
|
|
|
|||||||||||||||||||||||||||||||
k |
1 |
R |
T 1 |
T 2 |
k |
1 |
R |
T 1 |
T 2 |
|
|
||||||||||||||||||||||||||||
|
|
E T −T |
|
|
10 |
|
|
|
|
|
|
|
E T −T |
|
10 |
|
|
|
E |
10 |
|
||||||||||||||||||
|
|
|
T |
−T |
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
γ =exp[ |
|
a |
|
2 1 |
] |
2 |
|
|
|
1 =exp[ |
|
a |
|
2 1 |
|
]=exp[ |
a |
|
] |
||||||||||||||||||||
|
R |
T 1 T 2 |
|
|
|
|
|
R |
T 1 T 2 |
T 2−T 1 |
R |
T 1 T 2 |
|||||||||||||||||||||||||||
|
|
|
|
|
|
|
lnγ = |
|
10 Ea |
Ea =0,1 R T 1 T 2 ln γ |
|
|
|
|
|
||||||||||||||||||||||||
|
|
|
|
|
|
|
RT 1 T 2 |
|
|
|
|
|
|||||||||||||||||||||||||||