

билеты 19
.pdfплотность Al2O3 4 г/см3, вязкость среды 10-3 Па·с, температура 25ºС.
P
Билет 7
1.Мономолекулярная адсорбция, форма изотермы адсорбции. Уравнение Генри. Основные положения теории Ленгмюра.
Закон Генри можно сформулировать следующим образом: при разбавлении системы (уменьшение давления) коэффициент распределения стремится к постоянному значению, равному константе распределении Генри. Относительно величины адсорбции А этот закон
запишется так: 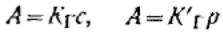
Эти уравнения представляют собой изотермы адсорбции вещества при малых концентрациях. В соответствии с ними закон Генри можно сформулировать так: величина адсорбции при малых давлениях газа (концентрациях вещества в растворе) прямо пропорциональна давлению (концентрации).
Отклонения от закона Генри, выражаемые изменениями коэффициентов активности в фазах, обычно не позволяют описать и прогнозировать ход изотерм с увеличением концентрации. (давления) адсорбата. Чтобы получить теоретическую изотерму адсорбции, описывающую более широкую область концентраций, необходимо использование представлений о механизме адсорбции и конкретных моделей.
Большую долю отклонений коэффициента активности адсорбата в поверхностном слое от единицы можно учесть, используя представление об адсорбции как о квазихимической реакции между адсорбатом и адсорбционными центрами поверхности адсорбента. В этом заключается основная идея адсорбционной теории Ленгмюра. Это положение уточняется следующими допущениями:
1)адсорбция локализована (молекулы не перемещаются по поверхности) на отдельных адсорбционных центрах, каждый из которых взаимодействует только с одной молекулой адсорбата; в результате образуется мономолекулярный слой;
2)адсорбционные центры энергетически эквивалентны — поверхность адсорбента эквипотенциальна;
3)адсорбированные молекулы не взаимодействуют друг с другом.
2.Современная теория строения ДЭС (теория Штерна) роль специфической адсорбции, перезарядка поверхности. Примеры образования ДЭС. Строение мицеллы (формулы ДЭС).
J
Современная теория строения двойного электрического слоя основана на представлениях Штерна. Она объединяет две предыдущие теории. Согласно современной теории слой противоионов состоит из двух частей.
Одна часть примыкает непосредственно к межфазной поверхности и образует адсорбционный слои (слой Гельмгсиьца) толщиной δ, которая равна радиусу гидратированных ионов, его составляющих. Другая часть противононов находится в диффузной части — диффузный слой (слой Гуи) с потенциалом φδ, толщина λ которой может быть значительной и зависит от свойств и состава системы. Потенциал в диффузной части двойного электрического слоя не может зависеть линейно от расстояния, так как ионы в нем распределены неравномерно. В соответствии с принятыми представлениями потенциал в слое Гельмгольца при увеличении расстояния от слоя потенциалопределяющих ионов снижается до потенциала диффузного слоя линейно, а затем, как будет показано, по экспоненте. Теория Штерна учитывает также специфическую (некулоновскую, химическую) составляющую адсорбции ионов на поверхности раздела фаз, которая существенным образом может влиять на изменение потенциала.
Пренебрежение размерами ионов приводит к тому, что не принимается во внимание толщина адсорбционною слон, и это, в свою очередь, вызывает большие погрешности при расчете параметров двойного электрического слоя. Кроме того, теория Гуи — Чепмена рассматривая только влияние концентрации и заряда ионов электролитов на изменение потенциала, не объясняет различного действия ионов разной природы, связанного со специфической адсорбцией их на межфазной поверхности.
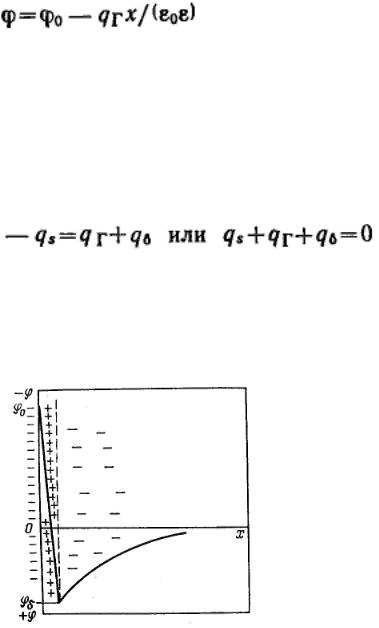
Штерн предложил рассматривать слой противоионов состоящим из двух частей: внутренней (плотный слой Гельмгольца) и внешней (диффузный слой). Таким образом, теорию Гуи — Чепмена можно использовать для описания только строения внешней части слоя, где можно пренебречь адсорбционными силами и размерами ионов. Внутреннюю (плотную) часть Штерн представил как адсорбционный моноионный слой, в котором противоионы примыкают к поверхности благодаря электростатическим силам и специфическому взаимодействию. Введенный Штерном потенциал φδ часто называют штерновским. В плотной части двойного электрического слоя потенциал уменьшается линейно от φ0 до φδ. Принимая текущими переменными φ и х вместо φδ и δ, получим:
Штерн попытался учесть влияние специфической адсорбции ионов на электрический потенциал, обусловленный действием ковалентных сил дополнительно к электростатическим силам. Так как радиус действия сил такой адсорбции соизмерим с размером ионов, это дает основание учитывать копалентные силы только для ионов, входящих в плотный слой Гельмгольца. Как видно из рисунка, плотность поверхностного заряда противоионов можно разделить на две части: плотность заряда qГ, обусловленного моноионным слоем, представляющим собой слой Гельмгольца, и плотность заряда qδ диффузного слоя Гуи. Общая поверхностная плотность заряда двойного электрического слоя равна сумме поверхностных плотностей зарядов плотного и диффузного слоев с обратным знаком:
По Штерну, заряд слоя Гельмгольца складывается из заряда ионов, адсорбированых как за счет электростатического адсорбционного потенциала Fzφ, так и за счет потенциала специфической адсорбции Ф. Было предположено, что поверхность имеет определенное число адсорбционных центров, каждый из которых взаимодействует с одним противоионом. Пример образования ДЭС:
Добавление в систему металл — вода раствора хлорида натрия приводит к избирательной адсорбции хлорид-анионов на поверхности металла. Появляется избыточный отрицательный заряд на поверхности металла и избыточный положительный заряд (ионы натрия) в близлежащем слое раствора, т. е. на межфазной поверхности образуется двойной электрический слой.
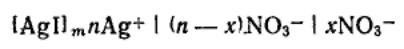
Сильно адсорбирующиеся ионы в плотном слое иногда способны не только полностью скомпенсировать поверхностный потенциал, но и создать избыточный заряд со знаком заряда противоионов. Это явление называется перезарядкой. Перезарядка приводит к смене противоионов в диффузном слое на ионы с зарядом другого знака.
На рисунке видно, что при перезарядке поверхностный потенциал φ0 и потенциал диффузного слоя φδ имеют разные знаки.
На формирование двойного электрического слоя существенное влияние оказывает природа поверхности конденсированной фазы, наличие определенных ионов в растворе, их концентрация. Рассмотрим систему водный раствор — поверхность иодида серебра. При избытке в растворе ионов серебра, например при добавлении нитрата серебра, эти ионы являются потенцналопределяюшими. В роли противоионов выступают нитрат-ионы, часть которых находится в плотном слое, а другая часть — в диффузном слое. Для такой системы формулу двойного электрического слоя можно записать следующим образом:
В дисперсных системах двойной электрический слой возникает на поверхности частиц. Частицу дисперсной фазы в гетерогенно-дисперсной системе вместе с двойным электрическим слоем называют мицеллой. Строение мицеллы можно показать той же формулой, что и строение двойного электрического слоя. Внутреннюю часть мицеллы составляет агрегат основного вещества. На поверхности агрегата расположены потенциалопре-деляющие ионы. Агрегат вместе с потенциалопределяющими ионами составляет ядро мицеллы. Ядро с противоионами плотной части двойного электрического слоя образуют гранулу. Гранулу окружают противоионы диффузного слоя. Мицелла в отличие от гранулы электронейтральна.
{(AgI)mnAg+|(n-x)NO3-}xNO3-
3.Рассчитайте избыточное давление внутри капель воды и равновесное давление пара при 20 ºС для аэрозоля с удельной поверхностью 108 м-1. Поверхностное натяжение воды 73 мДж/м2, плотность 1 г/см3, PS = 2306 Па.

Билет 8
1.Влияние дисперсности на термодинамическую реакционную способность. Вывод уравнения капиллярной конденсации Кельвина. Влияние дисперсности на растворимость, константу равновесия химической реакции и температуру фазового перехода.
Термодинамическая реакционная способность характеризует способность вещества переходить в какое-либо иное состояние, например переходить в другую фазу, вступать в химическую реакцию. Она указывает на удаленность данного состояния вешества или системы компонентов от равновесного состояния при определенных условиях. Термодинамическая реакционная способность определяется химическим сродством, которое можно выразить изменением энергии Гиббса или разностью химических потенциалов. Реакционная способность зависит от степени дисперсности вещества, изменение которой может приводить к сдвигу фазового или химического равновесия.
Соответствующее приращение энергии Гиббса dGд (благодаря изменению дисперсности) можно представить в виде объединенного уравнения первого и второго начал термодинамики:
Для индивидуального вещества V=Vм и при Т=const имеем:
Подставляя в это уравнение соотношение Лапласа, получим: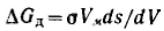
для сферической кривизны: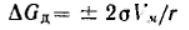 Если рассматривается переход вещества из конденсированной фазы в газообразную, то
Если рассматривается переход вещества из конденсированной фазы в газообразную, то
энергию Гиббса можно выразить через давление пара, приняв его за идеальный.

Дополнительное изменение энергии Гиббса, связанное с изменением дисперсности,
составляет: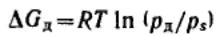
Подставляя данное выражение, получим:
Полученное соотношение называется уравнением Кельвина (уравнение капиллярной конденсации).(pд-равновесное давление адсорбата, ps-насыщ давление пара адсорбата) Для неэлектролитов его можно записать следующим образом:
Из этого уравнения видно, что с увеличением дисперсности растворимость растет, или химический потенциал частиц дисперсной системы больше, чем у крупной частицы, на величину 2σV/r.
Степень дисперсности может влиять также на равновесие химической реакции:
Сувеличением дисперсности повышается активность компонентов, а в соответствии с этим изменяется константа химического равновесия в ту или другую сторону, в зависимости от степени дисперсности исходных веществ и продуктов реакции.
Сизменением дисперсности веществ изменяется температура фазового перехода. Количественная взаимосвязь между температурой фазового перехода и дисперсностью вытекает из термодинамических соотношений.
Для фазового перехода: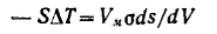 ,
,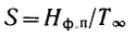
Для сферических частиц: 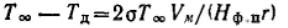
Видно, что с уменьшением размера частиц г температуры плавления и испарения вещества уменьшаются (Hф.п.>0).
2.Седиментационно-диффузное равновесие. Вывод уравнения(гипсометрический закон Лапласа). Мера седиментационной устойчивости. Факторы, влияющие на седиментационную устойчивость дисперсных систем.
В золях через определенное, иногда очень длительное, время оседания частиц может наступить момент, когда диффузионный поток станет равным седиментационному iдиф = iсед, т.е. наступит диффузионно-седцментационное равновесие. Так как такое равновесие наступает при определенном градиенте концентраций, в системе должно установиться соответствующее распределение дисперсной фазы по высоте. Чтобы определить закон этого распределения, воспользуемся данным соотношением (iдиф = iсед), учтя, что
J
и заменив x на h (расстояние по высоте):
J
После разделения переменных получим:
J
Интегрируя в пределах от ν0 до νh и соответственно от h = 0 до h, найдем:
J илиJ
Это уравнение носит название гипсометрического закона (от лат. hypsos — высота).

Если сравнить седиментацию при наличии диффузии и без нее, то обращает на себя внимание различие факторов, обеспечивающих устойчивость дисперсных систем к осаждению — седиментационную устойчивость. Эти факторы позволяют различать кинетическую седиментационную устойчивость (КСУ) и термодинамическую седиментационную устойчивость (ТСУ).
Мерой кинетической седиментационной устойчивости является величина, обратная константе седиментации:
J
Мерой ТСУ является гипсометрическая высота. Ее удобнее определить как высоту he, на протяжении которой концентрация дисперсной фазы изменяется в е раз.
J
Данная формула показывает, что гипсометрическая высота и соответственно термодинамическая седиментационная устойчивость тем больше, чем меньше размер частиц и разность между плотностями частиц и среды. Вязкость не влияет на ТСУ, в то (же время повышение температуры способствует устойчивости, так как усиливается тепловое движение. Кинетическая же седиментационная устойчивость с повышением температуры обычно снижается в связи с уменьшением вязкости среды
3.Рассчитайте разность уровней воды в двух сообщающихся капиллярах диаметрами 0,1 и 0,3 мм при 20 ºС. Поверхностное натяжение и плотность воды составляют соответственно 72,75 мДж/м2 и 0,998 г/см3.
&
Билет 9
1.Правило фаз Гиббса и дисперсность. Влияние кривизны поверхности (дисперсности) на внутреннее давление тел (вывод и анализ уравнения Лапласа). Капиллярные явления (уравнение Жюрена).
Дисперсность является самостоятельным и полноправным термодинамическим параметром системы, а для дисперсных систем правило фаз Гиббса принимает следующий вид:
F – количество степеней свободы, К – количество компонентов, Ф – количество фаз.
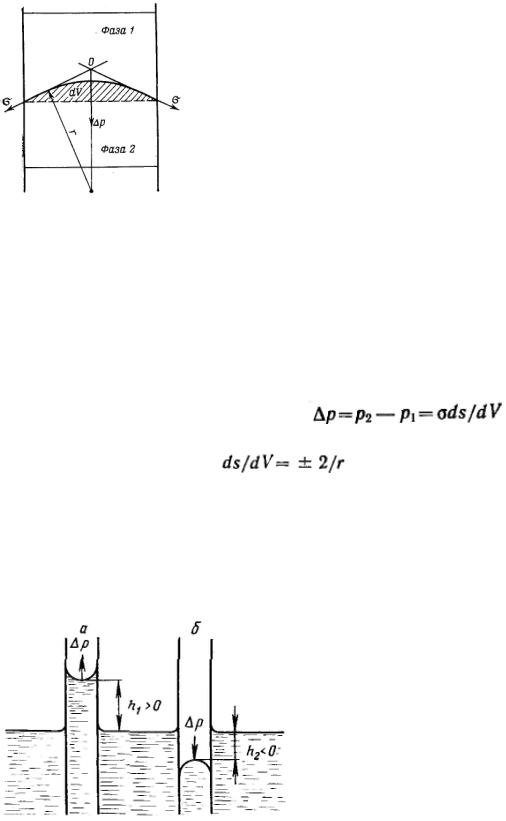
Рассмотрим результат влияния кривизны поверхности раздела между двумя несмешивающимися жидкостями на внутреннее давление в фазах.
Кривизна вызывает изменение площади и положения межфазной поверхности, что можно выразить приращением поверхностной энергии σds. Кроме того, изменяются объемы фаз V1 и V2 на dV1 и dV2. При условии постоянства объема всей системы dV1 = - dV2. Изменение объемов вызывает соответствующие изменения энергий фаз 1 и 2 на p1dV1 и p2dV2 (где p1 и р2 — давления внутри фаз). Соотношение между поверхностной энергией и «объемной» можно записать с помощью обобщенного уравнения первого и второго начал термодинамики
относительно энергии Гельмгольца F при T=const: 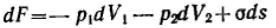
При равновесии между фазами |
F = 0, тогда |
Это уравнение называется уравнением Лапласа. |
|
Для сферической поверхности |
и уравнение принимает вид |
 .
.
Капиллярные явления наблюдаются в содержащих жидкость узких сосудах (капилляры, капиллярно-пористые тела), у которых расстояние между стенками соизмеримо с радиусом кривизны поверхности жидкости. Кривизна возникает в результате взаимодействия жидкости со стенками сосуда (адгезия, смачивание). Специфика поведения жидкости в капиллярных сосудах зависит от того, смачивает или не смачивает
Рассмотрим положение уровней жидкости в двух капиллярах, один из которых имеет лиофильную поверхность и поэтому стенки его смачиваются, у другого внутренняя поверхность лиофобизирована и не смачивается. В первом капилляре поверхность жидкости имеет отрицательную кривизну, поэтому дополнительное давление Лапласа стремится растянуть жидкость (давление направлено к центру кривизны) и поднимает ее в капилляре. Кривизна поверхности жидкости во втором капилляре положительна, дополнительное давление направлено внутрь жидкости, в результате жидкость в капилляре опускается
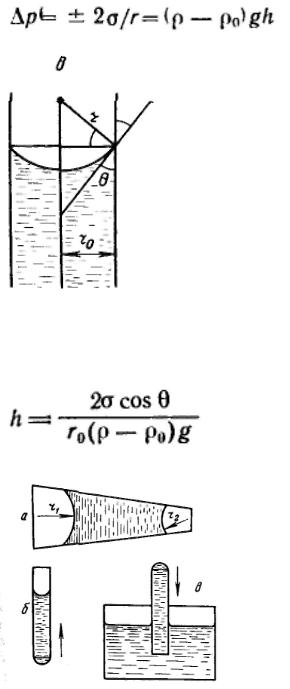
(отрицательное капиллярное поднятие). При равновесии лапласовское давление равно гидростатическому давлению столба жидкости высотойh:
Чтобы высоту капиллярного поднятия связать с характеристикой смачивания, радиус мениска необходимо выразить через угол смачивания θ и радиус капилляра r0. На рисунке показан мениск жидкости в капилляре. Видно, что r0 = r·соsθ, тогда высоту капиллярного поднятия можно представить в виде формулы Жюрена:
Нередко приходится наблюдать, как жидкость не может вытечь из капилляра под действием силы тяжести. Это объясняется проявлением действия капиллярного потенциала, направленного против силы тяжести, так как на нижнем конце капилляра жидкость образует мениск с положительной кривизной. Если часть капилляра, находящаяся над жидкостью, меньше высоты поднятия жидкости, жидкость из него не вытекает, так как кривизна мениска жидкости вверху капилляра становится положительной (положительный радиус кривизны), отвечающей гидростатическому давлению столба жидкости, равному размеру (высоте) капилляра, т. е. устанавливается равновесие.
2.Природа броуновского движения, понятие и определение среднеквадратичного сдвига по выбранному ?. Взаимосвязь между среднеквалратичным сдвигом и коэффициентом диффузии (вывод закона Эйнштейна-Смолуховского). Экспериментальная проверка закона.

Основой доказательства теплового молекулярного движения в телах явилось обнаруженное английским ботаником Робертом Броуном в 1827 г. с помощью микроскопа непрерывное движете очень мелких частичек — спор папоротника (цветочной пыльцы), взвешенных в воде. Более крупные частицы находились в состоянии постоянного колебания около положения равновесия. Колебания и перемещения частиц ускорялись с уменьшением их размера и повышением температуры и не были связаны с какими-либо внешними механическими воздействиями.
Теоретически обоснованная интерпретация броуновского движения — участие частиц дисперсной фазы ультрамикрогетерогенных систем в тепловом движении — была дана независимо друг от друга Эйнштейнии (1905 г.) и Смолуховским (1906 г.).
Проведенными исследованиями была окончательно доказана природа броуновского движения. Молекулы среды (жидкости или газа) сталкиваются с частицей дисперсной фазы, в результате чего она получает огромное число ударов со всех сторон.
Эйнштейн и Смолуховский для количественного выражения броуновского движения частиц ввели представление о среднем сдвиге частицы. Если при наблюдении движения частицы золя под микроскопом через определенные равные промежутки времени отмечать ее местонахождение, то можно получить ее траекторию движения. Так как движение происходит в трехмерном пространстве, то квадрат среднего расстояния, проходимого
частицей за любой промежуток времени, равен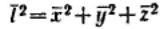 .
.
Под микроскопам наблюдают проекцию смещения частицы на плоскость за какое-то время,
поэтому 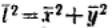 .
.
При равновероятных отклонениях частицы ее направление будет находиться между направлениями x и у, т. е. под углом 45° к каждой координате. Отсюда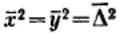 или
или
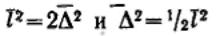 .
.
Из-за равновероятных отклонений среднеарифметическое значение сдвигов равно нулю. Поэтому используются среднеквадратичные расстояния, проходимые частицей:
Эйнштейн и Смолуховский, постулируя единство природы броуновского движения и теплового движения, установили количественную связь между средним сдвигом частицы (называемым иногда амплитудой смещения) и коэффициентом диффузии D.
Если броуновское движение является следствием теплового движения молекул среды, то можно говорить о тепловом движении частиц дисперсной фазы. Это означает, что дисперсная фаза, представляющая собой совокупность числа частиц, должна подчиняться тем же статистическим законам молекулярно-кинетической теории, приложимым к газам или растворам.