

2.5.7. Эксклюзионная хроматография
Эксклюзионная хроматография представляет собой вариант жидкостной хроматографии, в котором разделение происходит за счет распределения молекул между растворителем, находящимся внутри пор сорбента, и растворителем, протекающим между его частицами. Неподвижной фазой служит пористое тело или гель, а различное удерживание веществ обусловлено различиями в размерах молекул веществ, их форме и способности проникать в поры неподвижной фазы. В названии метода отражен механизм процесса, от английского термина "Size Exclusion", означающего "исключение по размеру". Эксклюзионная хроматография, в которой неподвижной фазой служит гель, называется гель-проникающая хроматография (ГПХ)
В отличие от остальных вариантов ВЭЖХ, где разделение идет за счет различного взаимодействия компонентов с поверхностью сорбента, роль твердого наполнителя в эксклюзионной хроматографии заключается только в формировании пор определенного размера, а неподвижной фазой является растворитель, заполняющий эти поры.
Принципиальной особенностью метода является возможность разделения молекул по их размеру в растворе в диапазоне практически любых молекулярных масс – от 102 до 108, что делает его незаменимым для исследования синтетических высокомолекулярных веществ и биополимеров.
Рассмотрим принципиаяьные основы метода. Объем эксклюзионной колонки можно выразить суммой трех слагаемых:
Vt=Vм +Vi+Vd, (2.41)
где Vм - мертвый объем (объем растворителя между частицами сорбента, иначе говоря, объем подвижной фазы; Vi− объем пор, занятый растворителем (объем неподвижной фазы); Vd - объем матрицы сорбента без учета пор.
Полный объем растворителя в колонке Vt представляет собой сумму обьемов подвижной и неподвижной фаз:
Vt= Vм +Vi. (2.42)
Удерживание молекул в эксклюзионкой колонке определяется вероятностью их диффузии в поры и зависит главным образом от соотношения размеров молекул и пор, что схематически показано на рис. 1.6. Коэффициент распределения Кd, как и в других вариантах жидкостной хроматографии, представляет собой отношение концентраций вещества в неподвижной и подвижной фазах:
Kd = С/С0. (2.43)
Так как подвижная и неподвижная фазы имеют одинаковый состав, то Кd вещества, для которого обе фазы одинаково доступны, равен единице. Эта ситуация реализуется для молекул с самыми малыми размерами (в том числе и молекул растворителя), которые проникают во все поры (см. рис. 1.20.), и поэтому движутся через колонку наиболее медленно. Их удерживаемый объем равен полному объему растворителя Vt. Все молекулы, размер которых больше размера пор сорбента, не могут попасть в них (полная эксклюзия) и проходят по каналам между частицами. Они элюируются из колонки с одним и тем же удерживаемым объемом, равным объему подвижной фазы Vм. Коэффициент распределения для этих молекул равен нулю.
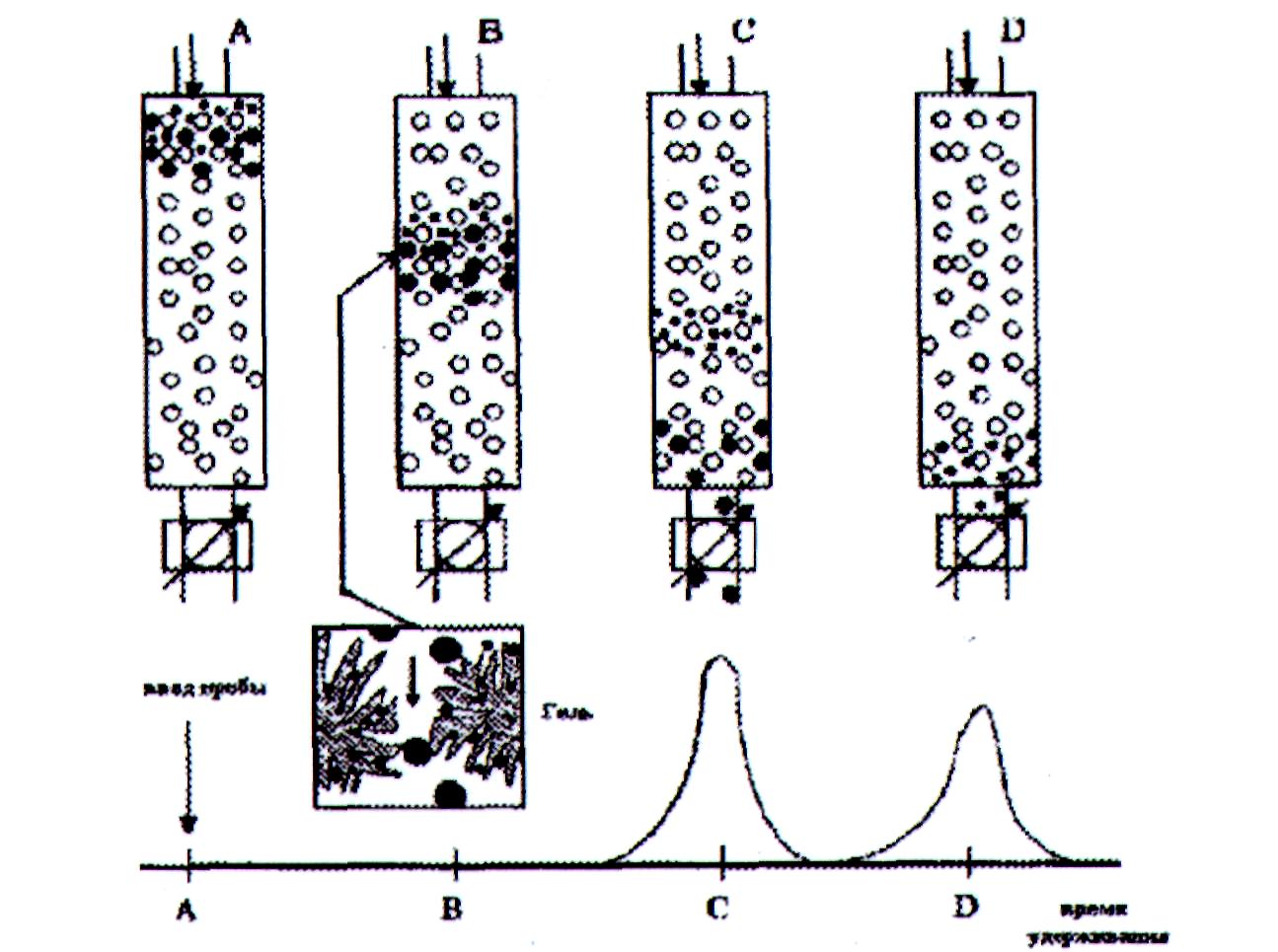
Рис. 1-20. Принцип разделения и детектирования пробы в эксклюзиоиной хроматографии.
А - ввод образца; В - разделение по размерам; С -выход крупных макромолекул; D - выход мелких макромолекул
Связь между удерживаемым объемом и молекулярной массой (или размером молекул) образца описывается частной калибровочной кривой (рис. 1.21.), т.е. каждый конкретный сорбент характеризуется своей калибровочной кривой, по которой оценивают область разделяемых на нем молекулярных масс. Точка А соответствует пределу эксклюзии, или мертвому объему колонки Vм. Все молекулы, масса которых больше, чем в точке А, будут элюироваться одним пиком с удерживаемым объемом Vм. Точка В отражает предел проникания, и все молекулы, масса которых меньше, чем в точке В, также будут выходить из колонки одним пиком с удерживаемым объемом V. Между точками А и В располагается диапазон селективного разделения. Соответствующий ему объем
Vi= Vt – Vм (2.45)
принято называют рабочим объемом колонки. Отрезок CD представляет собой линейный участок частной калибровочной кривой, построенной в координатах VR=IgM. Этот участок описывается уравнением
VR = С1 –С2 lgМ, (2.46)
где C1 - отрезок, отсекаемый на оси ординат продолжением отрезка CD, C2 - тангенс угла наклона этого отрезка к оси ординат.

Рис. 1.21. Эксклюзионная калибровочная кривая. Отрезок СD –линейный участок частной калибровочной кривой. VM – мертвый объем, Vi – рабочий объем колонки
Величину С2, называют разделительной емкостью колонки; ее выражают числом миллилитров растворителя, приходящегося на один порядок изменения молекулярной массы. Чем больше разделительная емкость, тем селективнее разделение в данном диапазоне масс. В нелинейных областях калибровочной кривой (участки АС и BD) в связи с уменьшением С2 эффективность фракционирования за- метно снижается. Кроме того, нелинейная связь между IgM и VR существенно усложняет обработку данных и снижает точность результатов. Поэтому стремятся выбирать колонку (или набор колонок) так, чтобы разделение анализируемого полимера протекало в пределах линейного участка калибровочной кривой.
Если какое-либо вещество элюируется с удерживаемым объемом больше Vi, то это указывает на проявление других механизмов разделения (чаще всего адсорбционного). Адсорбционные эффекты обычно проявляются на жестких сорбентах, но иногда наблюдаются и на полужестких гелях, видимо, из-за повышенного сродства к матрице геля. Примером, может служить адсорбция ароматических соединений на стиролдивинилбензольных гелях.
В эксклюзионной хроматографии стремятся полностью подавить адсорбционные и другие побочные эффекты, так как они, особенно при исследовании молекулярно-массового распределения (ММР) полимеров, могут существенно исказить результаты анализа. Одним из мешающих факторов является гидродинамический режим хроматографирования, в котором роль неподвижной фазы играют стенки колонки (канала). Разделение смеси макромолекул или частиц происходит вследствие различия скоростей протекания подвижной фазы вдоль оси канала и у его стенок, а также за счет распределения разделяемых частиц по сечению канала в соответствии с их размером.
Принципиальными отличиями зкеклюзионной хроматографии от других вариантов являются априори известная продолжительность анализа в конкретной используемой системе, возможность предсказания порядка элюирования компонентов по размеру их молекул, примерно одинаковая ширина пиков во всем диапазоне селективного разделения и уверенность в выходе всех компонентов пробы за достаточно короткий промежуток времени, соответствующий объему Vi. Данный метод применяют преимущественно для исследования ММР полимеров и анализа макромолекул биологического происхождения (белки, нуклеиновые кислоты и т.д.), но указанные особенности делают его чрезвычайно перспективным для анализа низкомолекулярных примесей в полимерах и предварительного разделения проб неизвестного состава. Получаемая при этом информация существенно облегчает выбор наилучшего варианта ВЭЖХ для анализа данной пробы. Кроме того, микропрепаративное эксклюзионное разделение часто используют в качестве первого этапа при разделении сложных смесей путем комбинации различных видов ВЭЖХ.
В эксклюзионной хроматографии полимеров предъявляются наиболее жесткие требования к стабильности потока подвижной фазы. Точность результатов в эксклюзионной хроматографии полимеров заметно зависит от температуры. При ее изменении на 10°С ошибка определения средних молекулярных масс превышает ±10%.

Выбор сорбентов, обеспечивающих оптимальные условия для решения конкретной аналитической задачи, проводят в несколько этапов. Матрица геля должна быть химически инертной, т.е. в ходе зкеклюзионной хроматографии не должно происходить химическое связывание разделяемых макромолекул. При разделении белков, ферментов, нуклеиновых кислот, при контакте с матрицей не должна происходить их денатурация. Первоначально на основе данных о химическом составе или растворимости анализируемых веществ устанавливают, какой вариант процесса следует применить хроматографию в водных системах или в органических растворителях, что в значительной степени определяет тип необходимого сорбента. Разделение веществ низкой и средней полярности в органических растворителях можно успешно осуществить как на полужестких, так и на жестких гелях. Исследование ММР гидрофобных пoлимеров, содержащих полярные группы, чаще проводят на колонках со стиролдивинилбензольными гелями, так как в этом случае практически не проявляются адсорбционные эффекты и не требуется добавка модификаторов к подвижной фазе, что значительно упрощает подготовку и регенерацию растворителя.
Для работы в водных системах используют главным образом жесткие сорбенты, иногда – полужесткие гели специальных типов. По калибровочным кривым или данным о диапазоне фракционирования выбирают сорбент нужной пористости с учетом имеющихся сведений о молекулярной массе образца. Если анализируемая смесь содержит вещества, отличающиеся по молекулярной массе не более чем на 2-2.5 порядка, то обычно удается разделить их на колонках с одним размером пор. При более широком диапазоне масс применяют наборы из нескольких колонок с сорбентами различной пористости. Ориентировочно калибровочную зависимость в этом случае получают сложением кривых для отдельных сорбентов.
Растворители, применяемые в эксклюзионной хроматографии, должны удовлетворять следующим основным требованиям:
полностью растворять образец при температуре разделения;
смачивать поверхность сорбента;
предотвращать адсорбцию (и другие взаимодействия) разделяемых веществ с поверхностью сорбента;
обеспечивать максимально высокую чувствительность детектирования;
иметь низкую вязкость и токсичность.
Кроме того, при анализе полимеров имеет существенное значение термодинамическое качество растворителя: весьма желательно, чтобы он был "хорошим" по отношению к разделяемому полимеру и матрице геля, т.е. были максимально выражены концентрационные эффекты.
Растворимость образца обычно является главным лимитирующим фактором, ограничивающим ассортимент пригодных подвижных фаз. Наилучшим органическим растворителем для эксклюзионной хроматографии синтетических полимеров по комплексу свойств является тетрагидрофуран (ТГФ). Он обладает уникальной растворяющей способностью, низкой вязкостью и токсичностью, лучше многих других растворителей совместим со стиролдивинилбензольными гелями и обеспечивает высокую чувствительность детектирования при использовании рефрактометра или УФ-детектора в области до 220 им. Для анализа высокополярных и нерастворимых в ТГФ полимеров (полиамиды, полиакрилонитрил, полиэтилентерефталат, полиуретаны и др.) обычно используют диметилформамид или м-крезол, а разделение полимеров низкой полярности, например, различных каучуков и полисилоксанов, часто проводят в толуоле или хлороформе. Последний является также одним из лучших растворителей при работе с ИК-детектором. о-дихлорбензол и 1,2,4-трихлорбензол применяют для высокотемпературной хроматографии полиолефинов (обычно при 135 0С), которые в других условиях не растворяются. Эти растворители имеют очень высокий показатель преломления, поэтому иногда их целесообразно использовать вместо тетрагидрофурана для анализа полимеров с низким коэффициентом преломления, что позволяет повысить чувствительность при детектировании рефрактометром.
Для предотвращения окисления растворителей и полужестких гелей в условиях высокотемпературной эксклюзионной хроматографии к о-дихлорбензолу и 1,2,4-трихлорбензолу добавляют антиокислители (иоиол, сантонокс R и др.).
Жесткие сорбенты совместимы с любыми подвижными фазами, имеющими рН<8-8.5. При более высоких значениях рН силикагель начинает растворяться и колонка необратимо теряет эффективность. Стиролдивинилбензольные гели совместимы в основном с элюентами умеренной полярности. Для работы на колонках с μ-стирогелем (от l000 Ǻ и выше) пригодны тетрагидрофуран, ароматические и хлорированные углеводороды, гексан, циклогексан, диоксан,трифторэтанол, гексафторпропанол и диметилформамид.
Степень набухания частиц геля в различных растворителях неодинакова, поэтому замена элюента в колонках с данными сорбентами может привести к снижению зффективности за счет изменения объема геля и образования пустот. При использовании неподходящих растворителей (ацетон, спирты) происходит столь сильная усадка геля, что колонка оказывается безнадежно испорченной. У сорбентов с малым размером пор (типа μ-стирогеля 100 Е и 500 Е) такая усадка наблюдается как в полярных, так и в неполярных растворителях, поэтому с ними, кроме того, нельзя работать в насыщенных углеводородах, фторированных спиртах и даметилформамиде. Удобным выходом из положения, является использование отдельных наборов колонок для каждого применяемого растворителя. Некоторые фирмы с этой целью выпускают колонки с одним и тем же размером пор, заполненные разными растворителями: тетрагидрофураном, толуолом, хлороформом и ДМФА.

Свои характерные особенности имеет эксклюзионная хроматография в водных средах. Из-за специфики многих разделяемых систем (белки, ферменты, полисахариды, полиэлектролиты и др.) и разнообразия применяемых сорбентов существует очень много вариаций состава ПФ для подавления различных нежелательных эффектов. В качестве сорбентов применяют декстрановые гели (сефадексы), полиакриламидные, оксиакрилметакрилатные гели, гели агарозы и др.
В процессе эксклюзионного хроматографирования поведение макромолекул определяется в первую очередь их гидродинамическими размерами, а характерной особенностью белков, ферментов и синтетических полиэлектролитов является зависимость размеров макромолекул от рН и ионной силы раствора. Чем меньше значение рН и ионной силы раствора, тем выгоднее становятся развернутые конформации макромолекул (так называемое полиэлектролитное набухание). В этом случае среднестатистические размеры растут, что приводит к уменьшению объемов удерживания в режиме эксклюзионной хроматографии. Общими приемами модификации является добавка различных солей и применение буферных растворов с определенным значением рН. В частности, поддержание рН<4 дает возможность подавить слабую ионообменную активность силикагелей, обусловленную присутствием на их поверхности кислых силанольных групп. Требуемая ионная сила подвижной фазы достигается при концентрации буферного раствора 0,05-0,6 М; оптимальную концентрацию подбирают экспериментально. Для предотвращения ионообменной сорбции катионных соединений наиболее часто используют такой активный модификатор, как тетраметиламмонийфосфат при рН=3. Однако при разделении некоторых белков могут проявляться гидрофобные взаимодействия, в свою очередь осложняющие эксклюзионный механизм разделения. Те же эффекты иногда проявляются и при работе с дезактивированными гидрофильными сорбентами. Для их устранения к растворителю добавляют метанол. Иногда в водную подвижную фазу вводят полярные органические растворители, полигликоли, кислоты, основания и поверхностно-активные вещества.
Важнейшей областью применения эксклюзионной хроматографии является исследование высокомолекулярных соединений. Применительно к синтетическим полимерам этот метод за короткий срок занял главенствующее положение для определения их молекулярно-массовых характеристик и интенсивно используется для изучения других видов неоднородности. В химии биополимеров эксклю-эионную хроматографию широко применяют для фракционирования макромолекул и определения их молекулярной массы.
Принципиальная черта эксклюзионной хроматографии высокомолекулярных синтетических полимеров заключается в невозможности разделения смеси на индивидуальные соединения. Эти вещества представляют собой смесь полимергомологов с различной степенью полимеризации и соответственно с разными молекулярными массами Мi. Молекулярную массу таких смесей можно оценить некоторой средней величиной, которая зависит от способа усреднения. Содержание молекул каждой молекулярной массы Мi определяют либо по их численной доле в общем числе полимерных молекул, либо по массовой доле в их общей массе. Обычно полимер характеризуют найденными этими способами средними величинами, которые называют соответственно среднечисленной Мn и среднемассовой Mw молекулярной массой. Значения Мn дают, например, криоскопия, осмометрия, эбулиоскопия, а значения Mw- светорассеяние и ультрацентрифугирование.
Если обозначить число молекул с молекулярной массой Мi через Ni то общую массу полимера можно выразить через ΣMiNi , численную долю молекул с массой Мi через Ni /ΣNi , а массовую долю молекул с массой Мi -через fi =МiNi /ΣМiМi . Чтобы определить часть общей массы полимера, соответствующую этим долям, их умножают на М.
Пpocсуммировав полученные значения для всех величин i, получают средние молекулярные массы:
![]() (2.47)
(2.47)
![]() (2.48)
(2.48)
Отношение Mw/Mn характеризует полидисперсность полимера.
На практике молекулярную массу полимеров часто определяют методом вискозиметрии. Средневязкостную молекулярную массу находят по уравнению Марка - Куна - Хаувинка:
[η]=KηMηa, (2.49)
где [η] − характеристическая вязкость; Кη , а - константы для данной системы полимер - растворитель при данной температуре.
Величина Мη описывается уравнением
Mη=(ΣMiafi ) 1/a. (2.50)
Как правило, величины средних молекулярных масс удовлетворяют неравенству
Мw >Мη >Мn. (2.51)
Обычно полимерный образец характеризуют комплексом значений Mw Mη Mn и Mw/Mη но этого может быть недостаточно. Наиболее полную информацию о молекулярно-массовой неоднородности образца дают кривые ММР. Типичная хроматограмма, полученная в процессе эсклюзионного разделения, представляет собой достаточно плавную кривую с одним или несколькими максимумами. Из этой кривой с использованием калибровочной зависимости и соответствующих расчетов определяют значения средних молекулярных характеристик и ММР полимера в дифференциальной или интегральной форме.