

Предмет, содержание и задачи курса «Физическая и коллоидная химия». Значение физической и коллоидной для технологии пищевых процессов.
Предмет и содержание: Физическая химия является основным теоретическим фундаментом современной химии, использующим теоретические методы таких важнейших разделов физики, как квантовая механика, статистическая физика и термодинамика, нелинейная динамика, теория поля и др. Она включает учение о строении вещества, в том числе: о строении молекул, химическую термодинамику, химическую кинетику и катализ. В качестве отдельных разделов в физической химии выделяют также электрохимию, фотохимию, физическую химию поверхностных явлений (в том числе адсорбцию), радиационную химию, учение о коррозии металлов, физико-химию высокомолекулярных соединений и др.
Задачи:
Может ли реакция протекать самопроизвольно?
Если реакция протекает, то как глубоко протекает (каковы равновесные концентрации продуктов реакции?)
Если реакция идет, то с какой скоростью?
Значение физической и коллоидной для технологии пищевых процессов.
Технология пищевых процессов – это науки о химических, физико-химических и физических способах переработки пищевого сырья в готовую продукцию (товарной продукции). Большое значение физическая и коллоидная химия имеет для пищевой технологии. Используемой в пищевой промышленности и общественном питании сырье и получаемые продукты в большинстве случаев являются коллоидными или высокомолекулярными системами. Такие процессы пищевых производств как уваривание, сепарация, дистилляция экстракция, кристаллизация, растворение и гидратирование могут быть обоснованы только законами физической химии. Все биохимические процессы, лежащие в основе пищевых производств также подчиняются законам физической химии.
На методах физической химии основан также контроль качества пищевых продуктов – определение кислотности, содержания сахаров, жиров, воды, витаминов и белков.
Таким образом, для рационального построения технологического процесса переработки сырья и объективной оценки качества полученной продукции специалисту пищевого производства необходимо знать и уметь применят на практике законы физической и коллоидной химии.
Элементы учения о строении вещества: поляризация, рефракция и межмолекулярные взаимодействия.
Поляризация — смещение электронов и атомов, а также ореинтацию молекул под действием внешнего электрического поля. Поляризация может быть:
Ориентационная – выражает ориентации молекул в электрическом поле.
Деформационная – характерна как для полярных, так и для неполярных молекул. Но зависит от температуры.
Следует различать поляризуемость молекул и поляризуемость вещества. Последняя характерна только лишь для диэлектриков.
Рефракция – это то же что и преломление света, т. Е. изменение направления световых волн при изменении показателя преломления среды, через которую эти лучи проходят.
Межмолекулярное взаимодействие – это относительно слабая связь молекул между собой, не приводящее к разрыву или образованию новых химических связей. (ванн-дер-вальсовые силы). Их основу составляет взаимодействие между электронами и ядрами и одной молекулы с ядрами и электронами другой. Полная энергия межмалекулярных взаимодействий складывается из следующих составляющих: Е=Еэл+Епол+Едисп. , где присутствуют энергии электростатического, поляризационного, дисперсионного взаимодействия.
Электростатическое – определяет кулоновскими силами притяжения м/у диполями полярных молекул и составляет в кристаллах, в газах и в жидкостях.
Поляризационное – связь диполя с другим индуцированным диполем, обусловленное деформацией электронной оболочки одной молекулы под влиянием электрического поля другой, что приводит к притяжению молекул.
Дисперсионное – возможно м/у любыми молекулами, как полярными так и неполярными.
Химическая термодинамика и её особенности, термодинамические системы и её параметры, термодинамические процессы.
Хими́ческая термодина́мика — раздел физической химии, изучающий процессы взаимодействия веществ методами термодинамики.
Основными направлениями химической термодинамики являются:
Классическая химическая термодинамика, изучающая термодинамическое равновесие вообще.
Термохимия, изучающая тепловые эффекты, сопровождающие химические реакции.
Теория растворов, моделирующую термодинамические свойства вещества исходя из представлений о молекулярном строении и данных о межмолекулярном взаимодействии.
Химическая термодинамика тесно соприкасается с такими разделами химии, как:
аналитическая химия;
электрохимия;
коллоидная химия;
адсорбция и хроматография.
Система – это часть окружающего мира, все остальное – окружающая среда.(например: содержимое колбы – система, а сама колба – среда.)
Система классифицируется по двум признакам:
по характеру обмена с окружающей средой энергией и веществом:
изолированные – мне могут обмениваться ни веществом ни энергией (пример: термос).
Закрытые – возможен обмен энергией, но не веществом (заполненная ампула с веществом).
Открытые – могут обмениваться и веществом и энергией. (кастрюля с едой).
По числу фаз системы делятся на:
Гомогенные
Гетерогенные – содержат несколько фаз, отделенных друг от друга поверхностями раздела (напитки со льдом).
Фаза – это совокупность систем, имеющие одинаковый состав и термодинамические свойства, отделенных от других частей поверхностями раздела.
По числу компонентов – индивидуальные химические вещества, составляющие систему и которые могут быть выделены из системы и существовать самостоятельно:
Однокомпонентные
Двухкомпонентные
Многокомпонентные – их системы могут быть как гомогенные, так и гетерогенные. (чай и кисель).
Термодинамический процесс – переход термодинамической системы из одного состояния в другое, который всегда связан с нарушением равновесия системы. Обычно при протекании процесса сохраняется постоянным какой-либо один параметр состояния:
Изотермический – при постоянной температуре (Т=const).
Изобарный – при постоянном давлении (P=const).
Изохорный – при постоянном объеме (V=const).
Адиабатический – при отсутствии теплообмена (Q=const).
При протекании процесса в неизолированных системах может происходить как поглащение, так и выделение теплоты:
Экзотермические (выделение теплоты)
Эндотермические (выделение теплоты)
Существует отдельный вид процессов, которые происходят сами по себе, и не требует для их осуществления энергии из вне – самопроизвольные процессы (газ заполняет весь объем сосуда). Несамопроизвольный процесс – требует привлечения энергии из окружающей среды.
4.Равновесное состояние – нулевое начало термодинамики.
Термодинамическое равновесие — состояние системы, при котором остаются неизменными по времени макроскопические величины этой системы (температура, давление, объем, энтропия) в условиях изолированности от окружающей среды. В общем, эти величины не являются постоянными, они лишь флуктуируют (колеблются) возле своих средних значений.
Известно четыре начала: 1.нулевое; 2.первое; 3.второе; 4.третье.
Нулевое – определяет условие равновесного состояния системы (часто нулевое начало термодинамики не фиксируется, а лишь рассматриваются условия положения системы в равновесном состоянии). Оно основано на постулатах, и практикой не опровергаются:
При неизменных внешних условиях система в равновесном состоянии не изменяется во времени;
Если система находится в состоянии равновесия, то в равновесии находятся и все части этой системы.
Равенство температуры во всех частях системы, находящейся в равновесии, называют нулевым началом термодинамики.
Нулевой закон термодинамики: если каждая из двух систем находится в тепловом равновесии с другой или другими системами, то эти две и более системы так же находятся в тепловом равновесии.
Если температура в системе одна и та же, то система находится в равновесном состоянии. Из нулевого начала следует ещё один вывод – аддитивность – какая-либо величина, характеризующая свойство системы в целом, равна сумме этих величин отдельных частей системы, независимо от того, каким образом система разбивается на части.
Функции состояния, первое начало термодинамики, теплоемкость.
Функция состояния в термодинамике — функция независимых параметров, определяющих равновесное состояние термодинамической системы; не зависит от пути (характера процесса), следуя которому система пришла в рассматриваемое равновесное состояние (т.е. не зависит от предыстории системы); к функциям состояния относят, в частности, характеристические функции системы:
внутренняя энергия;
энтропия;
энтальпия и др.
Термодинамическая работа и количество теплоты не являются функциями состояния, так как их значение определяется видом процесса, в результате которого система изменила своё состояние.
Первое начало термодинамики — один из трёх основных законов термодинамики, представляет собой закон сохранения энергии для термодинамических систем.
Первое начало термодинамики было сформулировано в середине XIX века в результате работ немецкого учёного Ю. Р. Майера, английского физика Дж. П. Джоуля и немецкого физика Г. Гельмгольца. Согласно первому началу термодинамики, термодинамическая система может совершать работу только за счёт своей внутренней энергии или каких-либо внешних источников энергии. Первое начало термодинамики часто формулируют как невозможность существования вечного двигателя первого рода, который совершал бы работу, не черпая энергию из какого-либо источника.
Формулировка:
Количество теплоты, полученное системой, идет на изменение ее внутренней энергии и совершение работы против внешних сил.
ТЕПЛОЕМКОСТЬ - кол-во теплоты, затрачиваемое для изменения темп-ры на 1 °С. Согласно более строгому определению, теплоемкость-термодинамич. величина, определяемая выражением:
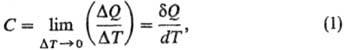
где DQ- кол-во теплоты, сообщенное системе и вызвавшее изменение ее т-ры на DТ. Отношение конечных разностей DQ/DТ наз. средней теплоемкостью, отношение бесконечно малых величин dQ/dT-истинной теплоемкостью. Поскольку dQ не является полным дифференциалом ф-ции состояния, то и теплоемкость зависит от пути перехода между двумя состояниями системы. Различают теплоемкость системы в целом (Дж/К), удельную теплоемкость [Дж/(г·К)], молярную теплоемкость [Дж/(моль·К)]. Во всех ниже приведенных ф-лах использованы молярные величины теплоемкости.
Тепловые эффекты химических реакций, определение тепловых эффектов.
Как известно, каждое физико-химическое превращение вещества сопровождается превращением энергии. Для сопоставления изменения энергии при различных реакциях в термодинамике используются понятие теплового эффекта, т. е. количества теплоты, которое выделяется или поглощается в химическом процессе при условии равенства начальной и конечной температуры. Тепловой эффект обычно относят к молю реагирующего вещества и выражают в джоулях.
Тепловые эффекты отличаются друг от друга, если процессы происходят в закрытом сосуде (при постоянном объеме V=const) или в открытом сосуде (при постоянном давлении P=const).
Тепловой эффект при постоянном объеме равен убыли внутренней энергии: QVT= –∆UT, а при постоянном давлении – убыли энтальпии:
QРT= –∆НT.
Тепловой эффект не зависит от промежуточных стадий, а определяется лишь начальным и конечным состоянием системы при условии, что единственной работой, совершаемой системой, является работа против внешнего давления и что давление или объем в течении всего процесса остаются неизменными (закон Гесса). С помощью закона Гесса производят различные термохимические расчеты.
Второе начало термодинамики, энтропия.
Второе начало термодинамики -- физический принцип, накладывающий ограничение на направление процессов передачи тепла между телами.
Второе начало термодинамики запрещает так называемые вечные двигатели второго рода, показывая, что невозможно всю внутреннюю энергию тела превратить в полезную работу.
Второе начало термодинамики является постулатом, не доказываемым в рамках термодинамики. Оно было создано на основе обобщения опытных фактов и получило многочисленные экспериментальные подтверждения.
Существуют несколько эквивалентных формулировок второго начала термодинамики:
Постулат Клаузиуса: «Невозможен процесс, единственным результатом которого являлась бы передача тепла от более холодного тела к более горячему» (такой процесс называется процессом Клаузиуса).
Постулат Томсона: «Невозможен круговой процесс, единственным результатом которого было бы производство работы за счет охлаждения теплового резервуара» (такой процесс называется процессом Томсона).
Термодинами́ческая энтропи́я(S), часто просто именуемая энтропия, в химии и термодинамике является функцией состояния термодинамической системы; её существование постулируется вторым началом термодинамики.
Понятие энтропии было впервые введено в 1865 году Рудольфом Клаузиусом. Он определил изменение энтропии термодинамической системы при обратимом процессе как отношение изменения общего количества тепла ΔQ к величине абсолютной температуры T:
![]()
где dS - приращение (дифференциал) энтропии, а δQ - бесконечно малое приращение количества теплоты.
Процессы в неизолированных системах: энергия Гиббса и энергия Гельмгольца, критерии равновесных и самопроизвольных процессов, максимальная уравнения Гиббса-Гельмгольца.
Свободная энергия Гиббса (или просто энергия Гиббса, или потенциал Гиббса, или термодинамический потенциал в узком смысле) — это величина, показывающая изменение энергии в ходе химической реакции и дающая таким образом ответ на принципиальную возможность протекания химической реакции; это термодинамический потенциал следующего вида:
![]()
где U — внутренняя энергия, P — давление, V — объем, T — абсолютная температура, S — энтропия.
Свобо́дная эне́ргия Гельмго́льца (или просто свобо́дная эне́ргия) — термодинамический потенциал, убыль которого в квазистатическом изотермическом процессе равна работе, совершённой системой над внешними телами.
Свободная энергия Гельмгольца для системы с постоянным числом частиц определяется так:
![]() ,
где U — внутренняя энергия, T — абсолютная
температура, S — энтропия.
,
где U — внутренняя энергия, T — абсолютная
температура, S — энтропия.
.В соответствии со вторым началом термодинамики критерием самопроизвольного процесса является рост энтропии. Если энтропийный фактор, определяющий возможность самопроизвольных процессов, соотносится с энтальпийным следующим образом: TdS≥dU (а для изобарного процесса TdS≥dH), - то из уравнений
dA=dU – TdS или ∆A=∆U-T∆S
dG=dH – TdS или ∆G=∆H - T∆S
следует: dA≤0, или ∆A≤0; dG≤0, или ∆G≤0.
Равенство означает равновесный процесс, знак «меньше» - самопроизвольный процесс. Эти соотношения являются основополагающими для расчетов и определения условий равновесных и самопроизвольных процессов для неизолированных систем.
Уравнения Гиббса – Гельмгольца – уравнения максимальной работы.
Они
позволяют установить связь между
максимальной работой равновесного
процесса и теплотой неравновесного
процесса: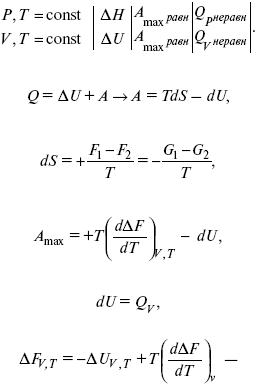
уравнение Гельмгольца (уравнение связывающее функции F и G с их температурными производными).
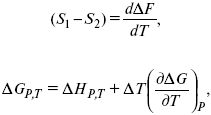
уравнение Гиббса (уравнение связывающее функции F и G с их температурными производными). Уравнения эти дают возможность рассчитать работу через температурный коэффициент функции Гельмгольца или через температурный коэффициент функции Гиббса.
Третье начало термодинамики.
Формулировка: По мере приближения температуры к 0 К энтропия всякой равновесной системы при изотермических процессах перестает зависеть от каких-либо термодинамических параметров и в пределе (при Т=0 К) для всех систем принимает одну и ту же, универсальную постоянную величину, которую можно принять равной нулю.
Или
«Приращение энтропии при абсолютном нуле температуры стремится к конечному пределу, не зависящему от того, в каком равновесном состоянии находится система».

где x — любой термодинамический параметр.
Третье начало термодинамики относится только к равновесным состояниям.
Третье начало термодинамики позволяет находить абсолютное значение энтропии, что нельзя сделать в рамках классической термодинамики (на основе первого и второго начал термодинамики).
Следствия:
Недостижимость абсолютного нуля температур.Из третьего начала термодинамики следует, что абсолютного нуля температуры нельзя достичь ни в каком конечном процессе, связанном с изменением энтропии, к нему можно лишь асимптотически приближаться, поэтому третье начало термодинамики иногда формулируют как принцип недостижимости абсолютного нуля температуры.
Поведение термодинамических коэффициентов. Из третьего начала термодинамики вытекает ряд термодинамических следствий: при должны стремиться к нулю теплоёмкости при постоянном давлении и при постоянном объёме, коэффициенты теплового расширения и некоторые аналогичные величины. Справедливость третьего начала термодинамики одно время подвергалась сомнению, но позже было выяснено, что все кажущиеся противоречия (ненулевое значение энтропии у ряда веществ при T = 0) связаны с метастабильными состояниями вещества, которые нельзя считать термодинамически равновесными.
Нарушения третьего начала термодинамики в моделях. Третье начало термодинамики часто нарушается в модельных системах. Так, при энтропия классического идеального газа стремится к минус бесконечности. Это говорит о том, что при низких температурах уравнение Менделеева — Клапейрона неадекватно описывает поведение реальных газов.
Таким образом, третье начало термодинамики указывает на недостаточность классической механики и статистики и является макроскопическим проявлением квантовых свойств реальных систем.
В квантовой механике, тем не менее, в модельных системах третье начало также может нарушаться. Таковы все случаи, когда применяется распределение Гиббса и основное состояние является вырожденным.
Несоблюдение третьего начала в модели, однако, не исключает того, что в каком-то диапазоне изменения физических величин эта модель может быть вполне адекватна.
Химический потенциал – фактор интенсивности физико-химических процессов.
Химический потенциал – это частная производная одной из характеристических функций (∆G, ∆A, ∆U, ∆H), чаще энергии Гиббса, по изменению числа молей одного компонента при неизменном числе молей остальных компонентов и неизменности соответствующих параметров состояния.
dG≤ - SdT + Vdp + (∂G/∂n1)T,p,n2,n3,..,nkdn1 + (∂G/∂ni)T,p,n1,..,ni-1,n+1,…,nk dni + (∂G/∂nk)T,p,n1,..,ni,nk-1dnk.
Знак «меньше» относится к самопроизвольному процессу, а знак «равенство» - к равновесному. Частные производные энергии Гиббса по одному из изменяющихся компонентов и есть химический потенциал μ.
Как определить значение химического потенциала? Для однокомпонентной системы можно записать: μ=G/n=GM
GM – мольная энергия Гиббса, или мольный термодинамический потенциал, компонента (индивидуального вещества). Таким образом, химический потенциал компонента тождественен мольной энергии Гиббса.
Константы равновесия, константы равновесий с учетом реальных условий.
Самопроизвольный процесс согласно второму началу термодинамики сопровождается убылью свободной энергии системы и заканчивается установлением равновесного состояния - химическое равновесие (константа равновесия).
Химическое равновесие выражается константами равновесия.
Константа равновесия равна отношению произведения концентрации продуктов реакции в числ. Произведения концентрации исходных веществ: Kc= Псiвi/ Псiаi
В
общем случае р-ции
![]()
где
vi и vj - стехиометрич. числа исходных в-в
Аi (i=1,2,..., q) и продуктов р-ции Аj (j=1, 2, ...,
r), активности к-рых соотв.
![]() и
и![]() константа
равновесия
константа
равновесия
Фугитивность – это такое давление реального газа, при котором газ ведет себя как идеальный. Применение фугитивности вместо парциальных давлений и использование соотношений и практикуется при относительно высоких давлениях и низких температурах, когда наблюдается значительное отклонение реальных газов от идеальных. Фугитивностью называется величина, которую необходимо подставить в выражение для химического потенциала идеального газа, чтобы получить значение химического потенциала реального газа.
Для перехода от идеальных растворов к реальным введено понятие активность – это такая концентрация, при использовании которой реальные растворы приобретают термодинамические свойства идеальных растворов. Активностью компонента в растворе называют величину, которую следует подставить в выражение для химического потенциала компонента в идеальном растворе, чтобы получить действительное выражение химического потенциала реального раствора.
Изотермы, изохора и изобара химической реакции, химическая переменная и химическое средство.
Изотермический – Т= const
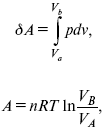 так
как
так
как
![]()
Изохорный – V = const
δА = 0,
δА = pdυ = 0,
δQ = dU + pdυ,
δQ = CvdT.
3. Изобарный – P = const
δА = pdυ,
A = pV2 – pV1.
4. Адиабатический – δQ = 0
1) δA = –dU,
A = –CV(T2 – T1), T2 > T1;
2) pdδ= –CvdT,
Химическая переменная – отношение изменения числа молей копмонента в химической реакции к его стехиометрическому коэффициенту, которое одинаково для всех компонентов и характеризует полноту реакции. Химическое сродство характеризует отклонение системы от состояния химического равновесия.
ХИМИЧЕСКОЕ СРОДСТВО (сродство реакции), параметр термодинамич. системы, характеризующий отклонение от состояния хим. равновесия. Если реакцию записать в виде ур-ния:
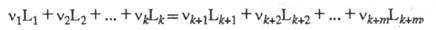
где L1, ..., Lk - исходные реагенты, Lk+1, ..., Lk+m - продукты р-ции, v1, ..., vk и vk+1 , ..., vk+m - стехиометрич. коэф.,
Принцип Ле Шателье-Брауна.
Формулировка: если на систему, находящуюся в устойчивом равновесии, подействовать извне, изменяя какое-нибудь из условий, определяющих положение равновесия, то в системе усилится то из направлений, которое ослабляет влияние произведенного воздействия, и положение равновесия сместится в том же направлении.
Уравнение изохоры и изобары позволяют определить смещение химического равновесия в зависимости от температуры. Если прямая реакция идет с выделением теплоты (∆U<0 или ∆Н<0), то рост температуры для экзотермической реакции приведет к уменьшению константы равновесия kc или kp. Смещение равновесия в зависимости от изменения температуры, когда ∆U<0 или ∆Н<0, можно представить в следующем виде:
увеличение
температуры

уменьшение температуры
Повышение температуры экзотермических реакций способствует увеличению количества исходных веществ, а рост температуры эндотермических реакций приводит к обогащению продуктами реакции по сравнению с равновесным состоянием до изменения температуры.
Согласно этому принципу увеличение концентрации приводит к смещению равновесия в сторону увеличения расхода тех компонентов, концентрация которых увеличивается.
Фазовое равновесие, правило фаз Гиббса.
Фазовое равновесие – одновременное существование термодинамически равновесных фаз в гетерогенной системе, например жидкости со своим насыщенным паром (система жидкость – газ), воды и льда при температуре плавления (система жидкость – твердое тело), двух несмешивающихся жидкостей (система жидкость – жидкость). Фазовое равновесие в зависимости от состава и параметров системы определяется правилом фаз Гоббса.
Систему или часть системы, однородную по физическому состоянию и химическому составу, называют фазой, а компонент представляет собой химически однородное вещество, которое может быть выделено и способно существовать самостоятельно.
Формулировка правила фаз Гоббса: Число степеней свободы равновесной термодинамической системы, на которую влияют n внешних факторов (или на которую изз внешних факторов влияют температура либо температура и давление) равно числу независимых компонентов системы минус число фаз плюс n (единица или два).
С=К-Ф+n, С=К-Ф+1, С=К-Ф+2
Правило фаз Гоббса распространяется на системы с ограниченным числом фаз и компонентов; различают одно-, двух- и трехфазные, одно-, двух- и трехкомпонентные системы. Число степеней свободы определяет вариантность системы. Системы могут быть моно-, ди- и тривариантными. Если С=0, то такие системы называют инвариантными.
Фазовое состояние системы в зависимости от внешних условий и состава системы определяется при помощи диаграмм состояния, или фазовых диаграмм.
Тепловые эффекты фазовых переходов.
Тепловой эффект – теплота, которая выделяется или поглощается в результате химической реакции при соблюдении следующих условий: отсутствие полезной работы, неизменность давления или объема системы, постоянство температуры до и после реакции, иными словами, реакция должна протекать в изобарно-изотермических или изохорно-изотермических условиях. Определить тепловой эффект реакции можно по закону Гесса: при постоянном давлении или объеме тепловой эффект химической реакции зависит только от вида и состояния исходных веществ и продуктов реакции, но не зависит от пути перехода. Если реакция происходит при постоянном давлении, то наблюдаемое изменение теплоты выражают через изменение энтальпии. Для реакций, протекающих при постоянном объеме, тепловой эффект отождествляют с изменением внутренней энергии.
Существуют такие вещества, у которых при фазовых превращениях - плавлении, испарении и кристаллизации - выделяется так называемая скрытая теплота фазового перехода, причем количество выделяющейся теплоты достаточно велико. Как видно из самого названия "скрытая теплота", в процессе фазового превращения вещества его температура не меняется, т.е. весь процесс идет при определенной температуре.
В настоящее время на практике используются два вида веществ для аккумуляторов данного тепла: хлорид кальция (CaCl2∙6Н20) и сульфат натрия (глауберова соль). Хлорид кальция имеет точку плавления 29°С, тепловой эффект фазового Перехода из твердого в жидкое состояние составляет 42 ккал/кг (при плотности 1,622 кг/м3). В лучшем случае в веществе, претерпевающем фазовый переход, аккумулируется такое же количество тепла, как в воде, занимающей 1/7 объема этого вещества при ее нагреве на 10°С.
Аккумуляторам, использующим скрытую теплоту фазовых переходов, как и воде, свойственно явление переохлаждения, и при применении таких аккумуляторов особенно важно его предотвращать.
Неравновесная термодинамика.
Неравновесная термодинамика (термодинамика необратимых процессов) – изучает общие закономерности, в которых протекают термодинамически необратимые процессы (передача теплоты, диффузия, химические реакции, перенос электрического тока).
Отличие неравновесной термодинамики от равновесной:
Термодинамические параметры системы изменяются во времени;
Эти параметры имеют разные значения в различных точках системы, т.е. зависят от координат;
Различают неравновесную систему:
А) Линейная – справделива при незначительных отклонениях реального процесса от равновесного
Б)нелинейная – при более существенных.
Общие сведения о неравновесной термодинамике
Как было указано выше, классическая термодинамика (ее три «начала») изучает термодинамические равновесные, обратимые процессы. Для неравновесных процессов она устанавливает лишь неравенства, которые указывают возможное направление этих процессов. Фундаментальными работами И.Р.Пригожина установлено, что вся термодинамика делится на три большие области: равновесную, в которой производство энтропии, потоки и силы равны нулю, слабо неравновесную, в которой термодинамические силы «слабы», и энергетические потоки линейно зависят от сил, и сильно неравновесную, или нелинейную, где энергетические потоки нелинейны, а все термодинамические процессы носят необратимый характер. Основная задача неравновесной термодинамики - количественное изучение неравновесных процессов, в частности определение их скоростей в зависимости от внешних условий. В неравновесной термодинамике системы, в которых протекают неравновесные процессы, рассматриваются как непрерывные среды, а их параметры состояния — как полевые переменные, то есть непрерывные функции координат и времени.
Слабо неравновесная (линейная) термодинамика рассматривает термодинамические процессы, происходящие в системах в состояниях, близких к равновесию. Таким образом, линейная термодинамика описывает стабильное, предсказуемое поведение систем, стремящихся к минимальному уровню активности. Первые работы в этой области принадлежат Ларсу Онсагеру, который в 1931 году впервые открыл общие соотношения неравновесной термодинамики в линейной, слабо неравновесной области - «соотношения взаимности». Суть их чисто качественно сводится к следующему: если сила «один» (например, градиент температуры) для слабо неравновесных ситуаций воздействует на поток «два» (например, на диффузию), то сила «два» (градиент концентрации) воздействует на поток «один» (поток тепла).
Таким образом, в слабо неравновесной области практически действуют законы равновесной термодинамики, система ни к чему не стремится и ее поведение в большинстве случаев вполне предсказуемо.
Сильно неравновесная термодинамика рассматривает процессы, происходящие в системах, состояние которых далеко от равновесия.
Когда термодинамические силы, действуя на систему, становятся достаточно большими и выводят ее из линейной области в нелинейную, устойчивость состояния системы и ее независимость от флуктуации значительно уменьшается.
В таких состояниях определенные флуктуации усиливают свое воздействие над системой, вынуждая ее при достижении точки бифуркации – потери устойчивости, эволюционировать к новому состоянию, который может быть качественно отличным от исходного. Происходит самоорганизация системы. Причем считается, что развитие таких систем протекает путем образования нарастающей упорядоченности. На этой основе и возникло представление о самоорганизации материальных систем.
Все материальные системы, от самых малых до самых больших, считаются открытыми, обменивающимися энергией и веществом с окружающей средой и находящимися, как правило, в состоянии, далеком от термодинамического равновесия.
Это свойство материальных систем позволило в свою очередь определить целый ряд новых свойств материи.
Вот некоторые из них.
- все процессы необратимы, так как они всегда сопровождаются потерями энергии;
- энтропия S в открытых системах имеет две составляющие: deS – характеризует обмен энтропией с внешним миром; diS – характеризует необратимые процессы внутри;
- материя обладает свойством самоорганизации.
Исследования И. Пригожиным живой материи как открытых материальных систем были в основном сосредоточены на сравнительном анализе организации структур живой и неживой материи, термодинамическом анализе реакций гликолиза и ряде других работ.
Элементы статистической термодинамики
В рамках статистической термодинамики состояние системы определяется не самими значениями физических величин, а вероятностными законами их распределения. Исходным для определения суммы по состояниями служит закон распределения Больцмана. Этот закон отражает неравнозначность энергии разных молекул и характеризует распределение молекул (частиц) по энергиям. Величину, которая объединяет молекулы по уровням их энергии, называют фактором Больцмана.
В случае невзаимодействующих частиц идеального газа каноническое распределение Гиббса превращается в распределение Больцмана.