

Enzymes (Second Edition)
.pdf60 STRUCTURAL COMPONENTS OF ENZYMES
3.4.2 The -Pleated Sheet
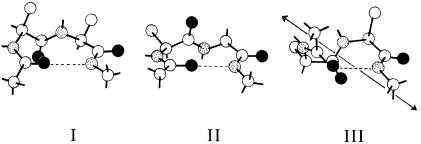
The -pleated sheet, another very abundant secondary structure found in proteins, is composed of fully extended polypeptide chains linked together through interamide hydrogen bonding between adjacent strands of the sheet (Figure 3.11). Figure 3.11A illustrates a -pleated sheet composed of two polypeptide chains. Note that the arrangement of peptide C O and N H groups permits this structure to be extended in either direction (i.e., to the left or right in the plane of the page as shown in Figure 3.11A) through the same type of interamide hydrogen bonding (Figure 3.11B).
For the moment, let us focus on a two-stranded sheet, as in Figure 3.11A: the two component polypeptide chains could come from two distinct polypeptides (an intermolecular sheet) or from two regions of the same contiguous
Figure 3.11 (A) A -pleated sheet composed of two segments of polypeptide held together by interchain hydrogen bonding. (B) An extended -pleated sheet composed of four segments of polypeptide.
SECONDARY STRUCTURE |
61 |
Figure 3.12 Three common forms of turn.
polypeptide (an intramolecular sheet); both types are found in natural proteins. If we imagine a sheet within the plane of this page, we could have both chains running in the same direction, say from C-terminus at the top of the page to N-terminus at the botton. Alternatively, we could have the two chains running in opposite directions with respect to the placement of their N- and C-termini. These two situations describe structures referred to as parallel and antiparallel -pleated sheets, respectively. Again, one finds both types in nature.
3.4.3 Turns
A third common secondary structure found in natural proteins is the turn (also known as a reverse turn, hairpin turn, or bend). The turns are short segments of the polypeptide chain that allow it to change direction — that is, to turn upon itself. Turns are composed of four amino acid residues in a compact configuration in which an interamide hydrogen bond is formed between the first and fourth residue to stabilize the structure. Three types of turn are commonly found in proteins: types I, II, and III (Figure 3.12). Although turns represent small segments of the polypeptide chain, they occur often in a protein, allowing the molecule to adopt a compact three-dimensional structure. Consider, for example, an intramolecular antiparallel sheet within a contiguous segment of a protein. To bring the two strands of the sheet into register for the correct hydrogen bonds to form, the polypeptide chain would have to change direction by 180°. This can be accomplished only by incorporating a type I or type II turn into the polypeptide chain, between the two segments making up the sheet. Thus turns play a very important role in establishing the overall three-dimensional structure of a protein.
3.4.4 Other Secondary Structures
One can imagine other regular repeating structural motifs that are stereochemically possible for polypeptides. In a series of adjacent type III turns, for
62 STRUCTURAL COMPONENTS OF ENZYMES
example, the polypeptide chain would adopt a helical structure, different from the helix, that is known as a 3 helix. This structure is indeed found in proteins, but it is rare. Some proteins, composed of high percentages of a single amino acid type, can adopt specialized helical structures, such as the polyproline helices and polyglycine helices. Again, these are special cases, not commonly found in the vast majority of proteins.
Most proteins contain regions of well-defined secondary structures interspersed with segments of nonrepeating, unordered structure in a conformation commonly referred to as random coil structure. These regions provide dynamic flexibility to the protein, allowing it to change shape, or conformation. These structural fluctuations can play an important role in facilitating the biological activities of proteins in general. They have particular significance in the cycle of substrate binding, catalytic transformations, and product release that is required for enzymes to function.
3.5 TERTIARY STRUCTURE
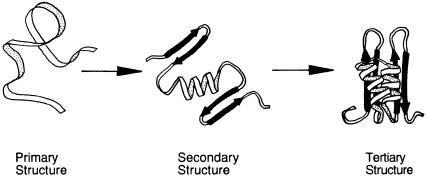
The term ‘‘tertiary structure’’ refers to the arrangement of secondary structure elements and amino acid side chain interactions that define the three-dimen- sional structure of the folded protein. Imagine that a newly synthesized protein exists in nature as a fully extended polypeptide chain — it is said then to be unfolded (Figure 3.13A) [Actually there is debate over how fully extended the polypeptide chain really is in the unfolded state of a protein; some data suggest that even in the unfolded state, proteins retain a certain amount of structure. However, this is not an important point for our present discussion.] Now suppose that this protein is placed under the set of conditions that will lead to the formation of elements of secondary structure at appropriate locations along the polypeptide chain (Figure 3.13B). Next, the individual elements of second-
Figure 3.13 The folding of a polypeptide chain illustrating the hierarchy of protein structure from primary structure or amino acid sequence through secondary structure and tertiary structure. [Adapted from Dill et al., Protein Sci. 4, 561 (1995).]
TERTIARY STRUCTURE |
63 |
ary structure arrange themselves in three-dimensional space, so that specific contacts are made between amino acid side chains and between backbone groups (Figure 3.13C). The resulting folded structure of the protein is referred to as its tertiary structure.
What we have just described is the process of protein folding, which occurs naturally in cells as new proteins are synthesized at the ribosomes. The process is remarkable because under the right set of conditions it will also proceed spontaneously outside the cell in a test tube (in vitro). For example, at high concentrations chemicals like urea and guanidine hydrochloride will cause most proteins to adopt an unfolded conformation. In many cases, the subsequent removal of these chemicals (by dialysis, gel filtration chromatography, or dilution) will cause the protein to refold spontaneously into its correct native conformation (i.e., the folded state that occurs naturally and best facilitates the biological activity of the protein). The very ability to perform such experiments in the laboratory indicates that all the information required for the folding of a protein into its proper secondary and tertiary structures is encoded within the amino acid sequence of that protein.
Why is it that proteins fold into these tertiary structures? There are several important advantages to proper folding for a protein. First, folding provides a means of burying hydrophobic residues away from the polar solvent and exposing polar residues to solvent for favorable interactions. In fact, many scientists believe that the shielding of hydrophobic residues from the solvent is one of the strongest thermodynamic forces driving protein folding. Second, through folding the protein can bring together amino acid side chains that are distant from one another along the polypeptide chain. By bringing such groups into close proximity, the protein can form chemically reactive centers, such as the active sites of enzymes. An excellent example is provided by the serine protease chymotrypsin.
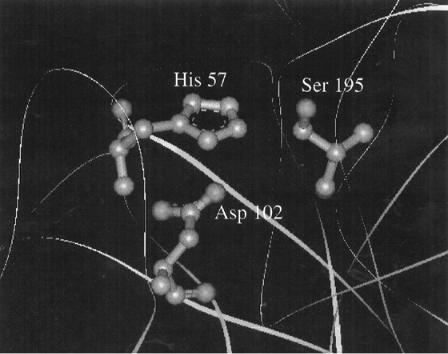
Serine proteases are a family of enzymes that cleave peptide bonds in proteins at specific amino acid residues (see Chapter 6 for more details). All these enzymes must have a serine residue within their active sites which functions as the primary nucleophile — that is, to attack the substrate peptide, thereby initiating catalysis. To enhance the nucleophilicity of this residue, the hydroxyl group of the serine side chain participates in hydrogen bonding with an active site histidine residue, which in turn may hydrogen-bond to an active site aspartate as shown in Figure 3.14. This ‘‘active site triad’’ of amino acids is a structural feature common to all serine proteases. In chymotrypsin this triad is composed of Asp 102, His 57, and Ser 195. As the numbering indicates, these three residues would be quite distant from one another along the fully extended polypeptide chain of chymotrypsin. However, the tertiary structure of chymotrypsin is such that when the protein is properly folded, these three residues come together to form the necessary interactions for effective catalysis.
The tertiary structure of a protein will often provide folds or pockets within the protein structure that can accommodate small molecules. We have already used the term ‘‘active site’’ several times, referring, collectively, to the chemically reactive groups of the enzyme that facilitate catalysis. The active site of
64 STRUCTURAL COMPONENTS OF ENZYMES
Figure 3.14 The active site triad of the serine protease -chymotrypsin. [Adapted from the crystal structure reported by Frigerio et al. (1992) J. Mol. Biol. 225, 107.] (See Color Plates.)
an enzyme is also defined by a cavity or pocket into which the substrate molecule binds to initiate the enzymatic reaction; the interior of this binding pocket is lined with the chemically reactive groups from the protein. As we shall see in Chapter 6, there is a precise stereochemical relationship between the structure of the molecules that bind to the enzyme and that of the active site pocket. The same is generally true for the binding of agonists and antagonists to the binding pockets of protein receptors. In all these cases, the structure of the binding pocket is dictated by the tertiary structure of the protein.
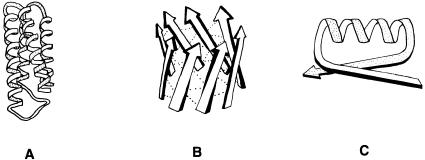
While no two proteins have completely identical three-dimensional structures, enzymes that carry out similar functions often adopt similar active site structures, and sometimes similar overall folding patterns. Some arrangements of secondary structure elements, which occur commonly in folded proteins, are referred to by some workers as supersecondary structure. Three examples of supersecondary structures are the helical bundle, the barrel, and the — — loop, illustrated in Figure 3.15.
In some proteins one finds discrete regions of compact tertiary structure that are separated by stretches of the polypeptide chain in a more flexible arrange-
SUBUNITS AND QUATERNARY STRUCTURE |
65 |
Figure 3.15 Examples of supersecondary structures: (A) a helical bundle, (B) a barrel, and
(C) a — — loop.
ment. These discrete folded units are known as domains, and often they define functional units of the protein. For example, many cell membrane receptors play a role in signal transduction by binding extracellular ligands at the cell surface. In response to ligand binding, the receptor undergoes a structural change that results in macromolecular interactions between the receptor and other proteins within the cell cytosol. These interactions in turn set off a cascade of biochemical events that ultimately lead to some form of cellular response to ligand binding. To function in this capacity, such a receptor requires a minimum of three separate domains: an extracellular ligand binding domain, a transmembrane domain that anchors the protein within the cell membrane, and an intracellular domain that forms the locus for protein—protein interactions. These concepts are schematically illustrated in Figure 3.16.
Many enzymes are composed of discrete domains as well. For example, the crystal structure of the integral membrane enzyme prostaglandin synthase was recently solved by Garavito and his coworkers (Picot et al., 1994). The structure reveals three separate domains of the folded enzyme monomer: a-sheet domain that functions as an interface for dimerization with another molecule of the enzyme, a membrane-incorporated -helical domain that anchors the enzyme to the biological membrane, and a extramembrane globular (i.e., compact folded region) domain that contains the enzymatic active site and is thus the catalytic unit of the enzyme.
3.6 SUBUNITS AND QUATERNARY STRUCTURE
Not every protein functions as a single folded polypeptide chain. In many cases the biological activity of a protein requires two or more folded polypeptide chains to associate to form a functional molecule. In such cases the individual polypeptides of the active molecule are referred to as subunits. The subunits may be multiple copies of the same polypeptide chain (a homomultimer), or
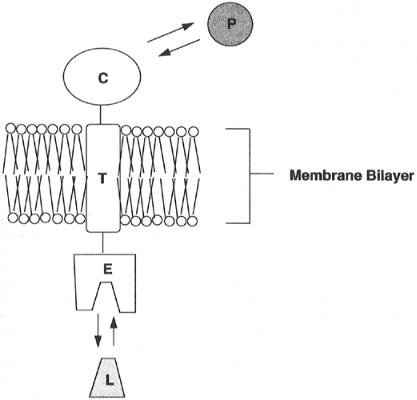
66 STRUCTURAL COMPONENTS OF ENZYMES
Figure 3.16 Cartoon illustration of the domains of a typical transmembrane receptor. The protein consists of three domains. The extracellular domain (E) forms the center for interaction with the receptor ligand (L). The transmembrane domain (T) anchors the receptor within the phospholipid bilayer of the cellular membrane. The cytosolic domain (C) extends into the intracellular space and forms a locus for interactions with other cytosolic proteins (P), which can then go on to transduce signals within the cell.
they may represent distinct polypeptides (a heteromultimer). In both cases the subunits fold as individual units, acquiring their own secondary and tertiary structures. The association between subunits may be stabilized through noncovalent forces, such as hydrogen bonding, salt bridge formation, and hydrophobic interactions, and may additionally include covalent disulfide bonding between cysteines on the different subunits.
There are numerous examples of multisubunit enzymes in nature, and a few are listed in Table 3.3. In some cases, the subunits act as quasi-independent catalytic units. For example, the enzyme prostaglandin synthase exists as a homodimer, with each subunit containing an independent active site that processes substrate molecules to product. In other cases, the active site of the enzyme is contained within a single subunit, and the other subunits serve to
SUBUNITS AND QUATERNARY STRUCTURE |
67 |
||
Table 3.3 Examples of multisubunit enzymes |
|
||
|
|
|
|
Enzyme |
Number of Subunits |
|
|
|
|
|
|
HIV protease |
2 |
|
|
Hexokinase |
2 |
|
|
Bacterial cytochrome oxidase |
3 |
|
|
Lactate dehydrogenase |
4 |
|
|
Aspartate carbamoyl transferase |
12 |
|
|
Human cytochrome oxidase |
13 |
|
|
|
|
|
|
stabilize the structure, or modify the reactivity of that active subunit. In the cytochrome oxidases, for example, all the active sites are contained in subunit I, and the other 3—12 subunits (depending of species) modify the stability and specific activity of subunit I. In still other cases the active site of the enzyme is formed by the coming together of the individual subunits. A good illustration of this comes from the aspartyl protease of the human immunodeficiency virus, HIV (the causal agent of AIDS). The active sites of all aspartyl proteases require a pair of aspartic acid residues for catalysis. The HIV protease is synthesized as a 99-residue polypeptide chain that dimerizes to form the active enzyme (a homodimer). Residue 25 of each HIV protease monomer is an aspartic acid residue. When the monomers combine to form the active homodimer, the two Asp 25 residues (designated Asp 25 and Asp 25 to denote their locations on separate polypeptide chains) come together to form the active site structure. Without this subunit association, the enzyme could not perform its catalytic duties.
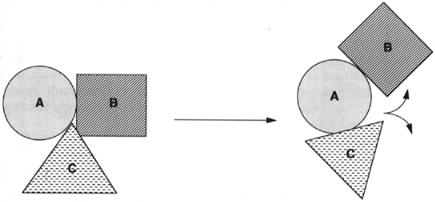
The arrangement of subunits of a protein relative to one another defines the quaternary structure of the protein. Consider a heterotrimeric protein composed of subunits A, B, and C. Each subunit folds into its own discrete tertiary structure. As suggested schematically in Figure 3.17, these three subunits could take up a number of different arrangements with respect to one another in three-dimensional space. This cartoon depicts two particular arrangements, or quaternary structures, that exist in equilibrium with each other. Changes in quaternary structure of this type can occur as part of the activity of many proteins, and these changes can have dramatic consequences.
An example of the importance of protein quaternary structure comes from examination of the biological activity of hemoglobin. Hemoglobin is the protein in blood that is responsible for transporting oxygen from the lungs to the muscles (as well as transporting carbon dioxide in the opposite direction). The active unit of hemoglobin is a heterotetramer, composed of two subunits and two subunits. Each of these four subunits contains a heme cofactor (see Section 3.7) that is capable of binding a molecule of oxygen. The affinity of the heme for oxygen depends on the quaternary structure of the protein and on the state of oxygen binding of the heme groups in the other three subunits (a
68 STRUCTURAL COMPONENTS OF ENZYMES
Figure 3.17 Cartoon illustrating the changes in subunit arrangements for a hypothetical heterotrimer that might result from a modification in quaternary structure.
property known as cooperativity). Because of the cooperativity of oxygen binding to the hemes, hemoglobin molecules almost always have all four heme sites bound to oxygen (the oxy form) or all four heme sites free of oxygen (the deoxy form); intermediate forms with one, two, or three oxygen molecules bound are almost never observed.
When the crystal structures of oxyand deoxyhemoglobin were solved, it was discovered that the two forms differed significantly in quaternary structure.
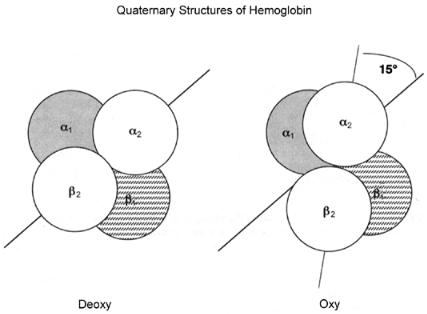
If we label the four subunits of hemoglobin , , , and , we find that at
the interface between the and subunits, oxygen binding causes changes in hydrogen bonding and salt bridges that lead to a compression of the overall
size of the molecule, and a rotation of 15° for the pair of subunits relative
to the pair (Figure 3.18). These changes in quaternary structure in part affect the relative affinity of the four heme groups for oxygen, providing a means of reversible oxygen binding by the protein. It is the reversibility of the oxygen binding of hemoglobin that allows it to function as a biological transporter of this important energy source; hemoglobin can bind oxygen tightly in the lungs and then release it in the muscles, thus facilitating cellular respiration in higher organisms. (For a very clear description of all the factors leading to reversible oxygen binding and structural transitions in hemoglobin, see Stryer, 1989.)
3.7 COFACTORS IN ENZYMES
As we have seen, the structures of the 20 amino acid side chains can confer on enzymes a vast array of chemical reactivities. Often, however, the reactions catalyzed by enzymes require the incorporation of additional chemical groups to facilitate rapid reaction. Thus to fulfill reactivity needs
COFACTORS IN ENZYMES |
69 |
Figure 3.18 Cartoon illustration of the quaternary structure changes that accompany the binding of oxygen to hemoglobin.
that cannot be achieved with the amino acids alone, many enzymes incorporate nonprotein chemical groups into the structures of their active sites. These nonprotein chemical groups are collectively referred to as enzyme cofactors or coenzymes; Figure 3.19 presents the structures of some common enzyme cofactors.
In most cases, the cofactor and the enzyme associate through noncovalent interactions, such as those described in Chapter 2 (e.g., H-bonding, hydrophobic interactions). In some cases, however, the cofactors are covalently bonded to the polypeptide of the enzyme. For example, the heme group of the electron transfer protein cytochrome c, is bound to the protein through thioester bonds with two modified cysteine residues. Another example of covalent cofactor incorporation is the pyridoxal phosphate cofactor of the enzyme aspartate aminotransferase. Here the cofactor is covalently linked to the protein through formation of a Schiff base with a lysine residue in the active site.
In enzymes requiring a cofactor for activity, the protein portion of the active species is referred to as the apoenzyme, and the active complex between the protein and cofactor is called the holoenzyme. In some cases the cofactors can be removed to form the apoenzyme and be added back later to reconstitute the active holoenzyme. In some of these cases, chemically or isotopically modified versions of the cofactor can be incorporated into the apoenzyme to facilitate structural and mechanistic studies of the enzyme.