

stennikova2
.pdfγ = kÒ+10 2 ÷ 4 . kÒ
Если известны константы скорости при температурах, отличающихся не на 10 К, то коэффициент Вант-Гоффа может быть определен из уравнения:
|
|
k2 |
|
|
Ò2 −Ò1 |
|||
|
|
|
= γ |
|
, |
|||
|
|
|
10 |
|||||
|
|
k |
|
|||||
|
|
|
|
|
|
|||
|
1 |
|
|
|
|
|||
где k2 и k1 – константы скорости при температурах Т2 и Т1. |
||||||||
Более строгую зависимость константы скорости от температуры дает |
||||||||
уравнение: |
|
|
|
|
|
|
|
|
d ln k = |
E |
|
, |
(3.6) |
||||
RT 2 |
||||||||
dT |
|
|
|
|
||||
которое получило название уравнения Аррениуса. Величина Е имеет размерность энергии и носит название энергии активации.
Энергию активации можно определить как тот избыток энергии по сравнению со средней энергией молекул при данной температуре, которым должны обладать молекулы, чтобы они могли легко вступить в химическую реакцию. Поскольку Е > 0, то производная (ln k)' >0, следовательно, lnk и k будут всегда возрастать с ростом Т.
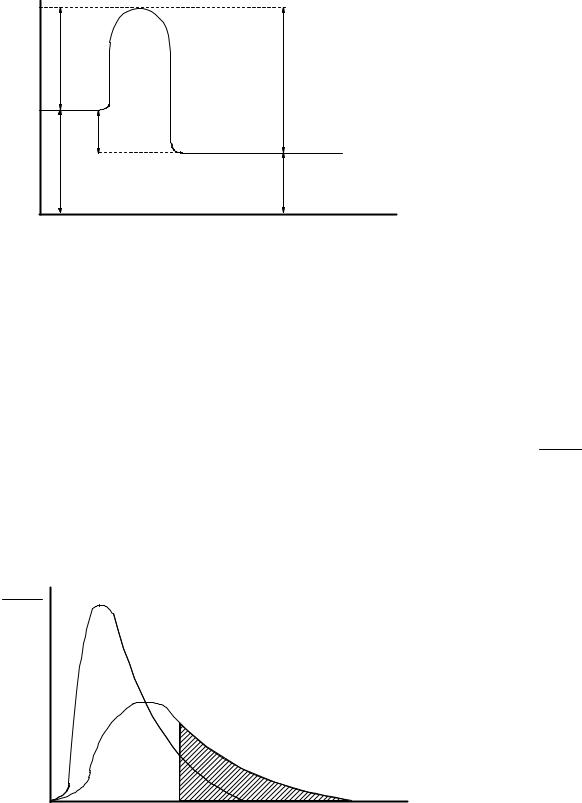
Связь энергии активации с тепловым эффектом можно проиллюстрировать с помощью представления об энергетическом барьере (рисунок 3.6). Химическую реакцию можно представить как переход системы из энергетического состояния 1 в состояние 2, сопровождающийся тепловым эффектом ∆Н (при Р = const). Из рисунка 3.6 видно, что переход из состояния 1 в состояние 2 возможен при затрате энергии Е, а обратный переход возможен при затрате энергии Е´. Поскольку Е´ > Е, то энергетический барьер обратной реакции будет больше, чем прямой. Следовательно скорость обратной реакции будет меньше скорости прямой.
В задачу химической кинетики входит поиск способов уменьшения энергетического барьера, энергии активации, для увеличения скорости. Одним из таких способов является применение катализатора.
При осуществлении реакции в прямом направлении выделяется количество энергии ∆Í = Å − Å′, а в обратном направлении затрачивается такое же количество энергии, что выделяется в прямом.
Уравнение (3.6) легко проинтегрировать. Считая, что Е ≠ f (T ) , получим
ln k = − |
E |
+ ln A , |
(3.7) |
RT
где lnA – константа интегрирования.
− E
k = Ae RT .
Отсюда видно, что величинами, характеризующими реакцию, являются предэкспоненциальный множитель А и энергия активации Е.
81
энергия |
Е |
|
|
A+B |
Е' |
||
|
|||
∆Н |
C+D |
||
|
|||
|
Н1 |
Н2 |
|
|
|
||
|
|
путь реакции |
Рисунок 3.6 – Диаграмма, иллюстрирующая связь Е и ∆Н |
|
|
Поскольку для вступления в химическую реакцию молекулы должны |
||
обладать некоторой избыточной энергией (Е), по сравнению со средней |
||
величиной (рисунок 3.7), то влияние повышения температуры на рост |
||
скорости реакции можно объяснить увеличением числа частиц, обладающих |
||
этим избытком энергии. |
dN |
|
Обратимся к рисунку 3.7, где изображена зависимость величины |
||
|
|
N0dε |
от энергии, так называемая кривая распределения молекул по энергиям. Где |
||
dN – число молекул, обладающих энергиями от ε до ε + dε ; N0 – общее |
||
число молекул. Пусть ε1 - энергия, превышающая среднюю на энергию |
||
активации. |
|
|
dN |
T1 |
|
N0dε |
|
|
|
|
|
|
T2 |
|
|
ε |
|
|
ε1 |
|
Рисунок 3.7 – Кривая распределения молекул по энергиям |
|
|
Площадь под кривой правее абсциссы ε1 , ограниченная кривой и осью абсцисс, определяет собой долю молекул, обладающих энергией,
82
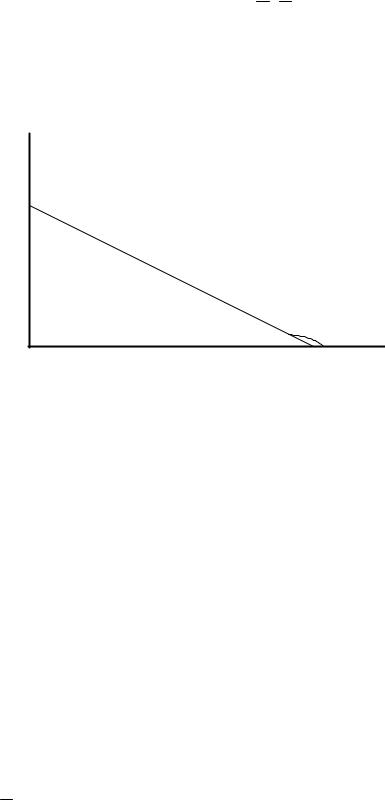
превышающей ε1 . С повышением температуры (Т2 >Т1) кривая сдвигается
вправо, и эта доля молекул быстро возрастает.
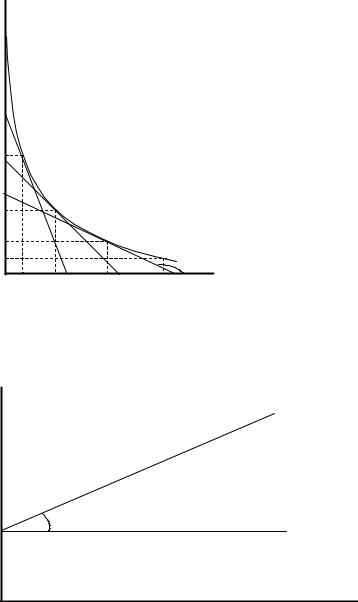
Величину энергии активации можно определить двумя методами. Первый метод – графический, с использованием уравнения Аррениуса в виде
(3.7):
ln k = − ER Ò1 + ln A .
Если построить график зависимости экспериментальных величин ln k от 1/Т, то получим прямую. По оси ординат отсекается отрезок, равный lnA.
Тангенс угла наклона на кривой равен − ЕR
lnk |
|
|
lnA |
|
|
|
tgα=-E/R |
|
|
α |
|
0 |
1 |
|
T |
||
|
Рисунок 3.8 – Зависимость логарифма константы скорости химической реакции от обратной температуры
Второй метод основан на измерении скорости химической реакции при двух температурах. Для этого из уравнения (3.6) получим:
|
k2 |
|
E |
|
1 |
1 |
|
||
|
|
|
|
||||||
ln |
|
|
= − |
|
|
|
− |
|
, |
k |
1 |
R |
T |
T |
|||||
|
|
|
|
|
2 |
1 |
|
||
E = |
RT1T2 |
ln |
k2 |
. |
||
T |
−T |
|
||||
|
|
k |
1 |
|
||
|
2 |
1 |
|
|
|
|
Постоянной энергия активации может быть только в простых реакциях. Для сложных реакций величина Е является переменной и не имеет такого простого физического смысла, как в случае простых реакций. Тем не менее, и в этом случае, принято величину Е называть энергией активации, но
определяют ее из дифференциального уравнения Аррениуса |
d ln k |
2 |
, |
||
Å = |
|
RT |
|
||
dT |
|
||||
|
|
|
|
|
|
используя зависимость k = f (T ) и ln k = f (T ) .
Для сложных реакций, следовательно, не будет линейной зависимости ln k = f (T1 ) .
83
3.4 Методы определения порядка реакции
Знание порядка реакции позволяет определить такие практически важные величины, как константа скорости, текущая концентрация, время достижения текущей концентрации. Это показано на примере реакций первого и второго порядков (раздел 3.2.2).
Для определения порядка реакции в целом необходимо определить частные порядки по каждому веществу, вступающему в реакцию, а затем суммировать их. Для определения порядка по данному веществу необходимо создать условия, при которых будет изменяться концентрация только этого вещества.
Укажем некоторые из таких условий:
1.В реакции принимает участие одно исходное вещество.
2.Скорость реакции зависит от концентрации одного реагента и катализатора, а концентрация катализатора постоянна по ходу реакции.
3.Все реагенты, кроме одного, берутся в большом избытке, в результате чего их концентрации мало меняются по ходу реакции и могут рассматриваться как постоянные.
4.Концентрация всех реагентов, кроме одного, поддерживается
постоянной каким-либо искусственным путем. Например, проводя реакции с участием иона ОН- как реагента, можно обеспечить присутствие буфера для поддержания концентрации ОН-.
При определении порядка реакции используется основное свойство
кинетического уравнения υ = kC Am C Bn , согласно которому константа
скорости и порядок реакции по ее компонентам остаются постоянными в течение всей реакции, а так же при изменении начальных концентраций реагентов.
Метод подстановки
По ходу реакции определяют текущие концентрации исходного вещества в различные моменты времени от начала реакции.
Предполагают, что реакция первого порядка. По уравнению для константы скорости первого порядка (раздел 3.2.2, с. 78) рассчитывают все возможные значения К. Если значения К постоянны (в пределах ошибки опыта), то реакция имеет первый порядок. Если же не постоянны значения К, или закономерно убывают или возрастают, то реакция не первого порядка. Аналогично проверяют на второй порядок. Если реакция не первого и не второго порядка, то для определения порядка используют другие методы.
Графический метод
Этот метод основан на том, что кинетическая кривая может иметь линейный вид (x) = f(t), где вид (x) соответствует конкретному порядку реакции:
84
для первого порядка |
ln C = f (t), |
для второго порядка |
1/С = f (t), |
для третьего порядка |
1/С2 = f (t), |
для нулевого порядка |
Спродукта = f (t). |
Поочередно строя такие зависимости, смотрят, в каких координатах получается прямая линия.
Этот метод и метод подстановки взаимосвязаны, и нет смысла его использовать, если метод подстановки не выявил порядок реакции.
Метод начальных скоростей
Скорость расходования вещества в начальный момент времени определяется по уравнению:
υ0 = k(C A0 ) n (CB0 ) m .
Для понижения порядка проводят ряд опытов, в которых берется вещество А с различной начальной концентрацией, C A0 , а начальная концентрация B во всех опытах берется одинаковой. Тогда
υ0 = k I (C A0 )n , где k I = k(CB0 )m , |
|
lnυ0 = ln k I + nln CA0 . |
(3.8) |
Получилось уравнение прямой в координатах ( lnυ0 , lnC ) с тангенсом
угла наклона, равным порядку реакции, n. Для определения начальных скоростей получают начальные участки кинетических кривых, поскольку лишь в начале реакции сохраняется постоянство концентрации вещества В.
Порядок реакции можно приблизительно определить по значениям двух начальных скоростей (υ10 è υ20 ), которые соответствуют двум начальным концентрациям реагента (Ñ10 è Ñ02 ), используя соотношение (3.8):
|
lnυ20 = ln k + nln C20 , |
|
|
|
|
|
|
|
||||||
|
lnυ0 |
= ln k + nln C0 , |
|
|
|
|
|
|
|
|||||
|
1 |
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
Вычитая |
|
второе |
уравнение |
из |
первого, |
получаем |
|||||||
lnυ0 |
− lnυ0 |
= n(ln C0 |
|
|
|
0 ) |
откуда n = |
ln(υ0 |
/υ0 ) |
. |
|
|
||
− ln C |
2 |
1 |
|
|
||||||||||
ln(C0 |
/ C0 ) |
|
|
|||||||||||
2 |
1 |
2 |
|
1 |
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
2 |
1 |
|
|
|
|
Для приближенного расчета скоростей можно использовать |
|||||||||||||
соотношение: |
dC |
|
≈ |
∆C |
|
, |
|
|
|
|
|
|
||
dt |
∆t |
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
||||
где ∆C - изменение концентрации реагента за промежуток времени ∆t .
Метод избытка (метод Вант - Гоффа)
Это метод определения порядка по изменяющейся во времени скорости реакции от концентрации реагента. Реакцию проводят при большом избытке всех реагентов, кроме одного, например А. Тогда
85
υ = kCAn , |
lnυ = ln k + n ln CA . |
В процессе реакции расходуется реагент и уменьшается скорость |
реакции. Скорости реакции находят из полной кинетической кривой, проводя |
графическое дифференцирование, определяя тангенсы углов наклона |
касательных к кинетической кривой (рисунок 3.9). |
CA |
0 |
tgα=v |
t |
Рисунок 3.9 – Графическое дифференцирование кинетической кривой |
lnv |
tgα = n |
lnC |
Рисунок 3.10 – Определение порядка реакции |
По найденным значениям υ строят график в координатах ( lnυ, ln CA ), согласно уравнению (3.8), и находят порядок, n (рисунок 3.10).
86
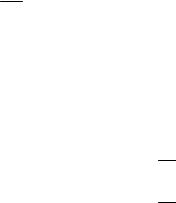
Метод нахождения общего порядка реакции
Для нахождения общего порядка реакции необходимо взять реагенты в стехиометрическом соотношении. Если стехиометрическое уравнение реакции:
|
аА + вВ → продукты |
|
|
|
|
|||||||||
то при СА : СВ = а : в, |
СВ = СА |
|
в |
. |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
а |
|
|
|
|
|
|
|||
Тогда кинетическое уравнение примет вид: |
|
|
|
|
|
|
||||||||
n m |
n |
â m |
â |
|
m |
(n+m) |
|
' |
(n+m) |
. |
||||
υ = kÑACB |
= kCA CA |
|
= k( |
|
) |
|
CA |
= k |
CA |
|||||
à |
à |
|
||||||||||||
Далее логарифмирование приведет |
к линейной |
зависимости lnυ от |
||||||||||||
и определению (n + m) как тангенс угла наклона (аналогично нахождению в методах 3 и 4).
Метод по доле непревращенного вещества к моменту времени t
Если а = С - доля исходного вещества, не вступившего в химическую
С0
реакцию к моменту времени t, то С = аС0 . Подставляя выражение для С в
уравнение
dCdt = −kC n
получаем:
dadt = kan (C0 )n−1 , adan = (C0 )n−1 dt.
Интегрирование в пределах (а = 1; а) и (t = 0; t) приводит к уравнению:
1 |
|
(a1−n −1) = k(C0n−1 )t . |
(3.9) |
|
n −1 |
||||
|
|
|||
Уравнение сложно для расчета «n», поэтому его применяют в двух частных случаях:
а) проводят ряд опытов, в которых взяты разные начальные концентрации, но во всех опытах реакция доведена до одинаковой доли а. В таких условиях левая часть уравнения (3.9) постоянна, обозначим ее В:
 = k(C0n−1 )t, |
|
ln B = ln k + (n −1) ln C0 + ln t, |
|
ln t = (1− n) ln C0 + ln B − ln k. |
(3.10) |
Полученное уравнение прямой в координатах (lnt; lnC0) позволяет графически найти «n», поскольку tgα =1− n .
Если проведено два опыта с одинаковым значением а, то, записав уравнение 3.10 для обоих опытов и вычитая одно из другого, получаем уравнение для n:
87

ln t2 = (1− n) ln(C20 ) + ln B − ln k
-
ln t |
1 |
= (1− n) ln(C 0 ) + ln B − ln k |
|
||||||
|
|
|
|
|
1 |
|
|
|
|
ln t |
2 |
− ln t |
1 |
= (1− n)(ln C 0 |
− ln C |
0 ) |
|||
|
|
|
|
2 |
1 |
||||
n =1+ |
ln(t2 / t1 ) |
|
|
||||||
ln(C 0 |
/ C 0 ) |
|
|
|
|||||
|
|
|
|
|
1 |
2 |
|
|
|
б) Можно определить порядок, располагая одной полной кинетической кривой. Выбирают на кривой два значения а, связанные между собой
соотношением: а2 = а12 и отвечающие этим значениям t2 и t1.
При этом скобка в левой части уравнения (3.9) для а2 будет иметь вид: (a12(1−n) −1) = (a11−n −1) (a11−n +1) .
Записав уравнение (3.9) для второго опыта и для первого и поделив уравнения одно на другое получаем, сократив общие множители:
n 1−1 (a11−n −1) (a11−n +1)= k(C 0 )n−1 t2 ,
n 1−1 (a11−n −1)= k(C 0 )n−1 t1 ,
a1−n +1 = t2 .
1 t1
После логарифмирования получаем:
|
|
|
||
|
|
t2 |
|
|
|
|
|||
|
ln t |
−1 |
||
n =1− |
1 |
|
. |
|
|
|
|||
|
ln a |
|||
|
|
|
1 |
|
3.5 Сложные реакции
Сложная реакция состоит из нескольких элементарных стадий, связанных друг с другом определенным образом через исходные вещества и промежуточные продукты.
В отношении сложных реакций в кинетике возникают две задачи. Прямая задача. Известны механизм и константы скорости каждой
элементарной стадии. Необходимо для конкретных условий (Т, Сi) описать кинетическое поведение каждого молекулярного продукта. Это достигается составлением и решением системы дифференциальных уравнений. Зная (вычислив) кинетические кривые для всех веществ, можно рассчитать скорость отдельных стадий для разных моментов времени протекания реакции.
Обратная задача. Экспериментально получают набор кинетических кривых и на основании полученных результатов проверяют предполагаемую схему реакции, определяют порядок и константу скорости каждой из стадий.
88

В сложных реакциях действует принцип независимости реакций,
согласно которому: если в системе одновременно протекает несколько
реакций, каждая из них независима от других реакций и будет протекать со скоростью, определяющейся своим дифференциальным уравнением и своей константой скорости.
Из этого принципа следует условие материального баланса: если в
результате протекания химических реакций исчезает или появляется какой-либо компонент, то скорость изменения концентрации этого вещества будет равна алгебраической сумме скоростей его образования и расходования во всех стадиях, то есть
dCi |
= ∑νiυi , |
(3.11) |
|
n |
|
dt |
1 |
|
где n – число элементарных стадий, в которых участвует i компонентов; νi – стехиометрический коэффициент i-го компонента в данной стадии, для
исходных веществ νi < 0, для продуктов νi > 0; υ – скорость реакции в
единичном объеме в данной стадии, определяется кинетическим уравнением
υ = k СаАСвВ
(А и В – исходные вещества в данной системе). Рассмотрим пример.
k1
|
|
|
|
|
k3 |
|
|
|
|
|
|
A + 1/2B |
|
|
|
C + 2D |
→ E. |
k2
Эта сложная реакция может быть расписана в две строчки
k1
A + 1/2B 
 C + 2D , k2
C + 2D , k2
C + 2D |
k3 |
→ E. |
Обе записи тождественны.
Запишем условие материального баланса для компонента D. Он участвует в трех стадиях (n = 3) с константами скоростей k1, k2 и k3
dcD = ∑3 νDυ.
dt 1
Таким образом, произведений νD . υ будет три. νD в реакции 1, где D является продуктом, равна +2; а в реакциях 2 и 3, где D является исходным
веществом, νD = -2.
ddtcD = 2k1CAC1B/ 2 – 2k2CC C 2D – 2k3CC C 2D .
Аналогично для вещества В:
dCB = ∑νBυ = - 1/2 k1CAC1B/ 2 |
+ 1/2 k2CC C 2D . |
|
2 |
|
|
dt |
1 |
|
89
3.5.1 Параллельные реакции
При параллельных реакциях одно и то же вещество (или вещества) участвует в нескольких реакциях, давая различные продукты. Например, разложение бертолетовой соли возможно по двум реакциям
К1
2KCl + O2
6 KClO3
3KClO4 + KCl
К2
Рассмотрим простейший случай – две параллельные реакции первого порядка. Получим уравнения для расчета констант реакции k1 и k2:
|
A |
|
|
|
|
B |
|
|
|
|
D |
||
|
|
|
|
|||
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
t = 0 |
a |
0 |
0 |
|||
t |
a - x |
|
x2 |
x1 |
||
Вначальный момент (t = 0) в системе присутствует только вещество А
сконцентрацией a, к моменту времени t концентрация a уменьшилась на x,
появилось x1 вещества В и х2 вещества D.
Составим дифференциальное уравнение для вещества А. Решая его,
найдем k1 и k2.
dCA |
= −k1CA − k2CA = −(k1 |
+ k2 )CA , |
|
|
|||
dt |
|||
|
|
|
d(a − x) |
= −(k1 + k2 )(a − x), |
|
|||||||||||
|
dt |
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
dx = (k1 + k2 )(a − x), |
∫ dx |
= ∫(k1 + k2 )dt, |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
x |
|
t |
|
dt |
|
|
|
|
|
|
|
x=0 a - x |
t =0 |
||||
− ln(a − x) + ln a = (k1 + k2 )t, |
|
|||||||||||||
k1 + k2 = 1 ln |
|
|
a |
. |
|
|
(3.12) |
|||||||
a − x |
|
|
||||||||||||
|
|
|
|
|
t |
|
|
|
|
|||||
Составим дифференциальные уравнения для веществ В и D. |
||||||||||||||
|
|
dcB |
|
|
= |
|
dx1 |
|
= k1 (a − x), |
|
|
(3.13) |
||
|
|
dt |
|
|
|
|
||||||||
|
|
|
|
dt |
|
|
|
|
|
|
|
|||
|
|
dcD |
|
= |
dx2 |
|
= k2 (a − x). |
(3.14) |
||||||
|
|
dt |
dt |
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|||||
Разделив уравнение (3.13) на (3.14), получим
90