

14. Второй закон термодинамики. Энтропия
Второе начало термодинамики — физический принцип, накладывающий ограничение на направление процессов передачи тепла между телами.
Второе начало термодинамики запрещает так называемые вечные двигатели второго рода, показывая, что невозможно всю внутреннюю энергию тела превратить в полезную работу.
Второе начало термодинамики является постулатом, не доказываемым в рамках термодинамики. Оно было создано на основе обобщения опытных фактов и получило многочисленные экспериментальные подтверждения.
Существуют несколько эквивалентных формулировок второго начала термодинамики:
Постулат Клаузиуса: «Невозможен процесс, единственным результатом которого являлась бы передача тепла от более холодного тела к более горячему» (такой процесс называется процессом Клаузиуса).
Постулат Томсона: «Невозможен круговой процесс, единственным результатом которого было бы производство работы за счет охлаждения теплового резервуара» (такой процесс называется процессом Томсона).
2-й закон термодинамики вводит в рассмотрение новую ф-цию состояния –
энтропию. S [кДж/моль*К] S=kln(W) W – НЕ работа W – термодинамическая вероятность системы

W – это число микросостояний, с помощью которых может быть реализовано данное макросостояние системы. Smin в холоде, с увеличением температуры, растет энтропия.
Sгаза>S жидкости
Энтропия – степень беспорядка системы (полный порядок, когда энтропия=0)
Формулировки 2ого закона.
Теплота наиболее холодного из участвующих тел не может быть источником работы.
Существует такая ФС системы, изменение которой следующим образом связано с подведенной теплотой.

В изолированной системе самопроизвольно могут протекать процессы, сопровождающиеся увеличением энтропии
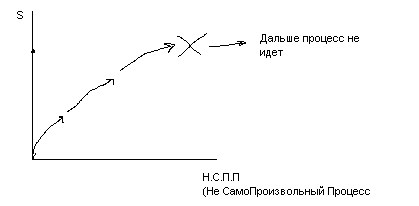
Предел протекания таких процессов – минимальное значение S
При абсолютном нуле – энтропия =0 (3й закон термодинамики)
Третье начало термодинамики (теорема Нернста) — физический принцип, определяющий поведение энтропии при абсолютном нуле температуры. Является одним из постулатов термодинамики.
Третье начало термодинамики может быть сформулировано так:
«Приращение энтропии при абсолютном нуле температуры стремится к конечному пределу, не зависящему от того, в каком равновесном состоянии находится система».
![]()
или
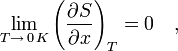
где x — любой термодинамический параметр.
Заметим, что третье начало термодинамики относится только к равновесным состояниям
Энтропия (от греч. ἐντροπία — поворот, превращение) — понятие, впервые введенное в термодинамике для определения меры необратимого рассеивания энергии. Термин широко применяется и в других областях знания: в статистической физике как мера вероятности осуществления какого-либо макроскопического состояния; в теории информации как мера неопределенности какого-либо опыта (испытания), который может иметь разные исходы, в исторической науке, для экспликации феномена альтернативности истории (инвариантности и вариативности исторического процесса).
Термодинамическая энтропия S, часто просто именуемая энтропия, в химии и термодинамике является функцией состояния термодинамической системы; её существование постулируется вторым началом термодинамики.
обобщение на случай произвольного квазистатического процесса выглядит так:
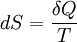
15. Энергия Гельмгольца. Ее изменение как критерий протекания процессов при V=const и T=const
Свободная энергия Гельмгольца (или просто свободная энергия) — термодинамический потенциал, убыль которого в квазистатическом изотермическом процессе равна работе, совершённой системой над внешними телами.
Определение
Свободная энергия Гельмгольца для системы с постоянным числом частиц определяется так:
![]() ,
где U
— внутренняя
энергия, T
— абсолютная температура,
S
— энтропия.
S
>=0 (Энтропия) , то в этом случае будет
самопроизвольный процесс.
,
где U
— внутренняя
энергия, T
— абсолютная температура,
S
— энтропия.
S
>=0 (Энтропия) , то в этом случае будет
самопроизвольный процесс.
Отсюда дифференциал свободной энергии равен:
![]() .
.
Видно, что это
выражение является полным дифференциалом
относительно независимых переменных
T
и V.
Поэтому часто свободную энергию
Гельмгольца для равновесного состояния
выражают как функцию
![]() .
.
Для системы с переменным числом частиц дифференциал свободной энергии Гельмгольца записывается так:
![]() ,
,
где μ — химический
потенциал, а N
— число частиц в системе. При этом
свободная энергия Гельмгольца для
равновесного состояния записывается
как функция
![]() .
.
Свободная энергия Гельмгольца и устойчивость термодинамического равновесия
Можно показать, что в системе с фиксированными температурой и объемом положение устойчивого равновесия соответствует точке минимума свободной энергии Гельмгольца. Другими словами, в этой точке (для такой системы) никакие изменения макроскопических параметров невозможны.
Свободная энергия Гельмгольца и максимальная работа
Свободная энергия Гельмгольца получила своё название из-за того, что она является мерой работы, которую может совершить термодинамическая система над внешними телами.
Пусть система переходит из состояния 1 в состояние 2. Поскольку работа не является функцией состояния системы, работа, совершенная системой в данном процессе будет зависеть от пути, по которому этот переход будет осуществляться.
Зададимся целью определить максимальную работу, которую система может совершить в этом случае.
Можно показать, что эта максимальная работа равна убыли свободной энергии Гельмгольца :
![]() .
Здесь индекс f
означает, что рассматриваемая величина
является полной работой системы в данном
процессе (см. ниже).
.
Здесь индекс f
означает, что рассматриваемая величина
является полной работой системы в данном
процессе (см. ниже).
Свободные энергии Гельмгольца и Гиббса
В приложениях «свободной энергией» иногда называют не свободную энергию Гельмгольца, а энергию Гиббса. Это связано с тем, что энергия Гиббса также является мерой максимальной работы, но в данном случае рассматривается только работа над внешними телами, исключая среду:
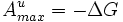 ,
где G
— энергия Гиббса.
,
где G
— энергия Гиббса.
AF=AU-TAs - свободная энергия Гельмгольца (V=const)
Если процесс протекает при V=const и T=const
F=U-Ts
dF=dU-TdS-SdT
dF=dU-TdS
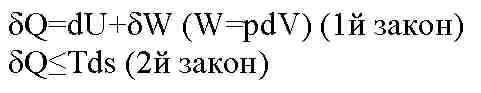
Объединяя выражения получаем: TdS>dU+pdV

В системах при V=const и T=const самопроизвольно протекают процессы, сопровождающиеся уменьшением энергии Гельмгольца
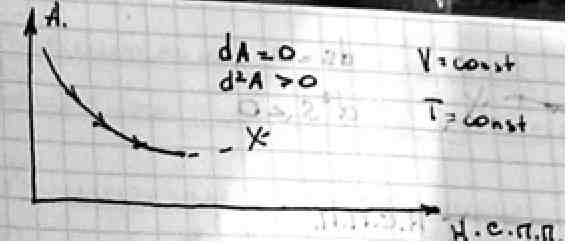
Предел протекания таких процессов – минимальное значение энергии Гельмгольца(А).
16. Энергия Гиббса. Ее изменение как критерий протекания процессов при P=const и T=const
Определение
Энергией Гиббса (или потенциалом Гиббса, или просто термодинамическим потенциалом в узком смысле) называют термодинамический потенциал следующего вида:
![]() ,
,
где U — внутренняя энергия, P — давление, V — объем, T — абсолютная температура, S — энтропия. Энергию Гиббса можно понимать как полную химическую энергию системы (кристалла, жидкости и т.д.)
Дифференциал энергии Гиббса для системы с постоянным числом частиц:
![]() .
.
Для системы с переменным числом частиц этот дифференциал записывается так:
![]() .
.
Здесь μ — химический потенциал, который можно определить как энергию, которую необходимо затратить, чтобы добавить в систему ещё одну частицу.
Можно показать, что химический потенциал есть отношение энергии Гиббса к числу частиц в системе:
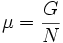 .
.
Химический потенциал применяется при анализе систем с переменным числом частиц, а также при изучении фазовых переходов.
Можно показать, что минимум потенциала Гиббса соответствует устойчивому равновесию термодинамической системы с фиксированными температурой, давлением и числом частиц.
Применение в химии
В химических процессах одновременно действуют два противоположных фактора — энтропийный (TΔS) и энтальпийный (ΔH). Суммарный эффект этих противоположных факторов в процессах, протекающих при постоянном давлении и температуре, определяет изменение энергии Гиббса (G):
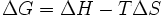 .
.
ΔG=ΔH-TΔs - свободная энергия Гиббса (p=const)
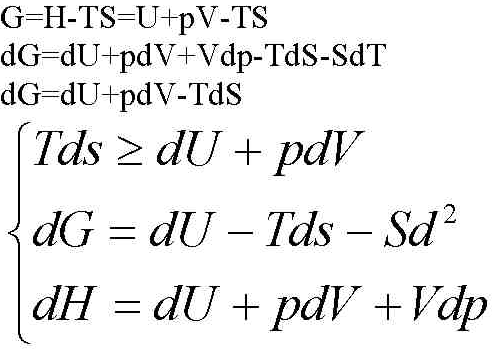
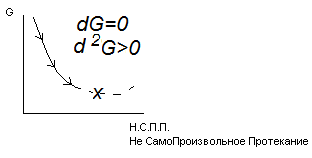
Совместно решая 1-3 и исключая dH, учитывая: dT=0, dV=0, получаем dG≤0
В системах при P=const и T=const самопроизвольно протекают процессы, сопровождающиеся уменьшением энергии Гиббса.
Предел протекания этих процессов – минимальное значение G Т.е. если ΔG>0 – прямая реакция невозможна
ΔG<0 – прямая реакция возможна
ΔG=0 – реакция обратима
из лекции:
Свободная энергия Гиббса.: G=H-TS(Дж\моль)
Процесс при P,V=const
TdS>=dU+pdV (*)
dG=dH-TdS-SdT(**)
dH=dU+pdV+Vdp (***)
**dG=dH-TdS-SdT, H=U+PV, то dH=dU+pdV+Vdp**
Решая систему уравнений из (*)(**)(***), учитывая, что dP=0.dT=0, получаем, что dG=0
В системе, при P,V=const самопроизвольно протекают процессы с уменьшением энергии Гиббса.
Предел протекания таких процессов – минимальное значение энергии Гиббса(G).