

Молекулярная биология клетки. Том 1
.pdf241
ДНК. В настоящее время это делают с помощью приборов, способных автоматически синтезировать любые последовательности из 80 нуклеотидов в течение ночи. Умение получать молекулы ДНК заданной последовательности дает возможность перестраивать гены, что является важным аспектом генной инженерии (гл. 5).
Существенной областью применения ДНК-олигонуклеотидов является дородовая (пренатальная) диагностика наследственных заболеваний. Более 500 наследственных болезней человека связаны с нарушением какого-то одного гена. В большинстве случаев эти мутации рецессивны. Это означает, что болезнь развивается, если человек получает дефектные копии гена сразу от обоих родителей. Одна из задач современной медицины состоит в том, чтобы выявлять такие аномальные эмбрионы до рождения, информировать об этом мать и дать ей возможность прекратить беременность. Например, для серповидноклеточной анемии известна точная нуклеотидная замена в мутантном гене (последовательность GAG заменена на GTG в цепи ДНК, кодирующей β-цепь гемоглобина). В данном случае синтезируют два олигонуклеотида. Один из них соответствует последовательности нормального гена в участке предполагаемых мутаций, другой несет замену, обусловливающую болезнь. В условиях когда эти последовательности достаточно коротки (примерно 20 нуклеотидов) и при температуре гибридизации, при которой стабильность сохраняют лишь точно совпадающие цепи, можно использовать радиоактивные зонды. Тест состоит в том, что из эмбриональных клеток, содержащихся в амниотической жидкости (ее получают в ходе процедуры, называемой амниоцентезом), выделяют ДНК и используют ее для Саузерн-блоттигна с радиоактивными ДНК-зондами. Дефектный эмбрион легко опознается, поскольку его ДНК будет гибридизоваться только с олигонуклеотидом, комплементарным мутантной последовательности ДНК. К сожалению, для большинства наследственных болезней дефект на уровне ДНК еще не расшифрован, однако круг заболеваний, для которых применяется дородовая диагностика, постоянно расширяется. Это стало возможно благодаря использованию феномена полиморфизма длины рестрикционных фрагментов. В данном случае с помощью гибридизации выявляют наличие или отсутствие определенных сайтов рестрикции, тесно сцепленных с дефектными генами.
4.6.10. Гибридизация позволяет выявлять и отдаленно родственные гены [46]
Возникновение новых генов в ходе эволюции связано с дивергенцией и дупликацией старых генов, а также объединением участков генов в новых комбинациях. По этой причине большинство генов имеют в геноме семейства родственные последовательности, часть которых, повидимому, обладает и близкой функцией. Выделение ДНК-клона, соответствующего первому из членов такого генного семейства, - процедура весьма трудоемкая (разд. 5.6.5). Однако выделение остальных генов этого семейства упрощается, поскольку первый ген может быть использован в качестве зонда. Поскольку в родственных генах маловероятно присутствие идентичных последовательностей, гибридизацию с ДНК-зондами обычно выполняют в менее строгих условиях. Благодаря этому даже неполное соответствие последовательности зонда позволяет сформировать стабильную двойную спираль (рис. 4-73).
Хотя использование нестрогой гибридизации сопровождается повышением вероятности получения фальшивых сигналов от случайных участков гомологии коротких последовательностей в неродственных участках ДНК, такая гибридизация представляет собой одно из наиболее удачных применений технологии рекомбинантных ДНК. Например, этот

242
Рис. 4-73. Сравнение нескольких модификаций метода гибридизации, отличающихся различной жесткостью условий. В реакции слева (жесткие условия) температура раствора поддерживается лишь на несколько градусов ниже температуры денатурации полностью комплементарной спирали ДНК (ее температуры плавления). В этих условиях спирали, совпадающие не полностью и формирующиеся при пониженной жесткости (см. справа), оказываются нестабильными. Справа - указаны условия гибридизации, используемые для поиска родственных генов, не полностью идентичных гену А.
подход привел к выделению всего семейства ДНК-связывающих белков, которые функционируют в качестве ключевых регуляторов экспрессии генов в ходе раннего эмбрионального развития Drosophila (см. разд. 16.5.19). Этот же подход был использован для идентификации родственных этому семейству генов из других организмов, включая человека.
4.6.11. Для локализации специфических последовательностей нуклеиновых кислот в хромосомах и клетках используют гибридизацию in situ [47]
Все макромолекулы клетки, и в том числе нуклеиновые кислоты, занимают в тканях и клетках строго определенное положение. Экстрагирование этих молекул из тканей или клеток путем гомогенизации приводит к потере той части информации, которая относится к расположению нуклеиновых кислот в клетках. Поэтому были разработаны методы для локализации специфических последовательностей нуклеиновых кислот in situ - в изолированных хромосомах, в определенных типах клеток, где ДНК- и РНК-зонды используются примерно так же, как меченые антитела. Этот метод называют гибридизацией in situ. Его применяют для анализа ДНК в хромосомах и РНК в клетках. Высокорадиоактивные зонды нуклеиновых кислот гибридизуют с хромосомами после кратковременного воздействия высоким рН с целью разделения пар оснований ДНК. Участки хромосом, связывающие радиоактивные зонды в процессе гибридизации, выявляются радиоавтографией. Пространственное разрешение этого метода может быт повышено при использовании не радиоактивно меченных, а химически меченных зондов ДНК. Как правило, при синтезе зондов используют нуклеотиды, содержащие боковую цепь биотина, и гибридизовавшиеся пробы выявляются при окраске стрептавидином (молекулы которого расположены в виде сети) или с помощью иных маркерных молекул (рис. 4-74). Были также разработаны методы гибридизации in situ, позволяющие судить о распределении специфических молекул РНК в клетках внутри тканей, В этом случае ткани не подвергают воздействию высоких значений рН, так что хромосомная ДНК остается в двухцепочечном состоянии и не может связывать зонд. Однако если ткань подвергнуть слабой фиксации,
243
РНК, содержащаяся в ней, при инкубации ткани с комплементарным ДНК-зондом обеспечивает возможность гибридизации. Таким способом удалось наблюдать, как реализуется дифференциальная активность генов Drosophila (рис. 4-75). Этот подход позволил существенно продвинуться в изучении молекулярных механизмов дифференцировки различных клеток эмбриона.
4.6.12. Методы рекомбинантных ДНК дают возможность изучать даже минорные белки клеток [48]
До недавнего времени изучение клеточных белков было ограничено лишь мажорными фракциями, т.е. белками, содержащимися в клетках в относительно большом числе. С помощью обычных методов хроматографии и электрофореза из нескольких сот граммов клеточной массы можно получить примерно 0,1 г (100 мг) одного из мажорных белков, составляющих 1% или более от всего количества клеточных белков. Этой массы белка вполне достаточно для изучения его аминокислотной последовательности, детального анализа биологической или ферментативной (если таковая имеется) активности и получения антител, которые могут быть использованы для локализации белков в клетках. Более того, когда удается вырастить подходящие кристаллы, то с помощью рентгеноструктурного анализа можно установить трехмерную структуру молекулы. Именно таким образом была определена структура и функция многих распространенных белков, в том числе гемоглобина, трипсина, иммуноглобулина и лизоцима.
Эукариотическая клетка содержит тысячи различных белков, но значительное большинство этих белков, и в том числе наиболее интересные, присутствуют в небольшом количестве. Некоторые из них иногда чрезвычайно трудно, если не невозможно, получить в чистом виде в количестве, превышающем несколько микрограмм. Разработка методов рекомбинантных ДНК сделала доступными любые клеточные белки (включая минорные белки) в больших количествах. Для этого клонируют ген нужного белка и затем встраивают его в специальную плазмиду, именуемую клонирующим вектором. Этот вектор сконструирован таким образом, что будучи введенным в бактерии, дрожжи или клетки млекопитающих соответствующего типа, он обеспечивает крупномасштабный синтез этого белка. Таким образом, если раньше для детальных структурных или функциональных исследований были доступны лишь немногие белки, в настоящее время практически все белки клетки могут быть предметом подобных исследований.
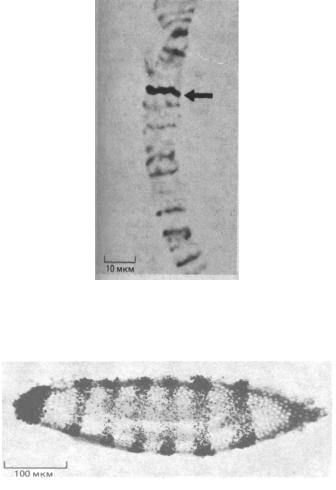
Рис. 4-74. Локализация гена на политенной хромосоме Drosophila с помощью гибридизации in situ с клонированным ДНК-зондом, меченным биотином. ДНК этой гигантской хромосомы частично денатурирована для гибридизации с ней зонда. После гибридизации и отмывки зонда, хромосомы обрабатывают ферментативным комплексом, в составе которого пероксидаза хрена конъюгирована со стрептавидином (см. рис. 4-58, Б). Чтобы выявить участок связывания зонда, препарат обрабатывают перекисью водорода и окрашивают; при этом пероксидаза обнаруживается в виде темной полосы на препарате (стрелка). (С любезного разрешения Tod Leverty, Gerald Rubin.)
Рис. 4-75. Радиоавтограф среза очень раннего эмбриона Drosophila, подвергнутого гибридизации in situ с использованием радиоактивного ДНКзонда, который комплементарен гену, принимающему участие в формировании сегментов ftz. Зонд гибридизуется с РНК эмбриона, и расположение зерен серебра после проявления указывает на то, что РНК, синтезируемая геном ftz, локализована в регулярных полосах, тянущихся поперек всего эмбриона. Ширина полос соответствует трем-четырем клеткам. На этих стадиях развития (клеточная бластодерма) эмбрион состоит примерно из
6000 клеток. (Из Е. Hafen, A. Kuroiwa, W. J. Gehring. Cell, 37; 833-841, 1984.)
244
4.6.13. Функция генов наиболее ярко проявляется в организме мутантов [49]
Предположим, что кому-то удалось клонировать ген, кодирующий вновь открытый белок. Как установить внутриклеточную функцию данного белка? Задача эта весьма непроста, поскольку ни трехмерная структура белка, ни полная нуклеотидная последовательность гена этого белка не позволяют судить о его функции. Кроме того, многие белки, например, структурные белки и белки сложных мультиферментных комплексов, будучи отделены от других компонентов сложной функциональной единицы, в состав которой они входят, не проявляют своей обычной активности.
Один из подходов, которые мы уже рассматривали (см. разд. 4.5.6) состоит в том, чтобы инактивировать определенный белок с помощью специфических антител и пронаблюдать за тем, какие клетки затронуты вследствие этого. В некоторых случаях такой способ позволяет достаточно надежно определить функцию белка, однако в отношении внутриклеточных белков он не очень эффективен, поскольку микроинъецированные антитела подвергаются разведению в процессе пролиферации клеток либо разрушаются в результате внутриклеточной деградации. Более удачное решение этой проблемы возможно с помощью генетических подходов. Мутанты, у которых отсутствует один из белков или, что более удобно, синтезируется его температурочувствительная форма (инактивируемая при небольшом повышении или понижении температуры), чрезвычайно полезны, когда перед исследователем стоит вопрос о функции белка. С их помощью идентифицированы функции ферментов, участвующих в основных метаболических путях бактерий. Благодаря этому подходу были открыты многие генопродукты, отвечающие за упорядоченное развитие эмбрионов Drosophila. Как правило, этот метод используется для изучения организмов с коротким циклом репродукции, таких, как бактерии, дрожжи, круглые черви и плодовые мушки. Воздействуя на их организм веществами, вызывающими изменения в ДНК (мутагенами), можно быстро получить большое количество мутантов и выбрать среди них тех, которые несут определенный интересующий экспериментатора дефект. Например, из популяции бактерий, подвергнутых воздействию мутагенов, были выбраны клетки, которые прекращают синтез ДНК при изменении температуры окружающей среды от 30 ° до 42 °С. Среди них было выявлено значительное число температурочувствительных мутантов, мутации в которых затрагивали бактериальные белки, участвующие в процессе репликации ДНК. Эти мутанты позже были использованы для идентификации и определения белков, необходимых для репликации ДНК.
Цикл репродукции у человека очень продолжителен. К тому же никто не станет подвергать людей целенаправленному воздействию мутагенов. Более того, следует иметь в виду, что человеческий плод, имеющий серьезные дефекты жизненно важных процессов, например репликации ДНК, погибнет задолго до рождения. Однако многие мутации могут практически не оказывать влияния на жизнеспособность - например, тканеспецифические дефекты лизосом либо рецепторов клеточной поверхности, спонтанно возникающие в человеческой популяции. Анализ фенотипа этих больных, равно как и исследование их клеток в культуре, дают возможность уникального исследования важных клеточных функций. Хотя такие мутации крайне редки, тем не менее они эффективно выявляются, поскольку их носители обращаются за медицинской помощью.

245
4.6.14. Клетки и организмы, содержащие измененные гены, можно исправить [50]
Получить мутантов, у которых нарушена репликация ДНК или, например, развитие глаза, в принципе довольно просто. Однако, чтобы связать этот дефект с изменением конкретного белка, могут понадобиться годы. Технология рекомбинантных ДНК дала в руки исследователей совершенно иной подход: анализ начинается с белка и завершается созданием мутантной клетки или целого организма. Поскольку такой подход по сравнению с традиционным направлением генетического анализа от гена к белку представляется обратным, его обычно называют обратной генетикой.
Обратная генетика начинается с выделения из клетки нужного белка. Используя методы, описанные в гл. 5, ген этого белка клонируют и определяют его нуклеотидную последовательность; затем эту последовательность меняют биохимическими методами, создавая мутантный ген, кодирующий измененную форму белка. Затем такой ген вводят в клетку, где он может встроиться в хромосому в процессе гомологической рекомбинации и превратиться таким образом в постоянный элемент генома. Если встроенный ген экспрессируется, то несущая его клетка и все ее потомки будут синтезировать измененный белок. В том случае, когда измененный in vitro ген вводят в оплодотворенную яйцеклетку, получается многоклеточный мутантный организм. Некоторые из таких трансгенных организмов передадут этот ген своим потомкам в качестве постоянного элемента клеток зародышевой линии (рис. 4-76). Такая генетическая трансформация в настоящее время становится обычной процедурой для плодовых мушек или млекопитающих. В принципе на сегодняшний день совершенно реальна и трансформация человека, но такие эксперименты не выполняют из страха перед возможными генетическими нарушениями, которые нельзя исключить у лиц, ставших объектом таких исследований.
4.6.15. С помощью искусственных генов, кодирующих антисмысловые РНК, можно создавать специфические доминантные мутации [51]
При введении мутантных генов в клетки бактерий или дрожжей, которые, как правило, гаплоидны, такие гены будут достаточно часто рекомбинировать с нормальными гомологами. В результате можно отобрать клетки, в которых мутантный ген заменил единственную копию нормального гена (рис. 4-77, А), например клетки, синтезирующие определенный белок в мутантной форме. Функции нормального белка можно определить, как правило, по фенотипу мутантных клеток. Что же касается высших эукариот, таких, как млекопитающие или плодовые мушки, пока еще не созданы методы, позволяющие легко заменять
Рис. 4-76. Сравнение нормальной личинки Drosophila и двух мутантных личинок, содержащих дефектные гены ftz. Одна из дефектных ( ftz-) личинок была трансформирована после инъекции в яйцеклетку, из которой она была получена клонированной ДНК, содержащей нормальную последовательность гена ftz. Эта дополнительная последовательность ДНК встроилась в одну из хромосом мухи и затем нормально наследовалась и экспрессировалась. Ген ftz необходим для нормального развития и его добавление к дефектному геному, как следует из опыта, восстанавливает сегменты личинки, отсутствующие у организмов ftz-. Методы получения трансгенных животных обсуждаются далее (см. рис. 5-88). (С любезного разрешения Walfet Gehring.)
Рис. 4-77. Ген с измененной нуклеотидной последовательностью может быть введен в хромосому организма-хозяина. У бактерий и дрожжей можно отобрать мутанты, у которых (А) в результате генетической рекомбинации измененный ген занял место нормального. В этом случае в клетках сохраняются только мутантные гены. У высших эукариот вместо замены происходит добавление гена (Б). Трансформированные клетки или организмы у них содержат помимо нормальных мутантные гены. Полагают, что у организмов, для которых характерен избыток ДНК, замены генов происходят достаточно редко, поскольку для этого необходимо, чтобы мутантный ген «нашел» среди множества других последовательностей свой нормальный гомолог и спарился с ним.

246
Рис. 4-78. Использование стратегии антисмысловых РНК для получения доминантных мутаций. Были сконструированы мутантные гены, синтезирующие РНК, последовательность которых комплементарна РНК, синтезируемым нормальными генами. РНК этих двух типов способны объединяться в двухцепочечные молекулы. Если синтезируется значительный избыток антисмысловых РНК, они могут гибридизоваться и таким
образом инактивировать большую часть нормальных РНК, синтезируемых геном X. Полагают, что таким образом в перспективе удастся инактивировать любой ген. В настоящее время эта методика применима лишь по отношению к некоторым генам.
нормальный ген клонированным мутантным геном. Генетическая трансформация таких организмов приводит обычно к инсерции клонированного гена в случайные участки генома и при этом клетка или организм содержат мутантный ген наряду с его нормальной копией (рис. 4-77, Б).
Было бы крайне полезно создание специфических доминантных мутаций в клетках высших эукариот путем введения мутантных генов, которые бы элиминировали активность их нормальных аналогов в клетке. Для этого используют весьма хитроумный и многообещающий подход, основанный на специфичности реакций гибридизации двух комплементарных цепей ДНК. Известно, что в норме только одна из двух цепей ДНК в данном участке транскрибируется в РНК и это всегда одна и та же цепь для данного гена. Если же клонированный ген был сконструирован таким образом, что транскрибируется только противоположно направленная цепь ДНК, появляется антисмысловая РНК с последовательностью, комплементарной нормальным гранскриптам РНК. В том случае, когда такая антисмысловая РНК синтезируется в достаточно больших количествах, она будет с большой частотой гибридизоваться со «смысловой» РНК, синтезируемой нормальными генами, и ингибировать синтез соответствующего белка (рис. 4-78). И если этот белок является жизненно важным для клетки или организма, описанные здесь доминантные мутанты погибнут и исследовать функции белка будет невозможно. Чтобы этого не произошло, можно сконструировать гены, синтезирующие антисмысловую РНК по команде, например, в ответ на изменение температуры или в присутствии определенной сигнальной молекулы. Клетки или организмы, содержащие такие индуцибельные антисмысловые гены, будут лишены специфического белка в определенное время, и в этом случае можно проследить за возникающим эффектом. Конечно, этот метод технически до конца еще не проработан, однако уже сейчас ясно, что он весьма перспективен для определения функции белков высших организмов.
Заключение
Технология рекомбинантных ДНК произвела революцию в исследовании клетки. В настоящее время с помощью рестрицирующих нуклеаз можно вырезать любой участок клеточной ДНК и вставить его в самореплици-
247
рующийся генетический элемент (плазмиду или вирус) для получения «клона геномной ДНК». С другой стороны, для получения «клона к ДНК» можно использовать ДНК-копию любой молекулы РНК. Таким образом, можно получить неограниченное количество высокоочищенной ДНК, определить ее нуклеотидную последовательность (скорость этой операции составляет сотни нуклеотидов в день), а также аминокислотную последовательность кодируемого ею белка. Методы генной инженерии позволяют синтезировать мутантные гены и вводить их в хромосомы клеток, где они в последующем превращаются в постоянный элемент генома. И если для переноса генов в качестве реципиента использовать оплодотворенное яйцо, могут быть получены трансгенные организмы, экспрессирующие мутантный ген и передающие его своим потомкам. Для клеточной биологии исключительно важно, что описанные методы дают возможность изменять клетки строго направленным образом, что в свою очередь позволяет оценить влияние на клетку изменения структуры определенного белка.
Применение технологии рекомбинантных ДНК открывает широкие перспективы. С помощью этих методов клетки бактерий, дрожжей и млекопитающих могут быть преобразованы в «фабрики» для масштабного производства любого белка. Это дает возможность детально анализировать структуру и функции белков или использовать их в качестве лекарственных средств. Кроме того, на технологии рекомбинантных ДНК основано получение высокоспецифических ДНК-зондов, с помощью которых изучают экспрессию генов в тканях, локализацию генов в хромосомах, выявляют гены, обладающие родственными функциями.
Литература
Общая
Cantor С. R., Schimmel P. R. Biophysical Chemistry, 3 vols. New York, W. H. Freeman, 1980 (A comprehensive account of the physical principles underlying many biochemical and biophysical techniques.)
Freifelder D. Physical biochemistry, 2nd ed. New York, W. H. Freeman, 1982.
Van Holde K.E., Physical Biochemistry, 2nd ed. Englewood, NJ, Prentice-Hall, 1985.
Цитируемая
1.Bradbury S. An Introduction to the Optical Microscope. Oxford, U. K., Oxford University Press, 1984. Fawcett D.W. A Textbook of Histology, llth ed. Philadelphia, Saunders, 1986.
2.Spencer M. Fundamentals of Light Microscopy. Cambridge, U. K., Cambridge University Press, 1982.
3.Boon M. Т., Drijver J. S. Routine Cytological Staining Methods. London, Macmillan, 1986.
4.Ploem J. S., Tanke H. J. Introduction to Fluorescence Microscopy. Royal Microscopical Society Microscopy Handbook No. 10. Oxford, U. K., Oxford Scientific Publications, 1987.
Willingham M. C., Pastan I. An Atlas of Immunofluorescence in Cultured Cells. Orlando, Fl, Academic, 1985.
5.Alien R. D. New observations of cell architecture and dynamics by video-enhanced contrast optical microscopy. Annu. Rev. Biophys. Biophys. Chem. 14, 265-290, 1985.
6.Pease D. C., Porter K. R. Electron microscopy and ultramicrotomy. J. Cell Biol., 91, 287s-291s, 1981.
7.Wischnitzer S. Introduction to Electron Microscopy. 3rd ed. Elmsford, NY, Pergammon, 1981.
8.Everhart Т.Е., Hayes T.L. The scanning electron microscope. Sci. Am., 226(1), 54-69, 1972. Hayat M. A. Introduction to Biological Scanning
Electron Microscopy. Baltimore, University Park Press, 1978.
Kessel R.G., Kargon R.H. Tissues and Organs. New Fork, W. H. Freeman, 1979.
(Атлас тканей позвоночных, исследованных с помощью сканирующего электронного микроскопа).
248
9.Sommerville J., Scheer U. eds. Electron Microscopy in Molecular Biology. A Practical Approach. Washington, D. C, IRL Press, 1987.
10.Heuser J. Quick-freeze, deep-etching preparation of samples for 3-D electron microscopy. Trends Biochem. Sci, 6, 64-68, 1981. Pinto da Silva P., Branton D. Membrane splitting in freeze-etching. J. Cell Biol, 45, 598-605, 1970.
11.Chiu W. Electron microscopy of frozen, hydrated specimens. Annu. Rev. Biophys. Biophys. Chem., 15, 237-257, 1986.
Unwin P. N. Т., Henderson R. Molecular structure determination by electron microscopy of unstained crystalline specimens. J. Моl. Biol., 94, 425-440, 1975.
12.Glusker J.P., Trueblood K.N. Crystale Structure Analysis: A Primer. Oxford, U. K., Oxford University Press, 1985.
13.Alzari P.M., Lascombe M.B., Poljak R.J. Three-dimentional structure of antibodies. Annu. Rev. Immunol., 6, 555-580, 1980.
Kendrew J.C. The three-dimentional structure of protein molecule. Sci. Am., 205(6), 96-111, 1961. Perutz M.F. The hemoglobin molecule. Sci. Am., 211(5), 64-76, 1964.
14. Cooke R.M., Cambell I.D. Protein structure determination by NMR. Bioessays, 8, 52-56, 1988.
Schulman R.G. NMR spectroscopy of living cells. Sci. Am., 248(1), 86-93, 1986. Wuthrich K., Wagner G. Internal dynamics of proteins. Trends Biochem. Sci., 9, 152-154, 1984.
15.Amman D. Ion Selective Microelectrodes: Principles, Design and Application. Berlin, Springer-Verlag, 1986. Auerbach A., Sachs F. Patch clamp studies on single ionic channels. Annu Rev. Biophys. Bioeng., 13, 269-302, 1984.
16.Grynkiewicz G., Poenie M., Tsien R. Y. A new generations of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem, 260, 3440-3450, 1985. Tsien r. Y., Poenie M. Fluorescence ratio imaging: a new window into intracellular ionic signalling. Trends Biochem. Sci., 11, 450, 1986.
17.Celis J.E., Graessmann A., Logter A. eds. Microinjection and Organdie Transplantation Techniques. London, Academic Press, 1986.
Gomperts B. D., Fernandez J. M. Techniques for membrane permeabilization. Trends Biochem. Sci., 10, 414-417, 1985. Ostro M.J. Liposomes Sci. Am., 256(1), 102-111, 1987.
Ureta Т., Radojkovic J. Microinjected frog oocytes. Bioessays, 2, 221-225, 1985.
18.Freshney R.I. Culture of Animal Cells: A Manual of Basic Technique. New York, Liss, 1987.
19.Herzenberg L. A. Sweet R.G., Herzenberg L. A. Fluorescence-activated cell sorting. Sci. Am., 234(3), 108-116, 1976. Kamarck M. E. Fluorescence-activated cell sorting of hybrid and transfected cells. Methods Enzymol, 151, 150-165, 1987.
Nolan G.P., Fiering S., Nicolas J.F., Herzenberg L.A. Fluorescence-activated cell analysis and sorting of viable mammalian cells based on beta-D-galactosidase activity after transduction of E. coli lac Z. Proc. Nat. Acad. Sci. USA, 85, 2603-2607, 1988.
20.Harrison R.G. The outgrowth of the nerve fiber as a mode of protoplasmic movement. J. Exp. Zool., 9, 787-848, 1910.
21.Ham R.G. Clonal growth of mammalian cells in a chemically defined, synthetic medium. PNAS, 53, 288-293, 1965.
Loo D. Т., Fuquay J. /., Rawson C. L., Barnes D. W. Extended culture of mouse embryo cells without senescence: inhibition by serum. Science, 236, 200-202, 1987.
Sirabasku D.A., Pardee А. В., Saw G.H., eds. Growth of Cells in Hormonally Defined Media. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory, 1982.
22.Ruddle F. H., Creagan R. P. Parasexual approaches to the genetics of man. Annu. Rev. Genet., 9, 407-486, 1975.
23.Colowick S. P., Kaplin N. O., eds. Methods in Enzymology, vols. 1 -... San Diego, CA, Academic Press, 1955-1988. (Многотомное издание,
содержащее общие и специальные статьи с описанием множества методов).
Cooper T.G. The Tools of Biochemistry. New York, Wiley, 1977.
Scopes R.K. Protein Purification Principles and Practice, 2nd ed. New York, Springer-Verlag, 1987.
24. Claude A. A coming age of cell. Science, 189, 433-435, 1975. de Duve C., Beaufay H. A short history of tissue fractionation. J. Cell Biol., 91, 293s-299s, 1981.
Meselson M., Stahl F. W. The replication of DNA in Escherichia coli. Proc. Nat. Acad. Sci. USA, 47, 671-682, 1958. (Для демонстрации полуконсервативной репликации ДНК использовали центрифугирование в градиенте плотности).
Palade G. Intracellular aspects of the process of protein synthesis. Science, 189, 347-358, 1975. Sheeler P. Centrifugation in Biology and Medical Science. New York, Wiley, 1981.
249
25. Moore D.J., Howell K.E., Cook G.M. W., Ewans W.H. eds. Cell Free Analysis of Membrane Traffic. New York, Liss, 1986.
Nirenberg N. W., Mattaei J. H. The dependence of cell free protein synthesis in E. coli on naturally occuring or synthetic polyribonucleotides. Proc. Nat. Acad. Sci. USA, 47, 1588-1602, 1961.
Racker E. A. A New Look on Mechanisms of Bioenergetics. New York. Academic Press, 1976. (Бесклеточные системы в исследовании энергетического метаболизма).
Zamecnic P. С. An historical account of protein synthesis with current overtones – a personalized view. Cold Spring Harbor Symp. Quant. Biol., 34, 1-16, 1969.
26.Dean P. D. G., Johnson W. S., Middle F. A. Affinity Chromatography: A Practical Approach, Arlington, VA, IRL Press, 1985. Gilbert M. T. High Performance Liquid Chromatography. Littleton, MA, John Wright-PSG, 1987.
27.Andrews A. T. Electrophoresis, 2nd ed. Oxford, U. K., Clarendon Press, 1986.
Laemmli U. K. Cleavage of the structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680-685, 1970.
28.Celis J. E., Bravo R. eds., Two - Dimentional Gel Electrophoresis of Proteins. New York, Academic Press, 1983. O'Farrell P.H. High-resolution two-dimentional electrophoresis of proteins. J. Biol. Chem. 250, 4007-4021, 1975.
29.Cleveland D. W., Fisher S. G., Kirschner M. W., Laemmli U. K. Peptide mapping by limited proteolysis in sodium dodecyl sulphate and analysis by gel-electrophoresis. J. Biol. Chem. 252, 1102-1106, 1977.
Ingram К М. A special chemical difference between the globins of normal human and sickle anemia hemoglobin. Nature, 178, 792-794, 1956.
30.Edman P., Begg G. A protein sequenator. Eur. J. Biochem. 1, 80-91, 1967. (Первое описание автоматизированного секвенатора).
Hewlck R. M., Hunkapiller M. W., Hood L. E., Dreyer W.J. A gas-liguid-solid phase peptide and protein seguenator. J. Biol. Chem. 256, 7990-7997, 1981.
Sanger F. The arrangement of amino acids in proteins. Adv. Protein Chem. 7, 1 -67, 1952.
Walsh K. A., Ericsson L. H. Parmelee D. C., Titani K. Advances in protein sequencing. Annu. Rev. Biochem. 50, 261-284, 1984. 31. Chase G.D., Rabinowltz J.L. Principles of Radio Isotope Methodology, 2nd ed. Minneapolis, Burgess, 1962.
Dyson N. A. An Introduction to Nuclear Physics with Applications in Medicine and Biology, Chichester, U. K., Horwood, 1981.
32.Calvin M. The path of carbon in photosynthesis. Science, 135, 879-889, 1962. (Один из первых случаев применения радиоизотопов в биологии).
Rogers A. W. Techniques of Autoradiography, 3rd ed. New York, Elsevier/North Holland, 1979.
33.Anderton B. H., Thorpe R. C. New methods of analysis of antigens and glycoproteins in complex mixtures. Immunol. Today, 2, 122-127, 1980. Coons A.H. Histochemistry with labelled antibody. Int. Rev. Cytol. 5, 1-23, 1956.
34.Milstein C. Monoclonal antibodies. Sci. Am., 243(4), 66-74, 1980.
Yelton D. E., Schariff M. D. Monoclonal antibodies: a powerful new tool in biology and medicine. Annu. Rev. Biochem., 50, 657-680, 1981. 35. Mabuchi L, Okuno M. The effect of myosin antibody on the division of starfish blastomeres. J. Cell Biol., 74, 251-263, 1977.
Mulcahy L. S., Smith M. R., Stacey D. W. Requirement for ras proto-oncogene function during serum stimulated growth of NIH 3T3 cells. Nature, 313, 241-243, 1985.
36. Drlica K. Understanding DNA and Gene Cloning. New York, Wiley, 1984.
Sambrook J., Fritsch E. F., Maniatis T. Molecular Cloning: A Laboratory Manual, 2nd ed. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory, 1989.
Watson J. D., Tooze J. The DNA Story: A Documentary History of Gene Cloning. New York, W.H. Freeman, 1981.
37.Garoff H. Using recombinant DNA techniques to study protein targeting in the eucaryotic cell. Annu. Rev. Cell. Biol., 1, 403-445, 1985. Jackson lan J. The real reverse genetics: targeted mutagenesis in the mouse. Trends Genet., 3, 119, 1987.
Kelly J.H., Darlington G.J. Hybrid genes: molecular appoaches to the tissue-specific gene regulation. Annu. Rev. Genet., 19, 273-296, 1985.
38.Nathans D., Smith H. 0. Restriction nucleases in the analysis and reconstructuring of DNA molecules. Annu. Rev. Biochem., 44, 273-293, 1975. Smith H. 0. Nucleotide sequence specificity of restriction endonucleases. Science, 205, 455-462, 1979.
39.Cohen S.N. The manipulation of genes. Sci. Am., 233(1), 24-33, 1975.
Maniatis T. et al. The isolation of structural genes from libraries of eucaryotic DNA. Cell, 15, 687-701, 1978.
250
Nooick R.P. Plasmids. Sci. Am., 243(6), 102-127, 1980.
40.Cantor C. R., Smoth C. L., Mathew M. K. Pulsed-field gel electrophoresis of very large DNA molecules. Annu. Rev. Biophys. Biophys. Chem., 17, 287-304, 1988.
Maxam A. M., Gilbert W, A new method of sequencing DNA. Proc. Nat. Acad. Sci. USA, 74, 560-564, 1977. Southern E. M. Gel electrophoresis of restriction fragments. Methods Enzymol., 68, 152-176, 1979.
41.Rigby P. W., Dieckermann M., Rhodes C., Berg P. Labeling deoxyribonucleic acid to high specific activity in vitro by nick translation with DNA polymerase I. J. Мої. Biol, 113, 237-251, 1977.
42.Galas D. J., Schmitz A. DNAse footprinting: a simple method for the detection of protein-DNA binding specificity. Nucl. Acid Res., 5, 31573170, 1978.
Gilbert W. DNA sequencing and gene structure. Science, 214, 1305-1312, 1981.
Prober J. M., et al. A system for rapid DNA sequencing with fluorescent chainterminating dideoxynucleotides. Science, 238, 336-341, 1987. Tullius T. D. Chemical "snapshots" of DNA: using the hydroxyl radical to study the structure of DNA and DNA protein complexes. Trends Biochem. Sci., 12, 297-300, 1987.
43.Berk A.J., Sharp P. A. Sizing and mapping of early adenovirus mRNAs by gel electrophoresis of SI endonuclease digested hybrids. Cell, 12, 721-732, 1977.
Hood L.E., Wilson J.H., Wood W.B. Molecular Biology of Eucaryotic Cells: A Problems Approach, pp. 56-61, 192-210, Mento Park, CA, Benjamin-Cummings, 1975.
Wetmer J. G. Hybridization and renaturation kinetics of nucleic acids. Annu. Rev. Biophys. Bioeng., 5, 337-361, 1976.
44.Alwine J. C., Kemp D. J., Stark G. R. A method for detection of specific RNAs in agarose gels by transfer to diazobenzomethyl-paper and hybridization with DNA probes. Proc. Nat. Acad. Sci. USA. 74, 5350-5354, 1977.
Southern Е. М. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Мої. Biol., 98, 503-517, 1975.
45.Itakura K., Rossi J. J., Wallace R. B. Synthesis and use of synthetic oligonucleotides. Annu. Rev. Biochem., 53, 323-356, 1984.
Ruddle F. H. A new era in mammalian gene mapping: somatic cell genetics and recombinant DNA methodologies. Nature, 294, 115-119, 1981.
White R., Lalovel J. M. Chromosome mapping with DNA markers. Sci. Am., 258(2), 40-48, 1988.
McGinnis W., Garber R.L., Wirz J., Kuroiwa A., Gehring W.J. A homologous protein-coding seguence in Drosophila homeotic genes and its conservation in other metazoans. Cell, 37, 403-408, 1984.
47.Gerhard D. S., Kawasaki E. S., Bancroft F. C., Shabo P. Localization of a unique gene by direct hybridization. Proc. Nat. Acad. Sci. USA, 78, 3755-3759, 1981.
Huten E., Kuroiwa A., Gehring W.J. Spatial distribution of transcripts from the segmentation gene fushi tarazu during Drosophila embryonic development. Cell, 37, 833-841, 1984.
Pardue M. L., Gall J. G. Molecular hybridization of radioactive DNA to the DNA of the cytological preparations. Proc. Nat. Acad. Sci. USA, 64, 600-604, 1969.
48.Abelson J., Butz E., eds. Recombinant DNA. Science, 209, 1317-1438, 1980.
Gilbert W., Villa-Komaroff L. Useful proteins from recombinant bacteria. Sci. Am., 242(4), 74-94, 1980.
49.Lederberg J., Lederberg E. M. Replica plating and inderect selection of bacterial mutants. J. Bacteriol., 63, 399-406, 1952. Nusslein-Volhard C., Wieschaus E. Mutations affecting segment number arid polarity in Drosophila. Nature, 287, 795-801, 1980.
50.Palmiter R. D., Brinster R. L. Germ line transformation of mice. Annu. Rev. Genet., 20, 465-499, 1986.
Pellicer A., et al. Altering genotype and phenotype by DNA-mediated gene transfer. Science, 209, 1412-1422, 1980.
Rubin G. M., Spradling A. C. Genetic transformation of Drosophila with transposable element vectors. Science, 218, 348-353, 1982.
Shortle D., Nathans D. Local mutagenesis: a method for generating viral mutants with base substitutions in preselected regions of the viral genome. Proc. Nat. Acad. sci. USA, 75, 2170-2174, 1978.
Struhl K. The new yeast genetics. Nature, 305, 391-397, 1983.
51. Melton D.A., Rebagliati M.R. Antisense RNA injections in fertilized eggs as a test for the function of localized mRNAs. J. Embryol. Exp. Morphol., 97, 211-221, 1986.
Rosenberg V.B., Preiss A., Seifert E., Jackle H., Knipple D.C. Production of phenocopies by Kruppel anti-sense RNA injection into Drosophila embryos. Nature, 313, 703-706, 1985.
Weintraub H., Isant J. G., Harland R. M. Anti-sense RNA as a molecular tool for genetic analysis. Trends Genet., 1, 22-25, 1985.