

Хроматометрия
Основной полуреакцией методов хроматометрии является процесс окисления дихроматом калия в кислой среде:
Cr2O72- +
14H+ +6e- → 2Cr3+ +
7H2O;
![]() =1,33 В.
=1,33 В.
По основному реактиву эту группу методов иногда называют дихроматометрией. Как показывает величина стандартного окислительно-восстановительного потенциала, дихромат является менее сильным окислителем, чем перманганат, и его практическая применимость поэтому не столь широка.
Рабочим раствором хроматометрии является раствор дихромата калия K2Cr2O7. Кристаллический препарат K2Cr2O7 легко приготовить достаточно чистым, он устойчив при хранении на воздухе, поэтому титрованные растворы дихромата калия готовят по точной навескекристаллической соли. Раствор дихромата калия сохраняет неизменный титр длительное, неопределенно долгое время, его можно кипятить без разложения. Железо (II) титруется дихроматом в солянокислой среде без каких-либо побочных процессов и осложнений. Недостатком реактива является образование в результате реакции окрашенных ионов Cr3+ , затрудняющих своей окраской фиксирование точки эквивалентности. Поэтому часто возникает необходимость в применении индикаторов. В качестве индикаторов используют дифениламин, а также дифениламинсульфоновую кислоту, фенилантраниловую кислоту и др.
Наиболее важными практическими применениями хроматометрии являются определение железа в различных пробах после предварительного восстановления его до Fe(II), а также урана, который предварительно переводят в U(IV). Кроме того, реакция титрования железа(II) дихроматом является заключительным этапом различных аналитических методик, основанных на реакциях взаимодействия Fe(III) или Fe(II) с определяемым веществом. Восстановители анализируют по методу замещения. Например, Cu(I) реагирует с Fe(III) в кислой среде:
Cu+ + Fe3+ = Fe2+ + Cu2+.
В результате реакции в растворе появляется количество ионов Fe2+, эквивалентное количеству Cu(I) в исходной пробе. Окислители определяют методом обратного титрования. Например, хром в сталях окисляют до Cr2O72-, добавляют избыток титрованного раствора соли Мора(NH4)2Fe(SO4)2.6H2O, и не вошедшее в реакцию количество оттитровывают дихроматом.
Нитраты в растворе определяют, обрабатывая пробу раствором соли Мора при кипячении в инертной атмосфере в присутствии молибдата каккатализатора:
3Fe2+ + NO-3 + 4H+ = 3Fe3+ + NO + 2H2O.
После охлаждения раствор титруют дихроматом.
Некоторые катионы титруют дихроматом непосредственно (Sn2+, Sb3+ и др.).
Хроматометрическое определение органических веществ широкого практического применения не получило в связи с неполнотой протекания многих реакций и появлением побочных продуктов (СО наряду с СО2 и др.), количество которых контролировать не удается. Ряд органических веществ, однако, легко окисляется дихроматом до СО2 и Н2О, и их хроматометрическое определение имеет практическое значение. Это метанол,глицерин и некоторые другие вещества.
Иодометрия
Иодометрические методы основаны на применении стандартного раствора тиосульфата натрия для титрования иода, выделившегося при взаимодействии определяемого окислителя с избытком иодида калия (при титровании по замещению) или оставшегося в избытке при медленном взаимодействии определяемого восстановителя с фиксированным объемом стандартного раствора иода (в случае обратного титрования).
Основы метода
Иодид-ион является восстановителем умеренной силы, его применяют для определения большого числа окислителей. Прямое титрованиестандартным раствором KI не используют из-за трудностей индикации конечной точки титрования: прекращение образования свободного иода с помощью крахмала заметить нельзя. Поэтому для определения окислителей иодометрическим методом применяют способ титрования по замещению. Прямое титрование окислителей стандартным раствором тиосульфата натрия невозможно в связи с тем, что только I2 в нейтральной среде окисляет ион S2O32- быстро в соответствии со стехиометрией реакции
I2 + 2S2O32-→ 2I- + S4O62-.
Другие окислители обладают способностью полностью или частично окислять тиосульфат до серы, сульфата или тетратионата, например
4HOI + S2O32- + H2O → 2SO42- + 4I- + 6H+.
При титровании иода раствором тиосульфата наиболее благоприятна нейтральная либо слабокислая среда. Высокая кислотность раствора приводит к разложению тиосульфата:
S2O32- + 2H+→ H2SO3 + S
H2SO3 реагирует с I2 в мольном соотношении 1:1
H2SO3 + I2 + H2O → SO42- + 4H+ + 2I-,
тогда как на 1 моль Na2S2O3 расходуется ½ моль I2. В щелочной среде иодометрическое определение также не следует проводить из-за реакции диспропорционирования иода:
I2 + 2OH- → IO- + I- +H2O.
Приготовление и стандартизация раствора тиосульфата натрия
Растворы тиосульфата обычно готовят из кристаллического Na2S2О35Н2О, который при хранении постепенно теряет часть кристаллизационной воды. Свежеприготовленные растворы первое время медленно изменяют свои характеристики вследствие разложения тиосульфата натрия. По этим причинам готовят обычно раствор приблизительно необходимой концентрации и стандартизируют его по другому исходному веществу.
Важнейшими факторами, определяющими устойчивость раствора тиосульфата, являются значение рН, присутствие микроорганизмов и примесей, концентрация раствора, присутствие атмосферного кислорода и воздействие прямого солнечного света. Для приготовления растворов тиосульфата следует применять дистиллированную воду, не содержащую примесей ионов тяжелых металлов – катализаторов окисления тиосульфата кислородом воздуха
2Na2S2O3 + O2 → 2Na2SO4 + 2S.
В присутствии катализаторов (ионов Сu2+, Fe3+) реакция разложения Na2S2O3 ускоряется за счет образования неустойчивых на воздухе ионов металла в низших степенях окисления, например:
2Сu2+ + 2S2O32- → S4O62- + 2Cu+,
2Сu+ + ½ O2 + Н2О → 2Сu2+ + 2OН-.
Для приготовления растворов рекомендуется свежепрокипяченная вода, так как бактерии разлагают растворы тиосульфата. Активность бактерий при рН = 9–10 минимальна. Для подавления роста бактерий можно добавлять такие вещества, как хлороформ, бензоат натрия или НgI2. Кипячение воды для приготовления растворов тиосульфата также обеспечивает удаление растворенного СО2, под влиянием которого будут изменяться характеристики раствора, поскольку восстановителем вместо Na2S2O3 будет выступать NaНSO3, образующийся в реакции:
Na2S2O3 + СО2 + Н2О → NaНСO3 + NaНSO3 + S.
Добавление небольших количеств (0,1г/л) Na2СО3 способствует удалению СО2
Na2СО3 + СО2 + Н2О → 2 NaНСO3.
Скорость разложения тиосульфата возрастает с уменьшением его концентрации в растворе. Если в растворе появляется муть, то такой раствор следует заменить. Наиболее часто Na2S2O3 готовят в виде 0,05 н. растворов. Навеску рассчитывают, принимая fэ(Na2S2O3)=1/2.
Для приготовления раствора тиосульфата натрия (вторичного стандарта) дистиллированную воду для растворения предварительно кипятят 1 ч, охлаждают в колбе, закрытой пробкой с U-образной трубкой, наполненной твердым КОН. Взвешенную в бюксе на технических весах навескуNa2S2O3×5Н2О растворяют в свежепрокипяченной и охлажденной дистиллированной воде. Раствор хранят в темном месте в хорошо закрытой посуде. Стандартизацию раствора Na2S2O3 проводят обычно через 5-7 дней.
15.
В качестве первичного стандарта для растворов Na2S2O3 могут быть окислители КВrО3, КIO3, выделяющие при взаимодействии с избытком иодид-ионов эквивалентное количество иода, который титруют стандартизируемым раствором тиосульфата. Чаще используют бихромат калияК2Сr2О7, который можно легко получить в химически чистом состоянии перекристаллизацией; он негигроскопичен и не содержит кристаллизационной воды; растворы его устойчивы при продолжительном хранении. Способ стандартизации основан на реакциях
Сr2О72- + 6I- + 14Н+ → 2Сr3+ + 3I2 + 7Н2О,
I2 + 2S2O32- → 2I- + S4О62-.
Методика приготовления раствора К2Сr2О7 аналогична методике приготовления первичного стандарта Nа2С2О4. При расчете навески следует помнить, что fэ(К2Сr2О7)=1/6.
В коническую колбу для титрования переносят пипеткой раствор установочного вещества К2Сr2О7, добавляют 10 мл 2н. раствора Н2SО4 и 1 г кристаллического КI (или соответствующий объем его концентрированного раствора). Колбы закрывают стеклянными пробками или накрывают часовыми стеклами, содержимое перемешивают, дают постоять 5-10 мин в темном месте, пока не завершится реакция. Выделившийся I2 титруют раствором Na2S2O3 до тех пор, пока окраска не станет слабо-желтой, затем титруемый раствор разбавляют водой приблизительно в 2 раза, добавляют индикатор крахмал и заканчивают титрование в тот момент, когда синяя окраска раствора перейдет в светло-зеленую (ионы Сr3+).
17.


Точку эквивалентности в Т. а. определяют по изменению окраски титруемого раствора или индикатора, вводимого в начале или в процессе титрования, изменению электропроводности раствора, изменению потенциала электрода, погруженного в титруемый раствор, изменению величины тока, оптической плотности и др.
Одним из широко применяемых способов фиксации точки эквивалентности является индикаторный метод. Индикаторы — вещества, которые дают возможность установить конечную точку титрования (момент резкого изменения окраски титруемого раствора). Наиболее часто индикатор добавляют ко всему титруемому раствору (внутренний индикатор). При работе с внешними индикаторами периодически берут каплю титруемого раствора и смешивают с каплей раствора индикатора или помещают на индикаторную бумагу (что приводит к потерям анализируемого вещества).
Процесс титрования изображают графически в виде кривых титрования, которые позволяют наглядно представить весь ход титрования и выбрать индикатор, наиболее пригодный для получения точных результатов, т.к. кривую титрования можно сопоставить с интервалом изменения окраски индикатора.
Ошибки в Т. а. могут быть методическими и специфическими, обусловленными особенностями данной реакции. Методические ошибки связаны с особенностями метода титрования и зависят от погрешностей измерительных приборов, калибровки мерной посуды, пипеток, бюреток, неполного отекания жидкостей по стенкам мерной посуды.
Специфические ошибки обусловлены особенностями данной реакции и зависят от константы равновесия реакции и от точности обнаружения точки эквивалентности.
Методы Т. а. в зависимости от реакций, лежащих в их основе, подразделяются на следующие основные группы.
Методы нейтрализации, или кислотно-основного титрования, основаны на реакциях нейтрализации, т. е. на взаимодействии кислот и оснований. Эти методы включают ацидометрию (количественное определение оснований с помощью титрованных растворов кислот), алкалиметрию (определение кислот с помощью титрованных растворов оснований), галометрию (количественное определение солей с помощью оснований или кислот, если они реагируют с солями в стехиометрических соотношениях
Методы осаждения основаны на титровании веществ, образующих в определенной среде нерастворимые соединения, например соли бария, серебра, свинца, цинка, кадмия, ртути (II), меди (III) и др. К этим методам относят аргентометрию (титрование раствором нитрата серебра), меркурометрию (титрование раствором нитрата закисной ртути) и др.
Методы комплексообразования, или комплексометрия (меркуриметрия, фторометрия и др.), основаны на применении реакций, при которых образуются комплексные соединения, например Ag+ + 2CN- Û Ag (CN)2]. Методы комплексообразования тесно связаны с методами осаждения, т.к. многие реакции осаждения сопровождаются комплексообразованием, а образование комплексов — выпадением в осадок малорастворимых соединений.
Методы окисления — восстановления, или оксидиметрия, включают перманганатометрию, хроматометрию (бихроматометрию), йодометрию, броматометрию, цериметрию, ванадометрию и др.
18.
Определение меди. Стандартный потенциал пары I2/2I- (0,536 В) заметно превышает соответствующее численное значение пары Сu2+/Cu+ (0,159 В), поэтому окисление иодида ионами меди (II) представляется нереальным. Сопоставление стандартных потенциалов показывает, что скорее можно говорить об окислении иодом, чем об окислении иодида. Однако на самом деле реакция окисления иодида ионами Сu2+ происходит количественно:
2Сu2+ + 4I- → 2СuI (т) + I2.
В результате взаимодействия Сu2+ и I- происходит не только восстановление меди до Сu+, но и образуется малорастворимый СuI, что существенно изменяет потенциал пары Сu2+/Cu+. Было показано, что стандартный потенциал пары Сu2+/CuI становится равным 0,865 В, что намного превышает стандартный потенциал пары I2/2I-. Выделившийся I2 титруют тиосульфатом натрия.
Существенное значение для протекания указанной реакции имеет концентрация иодида, которая в 4-5 раз должна превышать требуемую постехиометрии, и кислотность раствора, хотя концентрация иона водорода и не входит в явном виде в уравнение реакции. Необходимо создание слабокислой среды, так как в нейтральных растворах ионы Сu2+ гидролизуются, а продукты гидролиза реагируют с иодидом очень медленно, что удлиняет процесс титрования и затрудняет фиксирование точки эквивалентности. Установлено, что при иодометрическом определении меди pH в растворе должен быть меньше 4. В сильнокислых растворах (+0,3 моль/л) происходит индуцированное медью окисление иодида кислородом воздуха, что приводит к получению завышенных результатов.
К занижению результатов приводит адсорбция ионов I3- (или I2I-) осадком СuI. Влияние этого фактора можно значительно уменьшить, если в раствор ввести тиоцианат калия, который реагирует с СuI:
CuI(т) + SCN- → CuSCN (т) + I-.
Растворимость тиоцианата меди (I) в 10 с лишним раз меньше растворимости СuI, что обеспечивает протекание реакции по крайней мере на поверхности осадка. Тиоцианат меди СuSCN уже не адсорбирует I3- и, кроме того, введение тиоцианата увеличивает скачок титрования. Однако добавлять тиоцианат в начале титрования не следует, так как он может окисляться иодом.
Иодометрическое определение меди имеет большое практическое значение. Оно используется при анализе бронз, латуней, медных руд и т.д. Мешающего влияния Fе(III) избегают введением в раствор фторид- или пирофосфат-ионов, образующих с Fе3+ прочные комплексы, которые уже не восстанавливаются иодидом. При соблюдении всех условий иодометрический метод определения меди по точности не уступаетэлектрогравиметрическому, но намного превосходит его по экспрессности.
19.

20.
Одним из широко применяемых способов фиксации точки эквивалентности является индикаторный метод. Индикаторы — вещества, которые дают возможность установить конечную точку титрования (момент резкого изменения окраски титруемого раствора). Наиболее часто индикатор добавляют ко всему титруемому раствору (внутренний индикатор). При работе с внешними индикаторами периодически берут каплю титруемого раствора и смешивают с каплей раствора индикатора или помещают на индикаторную бумагу (что приводит к потерям анализируемого вещества).
При выборе окислительно-восстановительных индикаторов к ним предъявляют следующие требования.
1 Окраска окисленной и восстановленной форм индикатора должна быть различна.
2 Интервал значений потенциалов, при котором происходит редокс-переход индикатора, а, следовательно, изменение его окраски, должен быть мал и находиться внутри скачка на кривой титрования.
3 Изменение цвета раствора в конечной точке титрования должно быть отчетливым при небольшом количестве ндиикатора.
4 Индикатор должен быть устойчив к воздействию окружающей среды.
21.
Методы анализа, основанные на расшифровке поляризационных кривых (вольтамперограмм), получаемых в электролитической ячейке с поляризующимся индикаторным электродом и неполяризующимся электродом сравнения, называют вольтамперометрическим. Вольтамперограмма позволяет одновременно получить качественную и количественную информацию о веществах, восстанавливающихся или окисляющихся на микроэлектроде (деполяризаторах), а также о характере электродного процесса.
В качестве поляризующегося микроэлектрода часто применяют ртутный капельный электрод, а сам метод называют в этом случаеполярографией, следуя термину, который предложил Я. Гейровский, разработавший этот метод в 1922 г.
При небольшом потенциале катода сила тока сначала медленно увеличивается с возрастанием потенциала – это так называемый остаточный ток, его значение имеет порядок 10-7 А. По достижении потенциала восстановления на катоде начинается разряд ионов, определяемый диффузией, и сила тока резко возрастает, а затем становится постоянной – это предельный диффузионный ток.
Принципиальная схема полярографической установки: анализируемый раствор 1 находится в электролизере 2, на дне которого имеется слой ртути 3, являющийся анодом. Катодом служит ртутный капельный электрод 4, соединенный с резервауром ртути 5. Через электролизер протекает ток, напряжение которого, подаваемое на электроды, можно плавно менять с помощью реохорда или делителя напряжения 7 и измерять при этом гальванометром 6 силу тока, проходящего через раствор.
Зависимость тока I от приложенного напряжения Е при обратимом электродном процессе передается уравнением полярографической волны:
Е = Е1/2 + (R T / n F) ln ( Id – I ) / I, (1)
Где Е1/2 – потенциал полуволны; Id – диффузионный ток.
При I = Id / 2 уравнение (1) переходит в
Е = Е1/2 . (2)
Это соотношение показывает независимость потенциала полуволны от тока и, следовательно, от концентрации восстанавливающегося иона.Потенциал полуволны является, таким образом, качественной характеристикой иона в растворе данного фонового электролита, и определение потенциала полуволны составляет основу качественного полярографического анализа.
Количественный полярографический анализ основан на уравнении Ильковича, которое связывает диффузионный ток Id с концентрацией ионас и рядом других величин:
Id = 605 z D1/2 m 2/3 t1/6 c (3)
Где z - заряд иона; D – коэффициент диффузии; m – масса ртути, вытекающей из капилляра за 1 с, мг; t – время образования капли (периода капания), с.
В практике количественного полярографического анализа коэффициент пропорциональности межу концентрацией вещества и силой диффузионного тока обычно устанавливают с помощью стандартных растворов. При постоянных условиях полярографирования D, m, и t постоянны, поэтому уравнение (3) переходит в
Id = k c . (4)
При анализе некоторых систем, для которых применимость уравнения (4) установлена вполне надежно, часто используют менее трудоемкий метод стандартных растворов. Так же широко распространен в количественной полярографии и метод добавок.
Особое место в полярографическом анализе занимает амперометрическое титрование.
Амперометрическое титрование представляет собой разновидность полярографического метода анализа. Амперометрическое титрование проводится следующим образом: часть исследуемого раствора помещают в электролизер, снабженный индикаторным электродом и электродом сравнения. Между электродами устанавливают напряжение на 0,3 – 0,5 В больше потенциала полуволны (или редокс-потенциала) исследуемого вещества и приступают к титрованию. В процессе титрования отмечают показания гальванометра, на основании результатов строят кривую амперометрического титрования, откладывая на оси ординат показания гальванометра, а на оси абсцисс – объем титранта. Точка перегиба соответствует объему титранта в точке эквивалентности. Содержание определяемого вещества вычисляют по объему титранта, израсходованному в точке эквивалентности. Концентрация титранта должна превышать концентрацию раствора титруемого вещества в 10-15 раз.
При амперометрическом титровании индикаторными электродами могут быть ртутный капельный электрод, платиновый вращающийся и другие электроды. В качестве электродов сравнения применяют насыщенный каломельный, хлорсеребряный и другие электроды.
Вид кривой амперометрического титрования будет зависеть от того, какой компонент реакции титрования вступает в электродную реакцию и при каком потенциале ведется титрование. Сама реакция титрования, естественно, будет протекать независимо от этих условий.
Амперометрическое титрование следует проводить при потенциале, отвечающем области диффузионного тока. Обычно титруют при потенциале на 0,2-0,3 В более отрицательном, чем потенциал полуволны полярографически активного соединения.
Полярографическая установка служит для получения полярограмм, т.е. кривых зависимости силы тока, протекающего через раствор, от потенциала, приложенного к рабочему электроду. Прибор состоит из трех основных узлов: электролитической ячейки с рабочим электродом и электродом сравнения, источника напряжения для поляризации рабочего электрода и устройства для регистрации тока. В качестве неполяризующегося электрода сравнения используется слой ртути на дне ячейки. Применяются также и другие электроды сравнения: каломельный, ртутно-сульфатный, хлорсеребряный и др. Рабочим электродом может быть также твердый микроэлектрод, изготавливаемый из платины, золота, графита и других материалов.
Установка для амперометрического титрования может быть собрана на основе любого полярографа. Обычно для этой цели используется самая простая полярографическая установка. При этом рабочим может быть как ртутный капающий, так и твердый микроэлектрод. В качестве источников тока могут применяться аккумуляторные батареи и различные выпрямительные устройства. В комплект установки для титрования входят также микробюретка и магнитная мешалка.
Форма кривых титрования Id=f(V)зависит от того, какое в-во электроактивно до и после точки эквивалентности, но всегда эти кривые имеют по крайней мере две ветви, которые пересекаются в конечной точке (к. т.) титрования.
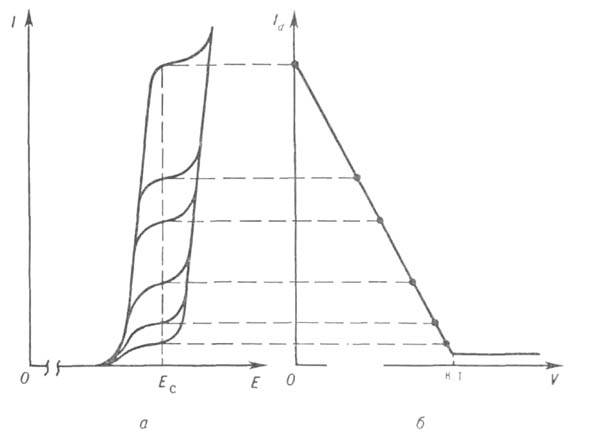
Вольтамперограммы (а) определяемого электроактивного вещества и кривая его амперометрич. титрования (6).
Обычно эти ветви прямолинейны. На рис. в кач-ве примера приведена кривая титрования электроактивного вещества нелектроактивным реагентом. При последоват. титровании неск. веществ на кривой титрования имеется соответствующее число точек перегиба. Важное преимущество амперометрическое титрование передвольтамперометрией - возможность определять не только электроактивные вещества, но и любые др. с применением электроактивных реагентов.
кач-ве
поляризующегося индикаторного электрода применяют
ртутный электрод, но чаще твердые
электроды из благородного металла
(обычно платины) или углеродного материала
(графита, стеклоуглерода и др.).
Вамперометрическое
титрование можно
использовать два
индикаторных электрода (без электрода сравнения),
выполненных из одного и того же материала
в виде пластин с одинаковыми относительно
большими поветями (1 х 1 см). Этот вариант
иногда неправильно наз. биамперометрич.
титрованием. Электроды погружают в
титруемый раствор, содержащий два
электроактивных в-ва и индифферентный
электролит. К электродам прикладывают
напряжение ![]() ,
обеспечивающее электродные процессы
одновременно на аноде и катоде двух
разных веществ или сопряженных (окисленной
и восстановленной) форм одного и того
же вещества. Величину
,
обеспечивающее электродные процессы
одновременно на аноде и катоде двух
разных веществ или сопряженных (окисленной
и восстановленной) форм одного и того
же вещества. Величину ![]() выбирают
так, чтобы ток соответствовал предельному
для обоих процессов. Роль индикаторного
играет тот электрод, на котором электродный
процесс обусловлен в-вом, присутствующим
в меньшей концентрации; второй электрод
практически служит электродом сравнения.
Иногда
выбирают
так, чтобы ток соответствовал предельному
для обоих процессов. Роль индикаторного
играет тот электрод, на котором электродный
процесс обусловлен в-вом, присутствующим
в меньшей концентрации; второй электрод
практически служит электродом сравнения.
Иногда ![]() выбирают
так, что оно мало и не соответствует
предельному току электроактивных в-в;
при этом ветви кривых титрования не
прямолинейны, однако в к.т. ток цепи
уменьшается до остаточного, или фонового,
обусловленного, например, примесями
(этот вариант ранее называли
"амперометрическое
титрование до
мертвой точки").
выбирают
так, что оно мало и не соответствует
предельному току электроактивных в-в;
при этом ветви кривых титрования не
прямолинейны, однако в к.т. ток цепи
уменьшается до остаточного, или фонового,
обусловленного, например, примесями
(этот вариант ранее называли
"амперометрическое
титрование до
мертвой точки").
Ниж. границы определяемых содержаний в амперометрическое титрование с одним индикаторным электродом 10-5М, с двумя - 10-6 М. амперометрическое титрование широко применяют для анализа орг. и неорг. веществ, определения растворимости осадков, устойчивости комплексных соед. и т.д.
23.
Потенциометрическое титрование основано на определении точки эквивалентности по результатам потенциометрических измерений. Вблизи точки эквивалентности происходит резкое изменение (скачок) потенциала индикаторного электрода. Это наблюдается, конечно, лишь тогда когда хотя бы один из участников реакции титрования является участником электродного процесса. Так, например, титрование по методу кислотно-основного взаимодействия может быть выполнено со стеклянным электродом. Определение хлорида - с хлорсеребряным и т.д. Так же, как и в других титриметрических методах, реакции потенциометрического титрования должны протекать строго стехиометрически, иметь высокую скорость и идти до конца.
Для потенциометрического титрования собирают цепь из индикаторного электрода в анализируемом растворе и электрода сравнения. В качестве электродов сравнения чаще всего применяют каломельный или хлорсеребряный.