

фізіологія Плиска остання
.pdfПлазміноген (профібринолізин, фібринолізин) — протеолітичний фермент (протеаза), подібний до трипсину. Він гідролізує фібриноген і фактори V, VII чи VIII, протромбін, XII, гальмуючи процеси зсідання. Інколи він є навіть причиною гіпокоагуляції. При пошкодженні тканин виділяється тканинний плазміногенактиватор (t-PA). Інгібітором є а2-антиплазмін. Сам плазмін — глобулінова фракція білків плазми крові.
Існують зовнішні й внутрішні активатори плазміногену. До зовнішніх відносять продукт життєдіяльності стрептококів — стрептокіназу. Внутрішні поділяють на тканинні (урокіназа, лужна фосфатаза, трипсин, калікреїн-кінінова система, комплемент С,) і кров'яні. Останні утворюються в процесі зсідання крові (це ХІІа). їхня дія виявляється лише в присутності лізокіназ. Урокіназа секретується і депонується нирками і печінкою. Крім стрептокінази і урокінази, використовують нікотинову кислоту, ксавін.
Фактор ХІІа взаємодіє з кініногеном та прекалікреїном (частина калікреїн-кінінової системи) і набуває властивості активувати плазміноген. Частина лізокіназ міститься в тканинах і має місцеву дію — фібриноліз у тканинах. Надалі спостерігається самоактивація плазміногену. Плазмін (протеаза подібна до трипсину) гідролізує фібрин до пептидів, а ті — пептидазами до амінокислот. Прохідність судини відновлюється, і відповідно відновлюються порушені функції того чи іншого органу. До цього часу спостерігається і регенерація пошкодженої ділянки судини.
Після активації плазміншвидкозникаєзкровотоку підвпливомантиплазміну. Внаслідок цього його дія є в основному місцевою. Активатори плазміногенумістятьсявусіхклітинахкровійособливовлейкоцитах. Останнітакожберутьучастьуфібринолізізадопомогоювласнихпротеаз, які гідролізуютьфібрин. Діяплазмінупригнічуєтьсяа2-антиплазміноміє-амі- нокапроновоюкислотою, щозастосовуєтьсявклінічнійпрактиці.
Процеси утворення невеликої кількості фібрину постійно відбуваються в крові. Однак утворений фібрин одразу фібринолізується плазміном, якийвінактивноадсорбує. Ціпроцесивнормі зрівноважені. При відсутності фібринолізу тромб організоувється (заміщується сполучною тканиною).
Незважаючи на значну кількість у крові прокоагулянтів, вона ворганізмівсе-такизалишаєтьсяврідкомустані. Цеобумовленотакими причинами; 1) гладенька стінка судин запобігає активації фактора XII і агрегації тромбоцитів; 2) однойменний заряд стінкисудин і фермових елементів спричиняє їх взаємовідштовхування; 3) фібрин покриває стінки судин і адсорбує активні фактори зсідання
243

(наприклад тромбін); 4) велика швидкість кровотоку запобігає накопиченню коагулянтів у достатній концентрації для запуску кас- кадно-комплексного механізму зсідання крові; 5) зсіданню крові запобігає наявність власних природних антикоагулянтів; 6) ендотелій синтезує простагландин — простациклін, який є сильним інгібітором агрегації тромбоцитів; 7) можливість ендотелію синтезувати і фіксувати антитромбін III.
Антикоагулянти поділяють на первинні або передіснуючі (антитромбіни III і IV, гепарин) і вторинні, які утворюються в процесах зсідання крові (фібрин чи антитромбін І) і фібринолізу.
Антитромбін III — альбуміноподібний білок (а2-глобулін плазми). Його роль полягає в інактивації тромбіну та факторів ХИа, ХІа, Ха і ІХа. Антитромбін III переводить тромбін у неактивний метатромбін, знижуючи його вмісту крові.
а2-Макроглобулін (360,000) — подібний до комплексу антитромбін — гепарин, який об'єднаний з протеолітичними коагулювальними факторами. Алейогоактивністьнеприскорюєтьсягепарином. Йогоголовнафункція — зв'язування агентів із сильною коагулювальною дією та запобігання їхній протеолітичній дії, поки вони можуть бути зруйновані різними шляхами, але не сс2-макроглобуліном.
Гепарин — молекула звисоконегативним зарядом, пов'язаним іззв'язаними полісахаридами. Власні антикоагулянтні властивості малі, але їх об'єднання з антитромбіном III збільшує такі останнього в 100-1000 разів. Тому в присутності гепарину антитромбін усуває тромбін із циркулюючої крові майже миттєво. Зазначений комплекс такожусуває деякі інші активовані коагулювальні фактори — XII, XI, IX і X. Гепарин утворюється вусіхорганахітканинахорганізмулюдини, аленайбільше— влегеняхіпечінці. Особливобагато його вбазофільних тучних клітинах, якімасоволокалізовані в перпкапілярній сполучній тканині по всьому тілі. Вони секретують у малих кількостях гепарин, який дифундує в циркулюючу кров. Базофіли крові (функціонально майже ідентичні з попередніми клітинами) також звільняють невелику кількість гепарину в плазму.
Гепарин гальмує всі три фази зсідання крові, зменшує адгезивність тромбоцитів і збільшує їхній негативний заряд, сприяє дезагрегації еритроцитів, покращує реологію. Реагує з позитивно зарядженими молекулами білка і NH2 групами, де б вони не знаходилися, гальмує понад 20 ферментативних реакцій. Активує ліполіз, реагує з токсинами, вірусами, антибіотиками. Антагоністи — серотонін, гістамін, альдостерон.
Гепаринактивнийтількизбілкомплазми(комплементом, абокофактором гепарину), утворюючи антитромбін II і сприяє фіксації останнього на поверхніендотелію, підвищуючийогоактивність. АнтитромбінII являєсобою сірковмісний ефір — мукополісахаридів. У такому разі гепарин гальмує всі фази коагуляції, пригнічуючи активність багатьох плазмових факторів і динамічні перетворення тромбоцитів. У малих дозах він стимулює фібриноліз і гальмує активність гіалуронідази. Останнє зменшує проникність
244
судинної стінки, гальмує реакції антиген — антитіло, має протибольовий та протизапальний ефекти.
Фібрин, адсорбуючи насвоїйповерхнітромбін, запобігає зсіданню. Тому з іншими антикоагулянтами він дістав назву антитромбіну І. Сам тромбін, відщеплюючи від протромбіну інгібітор фактора Ха, також має антикоагулянтні властивості. а2-Макроглобулін отримав назву антитромбіну IV і є другим за потужністю після антитромбіну III. Антикоагулянтну дію мають також пептиди, відщеплені від фібриногену тромбіном, та фактор XI після його взаємодії з XII і IX чинниками. Він починає гальмувати активність фактора XII. Усеце гальмує дію тромбіну, порушує агрегацію тромбоцитів.
Найпотужніші антикоагулянти — це волокна фібрину, що виникли в процесі тромбоутворення, та альфаглобулін (антитромбін III) або анти- тромбін-гепаринкофактор. 90% тромбіну, утвореногозпротромбіну, абсорбується фібриновими волокнами, що не дозволяє йому поширитися в крові й запобігає розвиткові тромбоутворення. Тромбін, що не адсорбувався фібриновими волокнами, швидко зв'язується з антитромбіном III, який блокує ефекти тромбіну на фібриноген та інактивує, зв'язуючи тромбін протягом наступних 15-20 хвилин.
Доприроднихантикоагулянтіввідносятьгірудин(міститьсявслині п'явки медичної), табанін (міститься в слині кровосисних) та деякі зміїні отрути. Всі вони мають антитромбінову активність. Для сповільнення процесів зсідання на практиці застосовують цитрат Na+, етилендиамінтетраацетат, які зв'язують вільні іони Са2+.
Динамічна рівновага коагулянтів і антикоагулянтів забезпечує необхідний агрегатний стан крові, а також захист організму від крововтрат при різних травмах. Забезпечення достатньої плинності крові відіграє велику роль у запобіганні виникненню таких захворювань, як інфаркт міокарда, тромбоз судин головного мозку. З віком у зв'язку з накопиченням у плазмі крові йсудинній стінці ліпідів зростає ризик такихускладнень.
Регуляція процесів зсідання здійснюється нервовими і гуморальними механізмами. Активація як симпатичної нервової системи, так і парасимпатичної, а також катехоламінів та ацетилхоліну викликає первинну гіперкоагуляцію (підвищену здатність до зсідання) внаслідок активації фактора XII, посилення гідролізу жирів зі збільшенням тромбопластичної активності і зростанням виділення фосфоліпідів з формових елементів. Це посилює протромбіназну активністькрові. Підвпливом адреналіну ізстіноксудинвиділяються також антикоагулянти й активатори фібринолізу. Важливу роль у цих процесах відіграють нирки та шлунково-кишковий тракт. Вони виводять з організму надлишок прокоагулянтів.
Гіпокоагуляцгя — процес завжди вторинний і спостерігається як наступний етап гіперкоагуляції. Як виняток первинна гіпокоагуляція
245
■I
зустрічається у разі недостатності первинних або вторинних факторів зсідання крові. Наприклад, гемофілія А (класична гемофілія) зумовлена генетичною недостатністю VIII чинника, гемофілія В — фактора IX, гемофілія С — фактора XI. Порушення нормальної роботи печінки призводить до недостатнього синтезу протромбіну, факторів VII, IX, X. Це так само утруднює процеси зсідання. Цьому також сприяє недостатність жиророзчинного вітаміну К, необхідного для синтезу тих самих чинників. Безумовно, і тромбоцитопенія призводить не тільки до порушень процесів зсідання, а й до підвищеної проникності судин, погіршення ретракції згустку.
Фактори VII і IX пов'язані з жіночою хромосомою як рецесивна ознака. Тому жінки частіше всього на гемофілію майже не хворіють, оскільки одназдвоххромосому нихнормальна.
Фактор VIII має два компоненти: великий з молекулярною масою близько 1 млн і менший з молекулярною масою близько 230 тис. Менший більш важливийдлявнутрішньоготромбоутворення. Йогодефіцит— причинакласичної гемофілії. Іншахвороба(кровотечі здосить різними характеристиками) називається хвороба Віллібранта, це — результат втрати великого компонента. Лікування класичної гемофілії — вливання очищеного чинника VIII або переливання крові.
Тромбоцитопенія— кровотечачастішезсудинмалогодіаметра. Хвороба тромбоцитопенічна пурпура. Виникає ламкість судинної стінки. Ідіопатична тромбоцитопенія — специфічні антитіла діють проти власних тромбоцитів, руйнуючи їх. Розвивається після гемотрансфузій від інших людей. Вважають, що розвивається автоімунний процес. Лікування— переливаннятромбоцитарної маси, спленектомія.
Підвищене тромбоутворення призводить до утворення таких тромбів, які відриваються з утворенням емболів, Останні з током крові можуть потрапляти внирки, мозок, іншіоргани, якщовониутворилисьу лівійполовині серця і великих артеріях. Тромбоутворення у венах призводить до емболізації судин легень.
При зменшенні діаметра судини шар плазми біля стінки збільшується, що зменшує в'язкість. У результаті змінюється і гематокрит. При зниженні температури зростає в'язкість. Так, призниженніїї до25 °Св'язкість зростає на 50 %. Однак можна зменшити гематокрит з 40 % до 20 % і тоді при перепаді температур в'язкість не зміниться. В'язкість зростає при гіперкапнії, й тому вона вища у венозній крові. Зростання в'язкості прискорює зсідання крові. Сповільнення швидкості кровообігу, підвищення кількості глобулінів сприяє агрегації клітин крові і, таким чином, тромбоутворенню, оскільки місцево зростає концентрація прокоагулянтів.
246
VI. СИСТЕМА КРОВООБІГУ. ФІЗІОЛОГІЧНІ ВЛАСТИВОСТІСЕРЦЕВОГОМ'ЯЗА_____
Усі клітини організму ніби плавають у міжтканинній рідині. Між кров'ю та міжклітинною рідиною відбувається постійний обмін їхніми складовими: водою, електролітами, газами, продуктами метаболізму, поживними речовинами.
Однак сам процес транспорту речовин ворганізмі можливийлише при наявності серцево-судинної системи. Вона одна з головних систем організму. Серцево-судинна система включає серце — м'язовийорган(помпа) ісудини(шляхитранспорту). Серце— цечотирикамерний порожнинний орган, який складається з двох передсердь ідвохшлуночків. Стінкаусіхвідділівскладаєтьсязендокарду(вистилаєвнутрішнюповерхнюіутворюєклапанитавсвоючергу складаєтьсязендотеліютапідендотелїальної основи— цесполучнатканина з кровоносними судинами та нервами), міокарда (м'язовий шар) та епікарда (сполучнотканиннй шар). Серце знаходиться в перикарді (осердя) — навколосерцевасумка. Міжпериіепікардом знаходитьсясерознарідина, яка зменшуєтертяприскороченняхміокарда,
Судини — це трубки, діаметр яких може змінюватися під впливом інтрата екстрасудинних нервових і гуморальних факторів. Як і всі інші системи, вона також має механізми регуляції (міогенні, нервові та гуморальні). Система кровообігу анатомічно складається з великого та малого кола кровообігів, які виконують дещо відмінні функції. Протевобохвипадкахсудиниєпровіднимишляхамитранспорту. Роль малого (легеневого) кола полягає, в основному, в газообмінійтепловіддачі; великого(системного) — врозподілійдоставці об'єму крові усім органам і тканинам відповідно до їхніх потреб і функціональногостану організму.
Судини, крім того, що вони є шляхами транспорту, беруть участь у підтриманні й регуляції кров'яного тиску врізних ділянках системикровообігу.
Головним чинником течії крові по судинах є робота серця. Цьому сприяє присмоктувальна дія грудної порожнини під час вдиху. Обумовлено це збільшенням негативного тиску в міжплевральному просторі й загалом у грудній порожнині. «М'язова помпа» — скорочення скелетних м'язів і вичавлювання з венозних судин крові у напрямку до серця. «Венозна помпа» — скорочення стінки венозних судин. Крім того, прискороченнішлуночківатріовентрикулярнаперегородка відтягується донизу, створюючи присмоктувальну тягу впередсердях.
247
Цілеспрямованій течії крові сприяють атріовентрикулярні клапани, півмісяцеві клапани аорти і легеневої артерії та клапани венознихсудин.
Само серце представлено двома видами клітин: типовими (робочими) й атиповими (пейсмекерами) кардіоміоцитами. Функція робочих кардіоміоцитів полягає, в основному, в забезпеченні насосної функції серця. Атипові кардіоміоцити бідніші міофібрилами з відповідним переважанням саркоплазми. Якщо в робочих кардіоміоцитах СР розвинутий менше, ніж у поперечносмугастих, то в атипових він розвинутий ще гірше. Вони також стійкіші до гіпоксії внаслідок високої швидкості анаеробного гліколізу. Все це наближає атипові кардіоміоцити до ембріональної тканини. У серці вони утворюютьспецифічну провідну систему тавиконуютьфункціїводіївритму.
Провідна система включає синоатріальний (1-го порядку) й атріовентрикулярний (2-го порядку) вузли, жмутик Пса з його правою і лівою ніжками та сітку волокон Пуркіньє. Крім синоатріального (номотопного), всі інші осередки виникнення збудження ектопічні. Швидкість проведення збудження робочими кардіоміоцитами у 5 разів менша, ніж атиповими (до 5 м/с). В окремих випадках (патологічні стани) робочі кардіоміоцити також набувають властивості генерувати потенціали дії.
Між окремими клітинами (атиповими і типовими) серця існують контакти — нексуси (вставні диски), опір яких, завдяки наявності іонних (повільні кальцієві) каналів, значно менший, ніж опір міжклітинної рідини. Це полегшує перехід збудження з клітини на клітину. Внаслідок цього міокард утворює функціональний синцитій, що не тільки покращує провідність, а й сприяє роботі серця як єдиного цілого. Тому виникнення потенціалу дії в одному місці обов'язково призведе до його поширення по всьому міокарду. При цьому збудження поширюється бездекрементно (без зміни амплітуди потенціалу дії).
Виходячизнаведеного(наявністьпровідноїсистемитанексусів), робота серця підпорядковується закону «все або нічого» на відміну від ізольованого збудження нервових волокон і поперечносмугастих м'язів. Робочі кардіоміоцити за допомогою вставних дисків утворюють функціональний синцитій у межах кожної камери серця. Вставні диски представлені десмосомами (забезпечують механічне зчеплення, яке протидіє розходженню кардіоміоцитів) та проміжними контактами (необхідні для прикріплення тонких актинових ниток найближчого саркомера до сарколеми кардіоміоцита). Щілинні контакти — це міжклітинні іонні (повільні кальцієві) канали, які проводять збудження від одного кардіоміоцита до іншого, що разом з провідною системою забезпечує синхронність скорочення
248
функціонального синцитій. Окремо виділяють ендокринні клітини, які синтезують атріопептид, який бере участь у регуляції артеріального тиску.
Міокард передсердь тонший, ніж у шлуночків, і має два шари (циркулярний та поздовжній). Циркулярний переважно оточує судини, які впадають у передсердя, перетискаючи їх при скороченнях. Міокард шлуночків товщий, особливо лівого шлуночка, і має три шари. Середній, окремий для кожного шлуночка (циркулярний) і зовнішній та внутрішній (спіралеподібні) спільні. М'язові волокна передсердь і шлуночків прикріплюються до фіброзного кільця, розташованого між ними. Сюди ж прикріплюються й атріовентрикулярні клапани. Розміри кардіоміоцитів передсердь менші за такі шлуночків. Місткість СР в міокарді менша, ніж у смугастих м'язах. В основному в передсердях утворюються дігіталісоподібні фактори — похідні арахідонової кислоти та гормони (атріонатрійуретичний фактор, ангіотензин).
У нормі від «головки» синоатріального (синусно-передсердного, синусового, Кіс-Флака) вузла передсердними волокнами (передні жмутики Бахмана) збудження передається до лівого передсердя. Від його «хвостової» частини жмутиками Венкебаха (середній) та Тореля (задній) і частково жмутиками Бахмана збудження передається до атріовентрикулярного вузла, який міститься в товщі міжшлуночкової перегородки, між передсердями та шлуночками.
Затримка збудження відбувається в атріовентрикулярному (Ашоффа-Тавара) вузлі та в місцях контакту з волокнами Пуркіньє. Більша швидкість проведення провідною системою обумовлена наявністю в її клітинах великої кількості швидких натрієвих каналів.
В окремих випадках збудження від синоатріального вузла передається до міокарда шлуночків жмутиком Кента (лівий, правий — тип А, тип Б постійного або непостійного синдрому WPW) швидше, ніж передсерднршлуночковим з'єднанням. Тому частина волокон активується передчасно. Якщо збудження поширюється жмутиком Джеймса, то воно досягає пучка Пса, минаючи атріовентрикулярний вузол, що знову ж таки передчасно активує частину міокарда шлуночків. При функціонуванні жмутика Махайма збудження також оминає пучок Пса і швидше спричиняє скорочення частини міокарда. Вусіх цих випадках виникає комбіноване скорочення міокарда — через звичайне і «ненормальне» (швидше) проведення збуження до міокарда. Це супроводжується порушенням роботи серцевого м'яза: скоротливості та ритму (синдроми WPW, CLC).
До фізіологічних властивостей кардіоміоцитів відносять автоматію (самовільна генерація потенціалу дії), збудливість, провідність, рефрактерність і скоротливість зі здатністю до розслаблення. Автоматія формує частоту і ритм скорочень серця. Збудливість і провідність визначають послідовність і синхронність скорочень як
249

окремих камер серця, так і передсердь з шлуночками в цілому. Рефрактерність запобігає явищу тетанусу в серцевому м'язі (рівнозначно зупинці серця у фазі систоли й припинення кровообігу), позачерговим (екстрасистоличні) передчасним скороченням за рахунокшвидкого повторного збудження та рециркуляції збудження в міокарді. Екстрасистоличні скорочення несуть у собі виникнення загрозливих (термінальних) для життя аритмій.
Розглянемо природу автомати. В атипових кардіоміоцитах існують повільні потенціалзалежні кальцієві канали L-типу, які відкриваються при мембранному потенціалі, що дорівнює -60 мВ. Зрозуміло, що тільки-но мембранний потенціал досягає цього рівня, канали відкриваються, й усередину клітини за концентраційним (хімічним) і електричним градієнтами входять іони Са2+. Канали не суворо специфічні і пропускають також невелику кількість іонів Na+. Це спричиняє повільну деполяризацію клітинної мембрани. Ця деполяризація має назву — повільна спонтанна діастолічна дег поляризація. Водночас також спостерігається закриття калієвих каналів, і вихідний струм, який міг би шунтувати вхід іонів Са2+, зникає. При досяганні Е^ (від -45 до -50 мВ) відкриваються кальцієві канали іншого типу та швидкі потенціалзалежні натрієві, запускається лавиноподібна, регенераторна відповідь і розвивається швидка деполяризація клітинної мембрани внаслідок появи вхідного кальцієвого та натрієвого струмів,
Проте існування швидких натрієвих каналів беззастережно визнається не всіма дослідниками. Окремі авторивважають, що швидкі натрієві канали внаслідок низького мембранного потенціалу вже в попередньому стані інактивовані. На піку деполяризації (овершут відсутній— рис. 66) повільнікальцієвійшвидкінатрієвіканалипочинають інакивуватися, провідність калієвих — зростає. Розвивається процес реполяризації. На останніх етапах реполяризації швидкі натрієві й повільні кальцієві канали повертаються до попереднього стану, калієві канали закриваються. При досяганні мембранним потенціалом рівня -60 мВ знову створюються умови для
розвиткуспонтанноїдіастолічної депо-
МВ |
ляризації, і весь процес починається |
|
|
|
спочатку. Однак можливість кожної |
|
клітини провідної системи до само- |
|
вільної генерації потенціалу дії змен- |
|
шується в міру того, як вона відда- |
|
ляється від синоатріального вузла. Це |
явищеотрималоназвуградієнтаавто-
Рис. 66. Потенціал дії ати- мати. Його фізіологічне значення по- |
|
пових кардіоміоцитів |
лягає в тому, що при ураженні високо |
L 250
розміщених ділянок провідної системи роль водія ритму беруть на себе нижче розташовані. В нормальних умовах автоматія всіх інших відділів провідної системи пригнічується більш високою частотою виникнення потенціалів дії у синоатріальному вузлі. Збудження останнього ніби розряджає інші, нижче розміщені ділянки. Синоатріальний вузол (пейсмекер першого порядку) генерує потенціали дії з частотою близько 60-80 уд~\ атріовентрикулярний (пейсмекер другого порядку) — 40—50 уд~\ жмутик Пса (пейсмекер третього порядку) — 30-40 уд"1 і волокна Пуркіньє — 20 уд"1. Синоатріальний вузол — номотопний (нормально розміщений), інші — гетеротопні (ненормально розміщені) центри генерування збудження. При частоті скорочень серця 30-40 уд"1 кровообіг життєво важливих органів стає недостатнім, і людина втрачає свідомість, виникають судоми (приступ Едемса-Стокса). Для запобігання незворотним змінам у корі головного мозку потрібно штучно збільшити частоту серцевих скорочень. Ділянка міокарда, яка активується одним волокном Пуркіньє, дорівнює 1 см. Ця відстань отримала назву довжинивільного пробігу.
Як зазначалося вище, швидкість проведення збудження атиповимикардіоміоцитами більша, ніж робочими. Внаслідок цього збудження практично одночасно досягає всіх робочих кардіоміоцитів передсердь або шлуночків. Відповідно сила скорочень зростає. Однак в атріовентрикулярному вузлі через наявність особливостей будови і контактів атипових клітин (особливості геометричної будови) та специфіки розвитку потенціалу дії спостерігається затримка проведення збудження. За цей час обидва передсердя встигають повністю скоротитись і додатково наповнити кров'ю шлуночки на ЗО %. Затримка збудження в атріовентрикулярному вузлі забезпечує почерговість скорочень передсердь і шлуночків. В окремих випадках збудження може виникати в інших ділянках провідної системи, і воно також спричинятиме виникнення скорочення міокарда. Можливий також варіант скорочення при збудженні з двох різних ділянок. Саме так виникає подвійний тон Образцова-Стражеска, коли передсердя і шлуночки скорочуються одночасно: передсердя завдякизбудженнюзсиноатріальноговузла, шлуночки— зжмутика Пса або інших ділянок провідної системи. Це зустрічається при повній атріовентрикулярній блокаді.
Наступна затримка проведення збудження спостерігається при його переході з волокон Пуркіньє на робочі кардіоміоцити. Вона забезпечує синхронізацію збудження та одночасне скорочення усього робочого міокарда.
Існують особливості проведення збудження одним кардіоміоцитом і ділянками міокарда. Оскільки потенціал дії кардіоміоцитів
251
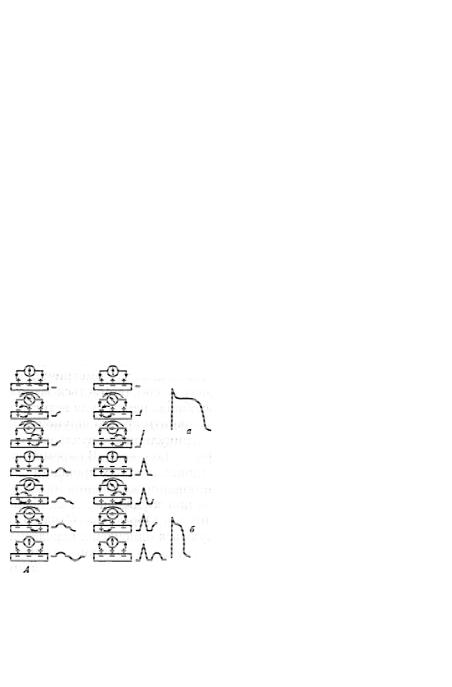
більш тривалий, ніж у скелетних м'язах, то реполяризація починається лише після повної деполяризації клітини. При цьому напрямок її поширення у шлуночках буде протилежний напрямку поширення деполяризації. Відповідно напрямок струму в зовнішньому реєструвальному колі залишиться попереднім, і записана електрограмаматиме дваоднаковонаправленізубці(рис. 67, Б). Обумовлено це тим, що процеси реполяризації розвиваються значно повільніше, ніж деполяризації, швидкість самої реполяризації неоднакова в різних ділянках міокарда (на вершині й у субепікарді раніше, ніж біля основи і в субендокарді) та тим, що її поширення не пов'язано з провідноюсистемою.
Сповільнення процесів реполяризації в субепікарді обумовлено підвищенням тиску в шлуночках і, як наслідок, значним зменшенням коронарного кровотоку в епікарді.
Подібні явища спостерігаються у шлуночках. У передсердях реполяризація починається за деполяризацією, і тому на електрограмі другийзубецьматиме протилежнийнапрямок (рис. 67, А). Цеможна зареєструвати в разі припинення скорочення шлуночків.
Потенціал спокою робочих кардіоміоцитів дорівнює -90 мВ. Розвиток потенціалу дії (рис. 68, Б) у робочих кардіоміоцитах у фазу 0 маєтакусамуприроду, яківскелетнихм'язах: лавиноподібний, регенераторний вхід Na+ в клітину через швидкі потенціалзалежні натрієві канали. Водночас знижуєтьсяпровідністькалієвихка-
|
|
налів«аномальноговипрямлен- |
|
|
ня». Виникає овершут (+30 мВ) |
|
|
мембранного потенціалу. Нада- |
|
|
лі виникає короткочасна швид- |
|
|
ка рання реполяризація (фаза |
|
|
1) внаслідок пасивного входу |
|
|
аніонів СІ" за електричним гра- |
|
|
дієнтом. Можливо, вона обу- |
|
|
мовлена інактивацією швидких |
|
|
потенціалзалежних натрієвих |
|
|
каналів або активацією каліє- |
Б |
в |
вих (швидких потенціалзалеж- |
Рис. 67. Поширення деполяризації |
них). Фаза плато або повільна |
|
та реполяризації, напрямок струмів |
реполяризація (фаза 2) обумов- |
|
у зовнішній і внутрішній частинах |
ленавідкриттямповільнихкаль- |
|
електричного кола, |
електрограми |
цієвих каналів Т-типу і входом |
м'язової смужки з серця: передсердь |
у клітину іонів Са2+. У цей час |
|
(А), шлуночків (Б). Тривалість по- |
||
тенціалу дії біля основи (В— а) і на |
швидкі натрієві канали інакти- |
|
верхівці(В— б) |
|
вовані, й скоротлива клітина |
252 |
|
|