

17. Современные представления о принципе Дейла.
Дейл (1875-1968) – английский физиолог, в 1914 г. им было выделено вещество ацетилхолин, позже, уже в тридцатые годы он вместе со своими сотрудниками показал, что ацетилхолин является нейромедиатором в нервно-мышечном синапсе. Полученные данные позволили Дейлу сформулировать предположение о том, что поскольку каждый нейрон представляет собой единую метаболическую систему то, следовательно, во всех его пресинаптических окончаниях должен высвобождаться один и тот же медиатор. Это предположение и было названо принципом Дейла. Однако, в последние годы получены данные, убедительно свидетельствующие о совместимости нейроактивных веществ в одном нейроне, о возможности синтеза, транспорта и выведения одним и тем же нейроном нескольких разных нейроактивных в-в, в том числе и нейромедиаторов.
В современном звучании его можно сформулировать как положение о метаболической зависимости аксона и его окончаний от тела клетки. Т. обр. в настоящее время представление о хим. проведении сигналов в нервной клетке основывается на принципе множественности хим. сигналов, это подразумевает, что в индивидуальном нейроне синтезируется более одного медиатора, при этом каждое пресинптическое окончание способно выделить несколько медиаторов , сочетание которых может быть неодинаковым для разных синапсов одного и того же нейрона. Поэтому привычное понятие “один нейрон- один медиатор” следует понимать как условное.
18. Отличие некроза от апоптоза. Роль стволовых клеток и апотоза в индивид. развитии и некоторых нервных заболеваниях.
Некроз (греч. nekros - мертвый) возникает в результате прямого воздействия патогенного фактора (микроорганизм, ишемия и др.), нарушающего целостность мембраны клетки. Это приводит к массивному выбросу индукторов воспаления и к миграции иммунных клеток к очагу поражения. В результате в зоне поврежденной клетки развивается септическое или асептическое (в зависимости от причины) воспаление. При этом происходят характерные изменения как в ядре, так и в цитоплазме. Ядро сморщивается, наблюдается конденсация хроматина (каропикноз), затем он распадается на глыбки (каризрексис) и растворяется (кариолизис). В цитоплазме происходят денатурация и коагуляция белков. Мембранные структуры распадаются. Нарушаются окислительно-восстановительные процессы и синтез АТФ в митохондриях, и вся клетка начинает страдать от нехватки энергии. Постепенно клетка распадается на отдельные глыбки, которые захватываются и поглощаются макрофагами. На месте в прошлом функционально активной клетки формируется соединительная ткань.
Апоптоз (греч. аро - отделение + ptosis - падение) по морфологическим признакам существенно отличается от некроза и имеет ряд специфических особенностей. Факторами, инициирующими апоптоз, являются возрастание экспрессии генов - индукторов апоптоза (или угнетение генов-ингибиторов) либо повышенное поступление кальция внутрь клетки. Клеточная мембрана при этом остается сохранной. Несмотря на внешнюю сохранность мембраны митохондрий, нарушаются окислительно-восстановительные процессы в основном за счет блокирования 1 митохондриального комплекса. Результатом описанных выше процессов является возрастание синтеза протеаз, которые начинают постепенно расщеплять внутриклеточные структуры. От мембраны клетки отщемляются небольшие везикулы, наполненные содержимым цитоплазмы (митохондрий, рибосомы и др.), окруженные мембранным липидным бислоем. Клетка соответственно уменьшается в объеме и сморщивается. Отщепившиеся везикулы поглощаются соседними клетками. Ядро сморщивается на завершающих стадиях процесса, хроматин частично конденсируется, что говорит о сохранной активности ряда участков ДНК. Оставшиеся от клетки элементы фагоцитируются тканевыми макрофагами без развития воспалительной реакции и формирования соединительной ткани.
|
Основные отличительные черты некроза и апоптоза
|
Результатом апоптоза является постепенное и медленное избавление от "ненужных" в функциональном отношении на данный момент клеток. При этом не развивается воспаление и не нарушается нормальное функционирование соседних клеток, а также не происходит соединительнотканного замещения, что позволяет сохранить структуру органа. Функциональные элементы клетки, находящейся в состоянии апоптоза, не разрушаются, а поглощаются другими клетками и могут использоваться дальше. Особенно большую роль апоптоз играет в эмбриогенезе, когда важно постепенно избавляться от выполнивших свою функцию клеток, а активное фагоцитирование с развитием реакции воспаления может нарушить созревание плода.
Среди заболеваний нервной системыособую роль апоптоз играет в развитии церебральных дегенерации. Триггерные факторы апоптоза ЦНС сегодня изучены недостаточно. Предполагаются влияние нейротропных, персистирующих внутриклеточно вирусов; нарушение считывания генетической информации; воздействие индукторов апоптоза. Все эти факторы пока еще не получили достаточного подтверждения.
Общим для всех дегенеративных заболеваний ЦНС является снижение устойчивости нервных клеток к стимуляторам апоптоза - эксайтоаминокислотам, вирусным белкам или ионам кальция (см. выше). Однако цепь событий, приводящих к апоптозу, имеет существенные различия при разных заболеваниях
Болезнь Паркинсона(БП). В патогенезе БП важную роль играет нарушение функции митохондрий за счет блокирования 1 митохондриального комплекса. Результатом этого является снижение содержания в клетках АТФ и последующее уменьшение образования глутатиона - универсального антиоксиданта ЦНС. Следующим этапом патогенеза является окислительный стресс, связанный с накоплением свободных радикалов. В условиях окислительного стресса происходит активация NMDA-рецепторов, приводящая к повышенному входу кальция внутрь клетки и дальнейшему развитию апоптоза; некоторые авторы считают также, что окислительный стресс может дополнительно вызывать экспрессию гена р53 с последующей стимуляцией дегенерации нервных клеток. Процесс избирательно поражает нейроны подкорковых образований мозга, в большей степени стриатума и компактной части черного вещества. Изучение медикаментозной терапии БП с этих позиций показало, что специфическая терапия препаратами леводопы может активировать апоптоз, поскольку усиливает окислительный стресс. Так, при обработке культуры нейронов раствором дофамина в физиологической концентрации (0,1-1 мМ) в течение 24 ч обнаружены признаки, характерные для апоптоза: фрагментация ДНК, дезинтеграция аксонов, сморщивание клеток. Такие же изменения наблюдались и при обработке клеточных культур раствором леводопы. Напротив, применение агонистов дофаминовых рецепторов и блокаторов моноаминоксидазы типа В приводит к увеличению выживаемости культуры стволовых клеток в эксперименте, что предположительно связывают с активацией нейротрофических факторов, ингибирующих апоптоз. W. Tatton сообщает, что нейропротективное действие препаратов - ингибиторов моноаминоксидазы (L-депренил) может также быть результатом возрастания экспрессии генов-ингибиторов апоптоза.
Болезнь Альцгеймера(БА). Патогенез БА до настоящего времени остается невыясненным. Одним из возможных патогенетических механизмов считается внутриклеточное отложение (3-амилоида и предшественника амилоидного белка (amyloid precursor protein - АРР). В первую очередь при этом страдают ацетилхолинергические нейроны базального ядра Мейнерта, результатом чего являются недостаточность холинергических терминалей и дегенерация клеток коры головного мозга, преимущественно теменно-височно-затылочной области.
В нейронах, хронически обрабатываемых раствором, содержащим компоненты амилоида, развиваются изменения, типичные для апоптоза: отпочковывание везикул от мембраны и фрагментация ДНК. Процессы, происходящие в культуре клеток, при этом равнозначны при воздействии как (3-амилоида, так и АРР. Помимо этого, отмечается внутриклеточное отложение амилоида, также препятствующее нормальной жизнедеятельности клетки.
Не исключено, что апоптоз при БА реализуется по механизму ускоренного старения с патологическим накоплением кальция внутри клетки за счет активации NMDA-рецепторов с последующей активацией протеаз и разрушением клеточных структур.
Синдром Дауна (СД).СД, или трисомия хромосомы 21, является одной из наиболее частых генетических причин умственной отсталости. Основными характерными признаками СД служат уменьшенное количество нейронов ЦНС и извращенное дифференцирование нервных клеток. Причины и ход нейродегенеративного процесса при СД до конца не выяснены. Показано, что кортикальные нейроны эмбрионов с трисомией-21 до определенного момента нормально развиваются в культуре, однако потом начинают подвергаться апоптозу. Представляет интерес тот факт, что при СД, как и при БА, отмечаются интрацеллюлярные отложения амилоида в церебральных нейронах, что, возможно, косвенно указывает на сходный патогенез этих заболеваний.
Стволовые клетки
Найдены не только в основных регенерирующих тканях, таких, как эпителий и кровь, но также в статических тканях, таких, как нервная система и печень, где они играют центральную роль в тканевом росте и сохранении. Механизм, с помощью которого стволовые клетки поддерживают популяции высоко дифференцированных короткоживущих клеток, как кажется, включает критический баланс между альтернативными путями (судьбами): дочерние клетки либо сохраняют идентичность стволовых клеток, либо инициируют дифференциацию. Недавние исследования на низших организмах выявили регуляторный механизм ассимитричных клеточных делений стволовых клеток. В этих моделях окружение, вероятно, обеспечивает ключевые инструктивные сигналы для выбора судьбы клеток. Наше понимание сейчас распространяется на внутренние механизмы клеточной полярности, которая влияет на ассиметричные деления стволовых клеток/12
Вот, что нашла. Из 1-ой лекции по анатомии.
Хотя сами по себе нервные клетки могут размножаться и восстанавливаться. Например, стволовые клетки. Какое-то количество дендритов не специализируется и в случае необходимости стволовые клетки получают сигнал о необходимости «прийти на помощь», они прибывают на место повреждение и начинают специализироваться. Например, при инсульте значительная часть клеток гибнет, а некоторая восстанавливается из стволовых клеток.
Билет № 19 Постсинаптические потенциалы
Постсинаптический потенциал - это изменение мембранного потенциала постсинаптической мембраны в ответ на импульс, поступивший от пресинаптического нейрона (относительно кратковременные колебания мембранного потенциала (чаще десятки миллисекунд, реже секунды), возникающие в результате местного воздействия медиатора на постсинаптическую мембрану ).
Различают возбуждающий постсинаптический потенциал и тормозной постсинаптический потенциал .
Амплитуда постсинаптических потенциалов зависит от количества выделенного медиатора. Взаимодействуя со специфическими рецепторами постсинаптической мембраны, медиаторы увеличивают её проницаемость для определённых ионов, которые входят в клетку или выходят из неё в соответствии с электрохимическим градиентом. Если этот процесс приводит к уменьшению трансмембранной разности потенциалов (деполяризации), постсинаптические потенциалы являются возбуждающими (ВПСП). Тормозные постсинаптические потенциалы (ТПСП) выражаются в гиперполяризации клетки, обусловленной действием тормозного медиатора.
Как правило, нервная клетка имеет большое число синаптических входов; приходящие к ней сигналы алгебраически суммируются. В клетках, спонтанно генерирующих потенциалы действия, ВПСП увеличивает, а ТПСП уменьшает частоту разрядов. В «молчащих» клетках ВПСП может вызвать одиночный или групповой разряд, а одновременно возникший ТПСП блокировать этот эффект.
Таким образом, с помощью постсинаптических потенциалов осуществляется управление возбудимостью нервных клеток.
20 Гиперполяризационное и деполяризационное торможение
Ткань может находиться в трех состояниях: покоя, возбуждения и торможения. Возбуждение и торможение - это два активных состояния, которые возникают под действием раздражителей и сопровождаются энергозатратами.
По механизму возникновения выделяют два вида торможения: деполяризационное и гиперполяризационное.
Деполяризационное торможение
В основе его лежит механизм деполяризации мембраны, что приводит к утрате или существенному снижению способности реагировать на другие стимулы ( клетка в состоянии возбуждения заторможена, т.е клетки не отвечают на действие других раздражителей, (рис. 4.1).

Рис.4.1. Фаза абсолютной рефрактерности на пике потенциала действия
Разновидность деполяризационного торможения - парабиотическое торможение (торможение по Н.Е. Введенскому). Оно развивается в клетке под влиянием определенного вещества - парабиотика, которое меняет функциональное состояние клетки, нарушая ее функциональную лабильность. Основной причиной этого торможения является уменьшение функциональной лабильности клеток. Снижение лабильности характеризуется увеличением времени проведения импульсов, удлинением времени деполяризации и особенно реполяризации мембраны и следовых потенциалов (рис. 4.2 а,б,в).

Рис. 4.2. Изменение функциональной лабильности клетки под влиянием парабиотика приводящее к парабиотическому торможению. А - уравнительная фаза Б - парадоксальная фаза В - тормозная фаза
Парабиотик нарушает энергетические функции клеток: синтез и ресинтез АТФ, что замедляет работу Na-K насоса. Это приводит к изменению проницаемости мембраны для ионов и стойкой деполяризации. Процесс перехода из нормального состояния в парабиотическое происходит в несколько фаз.
1. Уравнительная фаза характеризуется тем, что на частые и редкие импульсы возникает одинаковая реакция. Для каждой ткани характерна своя функциональная лабильность. Нервная ткань проводит ~ 500 импульсов в секунду, мышечная - 300 импульсов в секунду, самая низкая лабильность в синапсах ~ 120 импульсов в секунду. Например, при снижении функциональной лабильности в нервной клетке до 200 импульсов в секунду она будет отвечать в одинаковой степени на более частые (300-400-500) и более редкие импульсы (200) (рис. 4.2 а). Если убрать парабиотик, ткань постепенно восстанавливает свою лабильность, но если парабиотик будет продолжать действовать, то наступит вторая фаза.
2. Парадоксальная фаза характеризуется тем, что на редкие импульсы ткань будет отвечать, а на частые нет. Эта фаза возникает потому, что функциональная лабильность продолжает уменьшаться, удлиняется период рефракторной фазы и ткань перестает реагировать на частые импульсы (рис. 4.2 б), которые попадают в рефрактерный период.
3. Тормозная фаза, наступает при продолжении действия парабиотика (рис. 4.2 в), прекращается ответ ткани на любые по частоте и силе импульсы, поскольку в клетках не происходит реполяризации мембраны и она остается в состоянии абсолютной рефрактерности. Нарушается ресинтез АТФ, (причем повреждение насосов происходит быстрее, чем нарушение каналов). В клетку попадает Na (вслед за ним - H2O, клетка может разбухнуть и погибнуть). Восстановить клетку из этого состояния невозможно. Таким образом, по мере развития парабиотического торможения уменьшается функциональная лабильность ткани. Этот вид торможения развивается под влиянием анестетиков, спирта, других веществ, являющихся парабиотиками.
Гиперполяризационное торможение
Его суть заключается в том, что увеличивается потенциал мембраны по отношению к покою, в связи с чем снижается способность клетки реагировать на раздражители. Возникшее явление характеризуется понижением возбудимости (рис. 4.3). Одним из проявлений этого вида является положительный следовой потенциал, который возникает после потенциала действия и связан с тем, что еще часть ионов К+ не возвращена в клетку ( на наружной поверхности мембраны остается достаточно большое количество ионов К+ ). В этом состоянии наблюдается явление относительной рефрактерности, т.е уменьшение ответа клетки на раздражитель. Необходим более сильный стимул, чтобы клетку возбудить. Из состояния гиперполяризации труднее достичь критического уровня деполяризации, чем из состояния покоя (рис. 4.3). Такое торможение чаще всего развивается в связи с большим выходом К+ наружу и большим поступлением Cl- в клетку. Изменение проницаемости мембран для К+ и Cl-, которое приводит к гиперполяризации мембраны, происходит под влиянием гамма - аминомасляной кислоты (ГАМК), вещества которое вырабатывают специальные нервные клетки (клетки Реншоу); серотонина и некоторых других веществ. Эти вещества увеличивают проницаемость мембраны для К+ и Cl- и приводят к гиперполяризации мембраны (более -100 мв) - формируется тормозной потенциал. Клетка переходит в состояние пониженной возбудимости.

Рис. 4.3. Схема возникновения гиперполяризационного иорможения.
Работа нервной системы чаще всего связана с этим видом торможения. В зависимости от места возникновения торможения различают: пресинаптическое (А) и постсинаптическое (Б); центральное и периферическое (рис. 4.4).

Рис. 4.4. Пресинаптическое (А) и постсинаптическое (Б) торможение
При пресинаптическом торможении чаще всего возникает деполяризация пресинаптической мембраны, в результате чего уменьшается выделение медиатора и передача импульса в синапсе. Постсинаптическое торможение развивается обычно по механизму гиперполяризационного и связано с возникновением тормозного постсинаптического потенциала (ТПСП) под влиянием нейромедиаторов (ГАМК, серотонина).
Центральное торможение имеет место в ЦНС, и впервые было описано И.М. Сеченовым в 1840 г. Накладывая кристаллик NaCl на зрительные бугры (таламус) мозга лягушки, он обнаружил увеличение времени двигательной рефлекторной реакции на раздражитель. Это послужило основанием для заключения, что в ЦНС имеются специфические тормозные центры, возбуждение которых кристалликом соли вызывает торможение в центрах спинномозговых рефлексов (рис. 4.5). Позднее было доказано, что одни и те же центры могут находиться в состоянии возбуждения и в состоянии торможения; а специфическими тормозными центрами могут являться клетки Реншоу, выделяющие тормозной медиатор - ГАМК.
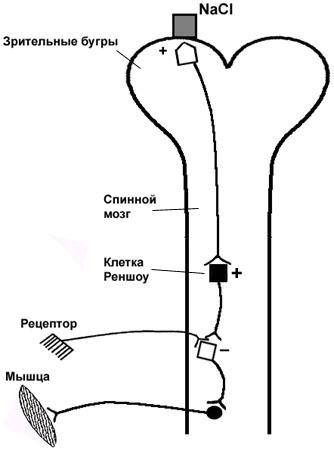
Рис. 4.5. Схема опыта "Сеченовское торможение"
Периферическое торможение было описано братьями Вебер на примере торможения деятельности сердца при раздражении блуждающего нерва. Периферическое торможение развивается вне ЦНС, в органах, и по механизму может быть как гиперполяризационное, так и деполяризационное.
Совокупность возбудительных и тормозных процессов в ЦНС и на периферии определяет специфическую биологически целесообразную деятельность нервной системы и различных органов.
Рецепторный потенциал
Возбуждение и торможение, которое возникает в нервной системе, создает все многообразие ответных реакций, обеспечивая процессы регуляции в организме. Существует специальная нервная структура, функция которой заключается в том, что она воспринимает различные раздражители и обеспечивает трансформацию любого вида энергии в нервный импульс. Эта структура называется рецептор. В рецепторе можно выделить две части: воспринимающую и трансформирующую.
Воспринимающая устроена таким образом, что стимул, действуя на нее, формирует в этих структурах генераторный (рецепторный потенциал). Это происходит вследствие растяжения мембраны клеток (проприорецепторы мышечной ткани), при действии механической волны (звуковые рецепторы), при изменении напряжения, давления (барорецепторы), при изменении электрохимического градиента (при действии химических веществ - хеморецепторы).
Если сила раздражителя и время действия увеличиваются, потенциал в этой части рецептора также увеличивается и удлиняется (рис. 4.6).

Рис. 4.6. Изменение рецепторного потенциала в зависимости от стимула.
При длительном действии раздражителя возникает явление привыкания (адаптации) рецептора, т.е уменьшается амплитуда потенциала.
Чем дольше действие раздражителя, тем быстрее наступает привыкание, чем сильнее раздражитель, тем хуже идет адаптация. Этот потенциал запускает во второй части рецептора формирование потенциала действия (ПД). Потенциал действия по уровню деполяризации везде одинаков. Он отличается по количеству импульсов, длительности интервалов между импульсами. Это явление называется трансформация рецепторного потенциала в потенциал действия. Рецепторный потенциал, как и местный, не распространяется (имеет локальный характер). Потенциал действия отвечает по принципу "все или ничего", обладает способностью к распространению. На выходе из рецепторов регистрируется ПД, который легко распространяется по нерву. В рецепторе создается частотный (сенсорный) код, который характеризует данный раздражитель. В зависимости от частоты и силы раздражителя, он будет отличаться количеством, частотой и длительностью импульсов, но не будет отличаться амплитудой (она одинакова). Любой по количеству стимул, действуя на соответствующий рецептор, трансформируется в ПД. Благодаря этому в НС по волокнам распространяются биопотенциалы с разной частотой и длительностью.