


48.Врожденные заболевания, диагностируемые с помощью неонатального скрининга. Клиническая картина, диагностика, принципы лечения фенилкетонурии.
Фенилкетонурия - наследственное аутосомно-рецессивное заболевание группы ферментопатий, характеризующееся дефицитом фенилаланингидроксилазы, связанное с нарушением метаболизма аминокислот, главным образом фенилаланина.
Этиология и патогенез
В результате мутации гена, контролирующего синтез 4-фенилаланингидроксилазы, развивается метаболический блок на этапе превращения фенилаланина в тирозин, который служит субстратом для синтеза биогенных аминов и меланина.
Следствием нарушения гидроксилирования являются накопление фенилаланина в крови и моче и снижение образования тирозина. Концентрация фенилаланина в плазме достигает уровня, достаточно высокого для активации альтернативных путей метаболизма с образованием фенилпирувата, фенилацетата, фениллактата и других производных, оказывающих токсический эффект на различные органы и ткани.
В наибольшей степени страдают структуры ЦНС.
Эпидемиология
В России частота ФКУ составляет 1:7000
Классификая
1 тип ФКУ: отсутствие или снижение активности фермента ФАГ
2 тип ФКУ: дефицит дигидроптеридинредуктазы
3 тип ФКУ: дефицит 6 пирувоилтетрагидроптеринсинтазы.
Форма заболевания |
Уровень ФА в сыворотке крови |
|
|
|
Мг/дл |
|
|
Здоровые дети |
1-2 |
|
|
Легкая гиперфенилаланинемия |
2-10 |
|
|
Легкая ФКУ |
10-15 |
|
|
Средняя ФКУ |
15-20 |
|
|
Классическая ФКУ |
>20 |
|
|
Клиническая картина
•Манифестация болезни происходит в возрасте 2–6 месяцев, отмечается вялость, отсутствие/потеря интереса к окружающему, иногда наоборот, повышенная раздражительность, беспокойство, гипертонус мышц, гиперрефлексия, экзематозная сыпь.
•«мышиный запах» пота и мочи.
•Во втором полугодии жизни наблюдается регресс в моторном и психоречевом развитии: дети перестают реагировать на обращенную речь, узнавать мать, не фиксируют взгляд и не реагируют на яркие игрушки, не переворачиваются на живот, не сидят.
•Физическое развитие нарушено в меньшей степени, однако, уменьшение окружности головы или микроцефалия, более позднее прорезывание зубов.
•У детей светлые волосы, голубые глаза. Кожа почти полностью лишена меланина и имеет повышенную чувствительность к инсоляции и травмам, наблюдаются тяжелая экзема, дерматит, фолликулярный кератоз, повышенная склонность к гнойничковым инфекциям.
•При отсутствии лечения развивается тяжелая умственная отсталость
Эпилептические приступы: генерализованные пароксизмы по типу инфантильных спазмов в виде «салаамовых судорог», кивков; могут наблюдаться абсансы. Приступы носят упорный характер и плохо поддаются антиконвульсантной терапии.
Диагностика
o неонатальный скрининг (определение концентрации фенилаланина в сухих пятнах крови) o методы флюориметрии или тандемной масс-спектрометрии
oПри отсутствии данных неонатального скрининга диагностику заболевания рекомендовано осуществлять на основании совокупности генеалогического анамнеза, результатов клинического и биохимического обследования (высокое содержание фенилаланина)
o ДНК-диагностика с целью выявления мутаций в генах РАН и РТРS o ЭЭГ
o МРТ с целью выявления очагов перивентрикулярной лейкопатии, кортикальной атрофии и других изменений у пациентов старше 12 лет
o УЗИ брюшной полости и почек o ФГДС
o психолого-педагогическое консультирование и логопедическое тестирование
Лечение
1. Гипофенилаланиновая диета
За счет исключения высокобелковых продуктов ― мясо, мясопродукты, рыбу, рыбопродукты, творог, яйцо, бобовые, орехи, шоколад и др., должна быть начата не позднее первых недель жизни ребенка.
Недостающее количество белка восполняется за счет специализированных лечебных продуктов, частично или полностью лишенных фенилаланина
2. Медикаментозная терапия - сапроптерина дигидрохлорид
49.Врожденные заболевания, диагностируемые с помощью неонатального скрининга. Клиническая картина, диагностика, принципы лечения галактоземии.
Галактоземия — наследственное аутосомно-рецессивное заболевание, в основе которого лежит нарушение обмена веществ на пути преобразования галактозы в глюкозу.
Этиология и патогенез
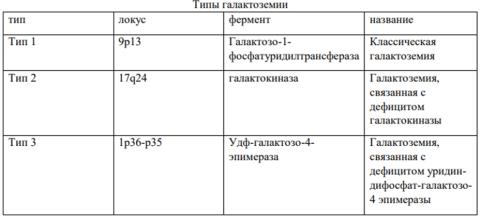
В основе заболевания лежит недостаточность одного из трех ферментов, участвующих в метаболизме галактозы: галактозо-1-фосфатуридилтрансферазы (ГАЛТ), галактокиназы (ГАЛК) и уридиндифосфат (УДФ)-галактозо-4-эпимеразы (ГАЛЭ)
Галактоза является источником энергии для клетки, играет пребиотическую роль, служит пластическим материалом для образования гликопротеидов, гликолипидов и других комплексных соединений, используемых организмом для формирования клеточных мембран, нервной ткани, нервных окончаний, процессов миелинизации нейронов и др.
В результате недостаточности любого из трех ферментов – ГАЛТ, ГАЛК или ГАЛЭ – в крови повышается концентрация галактозы, накапливается избыточное количество галактозо-1-фосфата.
Избыток галактозы в организме может метаболизироваться другими биохимическими путями: в присутствии НАДФ·Н (или НАД·Н) она может превращаться в галактитол. Накопление галактитола в
крови и тканях и повышение его экскреции с мочой; в хрусталике глаза избыток галактитола способствует формированию катаракты. Высокое содержание галактитола в тканях мозга способствует набуханию нервных клеток и формированию псевдоопухоли мозга.
Патологические процессы обусловлены не только токсическим действием указанных продуктов, но и их тормозящим влиянием на активность других ферментов, участвующих в углеводном обмене (фосфоглюкомутазы, глюкозо-6-фосфатдегидрогеназы), следствием чего является гипогликемический синдром.
Эпидемиология
В России составляет 1: 20 000
Классификация
Клиническая картина
Первые симптомы в первые дни жизни ребенка: нарушение функции печени (гепатомегалия, гипогликемия), иктеричность кожных покровов, рвота, диарея, гипотрофия, позже двусторонняя катаракта. Судорожный синдром, гипотония. Возможен ОПН, асцит, редко сепсис. При отсутствии лечениязадержка НПР, атаксия, тремор, нарушение пубертатного развития. Геморрагический синдром
Галактоземия, связанная с дефицитом галактокиназы: Быстрое развитие катаракты
Галактоземия, связанная с дефицитом уридин-дифосфат-галактозо-4 эпимеразы
1.Генерализованнаятяжелая клиника
2.Доброкачественнаятолько изменение формы эритроцитов
3.Отсутствуют психические и неврологические нарушения, поражение печени и почек
Диагностика
неонатальный скрининг (определение концентрации галактозы)
При + скрининге - определение активности фермента ГАЛТ и ДНК-диагностика УЗИ органов брюшной полости и почек ЭКГ Эхо-КГ
Офтальмоскопия КТ и МРТ головного мозга - по показаниям
Рентгенография для диагностики остеопороза (у детей старше 5 лет)