USMLE Step 2 CK λ Pediatrics
HEMOGLOBIN DISORDERS
Sickle Cell Anemia (Homozygous Sickle Cell or S-Beta Thalassemia)
A 6-month-old, African-American infant presents to the pediatrician with painful swollen hands and swollen feet.
•Occurs in endemic malarial areas: sub-Saharan Africa, Middle East, India; survival advantage with heterozygous trait provides protection against falciparum infection
−Hydrophobic valine residues → HbS polymerizes in the deoxygenated state, decreased pH; increased [HbS] in RBCs → characteristic sickle RBC shape (reversible)
−With repeated episodes → irreversible RBC sickling → become stiff and nondeformable → vasooclusion → tissue ischemia and intraand extravascular hemolysis.
•Single base pair change (thymine for adenine) at sixth codon of the beta gene (valine instead of glutamic acid)
−Sickle cell disease: up to 65% are SS, but there are also compound heterozygotes with Hg SC the most common, then HbSβ0 and then HbSβ+
−Hgb S-beta thal – 0 (α2β2s, α2β2Th-0): clinically same as Hb SS
−Hgb S –beta thal + (α2β2s, α2β2Th-+): variable depending on specific β-thalassemia mutation
−Hgb SC (α2β2s, α2β2c): same as Hb SS but less frequent events
•Sickle cell trait (Hb AS)
−Life span normal; serious complications rare
−CBC normal; normal RBC life span
−No limitation of activities
−Known complications: hematuria, renal papillary necrosis, hyposthenuria; splenic infarction at high altitude (>3000 m); exertional rhabdomyolysis, sudden death
•Clinical presentation
−Effects on blood: after transition to adult beta globin expression in 4–6 months; with health, maintains stable Hb at 6–9 g/dL; significant fluctuations occur with disease complications; also, typical leukocytosis (15–25,000) and mild thrombocytosis (400–475,000)
−Newborn usually without symptoms; development of hemolytic anemia over first 2–4 months (replacement of HbF); as early as age 6 months; some children have functional asplenia; by age 5, all have functional asplenia
−First presentation usually hand-foot syndrome (acute distal dactylitis)—sym- metric, painful swelling of hands and feet (ischemic necrosis of small bones)
−Infection: S. pneumonia with functional asplenia; peak in first 3 years of life; penicillin prophylaxis (orally 2x/day or monthly benzathine penicillin IM age
2 months –5 years) decreases rate by 84% and S. pneumoniae vaccine by another 70%
−Acute painful crises (vaso-occlusive):
°Severe, episodic pain
°Increased with age and peak age 20s
°Bone marrow ischemia, leading to possible infarction
204

°Triggers: infection, emotional stress, cold, wind, high altitude, dehydration
°Younger: mostly fingers and toes (acute distal dactylitis in infant beginning age 5–7 months), arms and legs; with increasing age: lower back, head, chest, abdomen
−More extensive vaso-occlusive crises → ischemic damage
°Skin ulcers
°Retinopathy
°Avascular necrosis of hip and shoulder
°Infarction of bone and marrow (increased risk of Salmonella osteomyelitis)
°Splenic autoinfarction
°Pulmonary: acute chest syndrome (along with sepsis, most common causes of mortality)
New pulmonary infiltrate on chest x-ray with ≥1 of the following: fever, tachypnea, dyspnea, hypoxia, chest pain
45% with no identifiable cause
30% infection: most recent statistics now show C. pneumoniae and M. pneumoniae are most common causes of acute chest syndrome in children; then viruses, and then S. pneumoniae
Also caused by pulmonary infarction and fat embolism
Treatment: oxygen, antibiotics, bronchodilators, analgesia, fluids, transfusion as needed; consider exchange transfusion if severe and progressive
°Stroke (peak age 6–9 yrs): most are ischemic of middle cerebral artery; treatment is rapid reduction in percent SS with RBC transfusion or partial automated exchange transfusion; resolution or marked decrease in 24–48 hrs; second stroke more likely without use of regular RBC transfusion program to suppress percent of SS (chronic transfusion regiment); best long-term treatment is stem cell transplant; current routine screening with annual transcranial Doppler study to detect cerebral blood flow velocity related to risk of stroke
°Priapism, especially in adolescence
−Acute splenic sequestration: rapid spleen enlargement, decreased [Hb], and decreased platelets; 30% by age 5 yrs (most age <2); teach family splenic palpation (early detection decreases mortality; remove spleen preventively if occurs again)
−Aplastic crisis: after infection with parvovirus B19; absence of reticulocytes during acute anemia; maturational arrest of RBC precursors in marrow for 10-14 days; because in SS disease, RBC lifespan is only 10-20 days instead of normal 120, there is profound anemia; need transfusional support until reticulocytes return; may hasten recovery with IVIG
−Cholelithiasis: symptomatic gallstones; sudden hemolysis → increased serum bilirubin → stores in gall bladder and can precipitate to form stones
•Labs
−Increased reticulocytes
−Mild to moderate anemia
−Normal MCV
−If severe anemia: smear for target cells, poikilocytes, hypochromasia, sickle RBCs, nucleated RBCs, Howell-Jolly bodies (lack of splenic function); bone marrow markedly hyperplastic
Published by dr-notes.com |
205 |
|
|
|
USMLE Step 2 CK λ Pediatrics
Note
Patients without a functioning spleen are predisposed to infection with encapsulated organisms. Pneumococcal vaccines 13 (PCV13) and 23 (PPSV23) are necessary.
−Renal: glomerular and tubular dysfunction; hyposthenuria in all; also gross hematuria, nephrotic syndrome, renal infarction, pyelonephritis, papillary necrosis, and end-stage renal disease requiring dialysis/transplant
•Diagnosis
−Every state with mandatory newborn screening program; identify newborns with the disease for prompt referral to providers with expertise and initiation of penicillin before age 4 months
−Most commonly used procedures are thin layer/isoelectric focusing and highperformance liquid chromatography
−Those with abnormal screens are retested at first clinical visit (and after age 6 months) to determine final hemoglobin phenotype; also a CBC and Hb phenotype determination is recommended for both parents to confirm the diagnosis and provide an opportunity for genetic counseling
•Treatment—prevent complications
−Immunize (pneumococcal regular plus 23-valent, meningococcal)
−Penicillin prophylaxis at 2 months until age 5
−Educate family (assessing illness, palpating spleen, etc.)
−Folate supplementation
−Aggressive antibiotic treatment of infections
−Pain control
−Transfusions as needed
−Monitor for risk of stroke with transcranial Doppler
−Hydroxyurea: only FDA-approved drug for sickle cell disease; inhibits polymerization in frequent painful crises by increasing expression of fetal Hb
−Stem-cell transplant: only curative option; reserved for those with severe and life-threatening complications
Clinical Recall
Which of the following infectious complications of sickle cell disease is correctly matched to its causative organism?
A.Osteomyelitis: Streptococcus
B.Pneumonia: Pseudomonas
C.Dactylitis: Coxsackie virus
D.Acute chest syndrome: Staphylococcus
E.Aplastic crisis: Parvovirus B19
Answer: E

THALASSEMIAS
Alpha Thalassemia
•The genes for alpha chains are duplicated: there are 2 pairs of alleles (4 genes) on chromosome 16. Mutations are caused by complete gene deletions, so there are 4 syndromes.
–Alpha thalassemia silent trait
°Common in African Americans
°One gene deletion; clinically silent
°Diagnosis requires molecular analysis: no abnormal hemoglobins, no increase in HbF (in contrast to beta thalassemias)
•HgB H disease: deletion of 3 genes; Hgb Barts >25% in newborn period and easily diagnosed with electrophoresis
–At least 1 parent has alpha-thalassemia trait; later beta-tetramers develop (Hgb H—interact with RBC membrane to produce Heinz bodies) and can be
identified electrophoretically; microcytosis and hypochromia with mild to moderate anemia; target cells present, mild splenomegaly, jaundice and cholelithiasis
–Typically do not require transfusions or splenectomy; common in Southeast Asians
•Alpha-thalassemia major: deletion of 4 genes; severe fetal anemia resulting in hydrops fetalis
–Newborn has predominantly Hgb Barts with small amounts of other fetal Hgb; immediate exchange transfusions are required for any possibility of survival; transfusion-dependent with only chance of cure (bone marrow transplant)
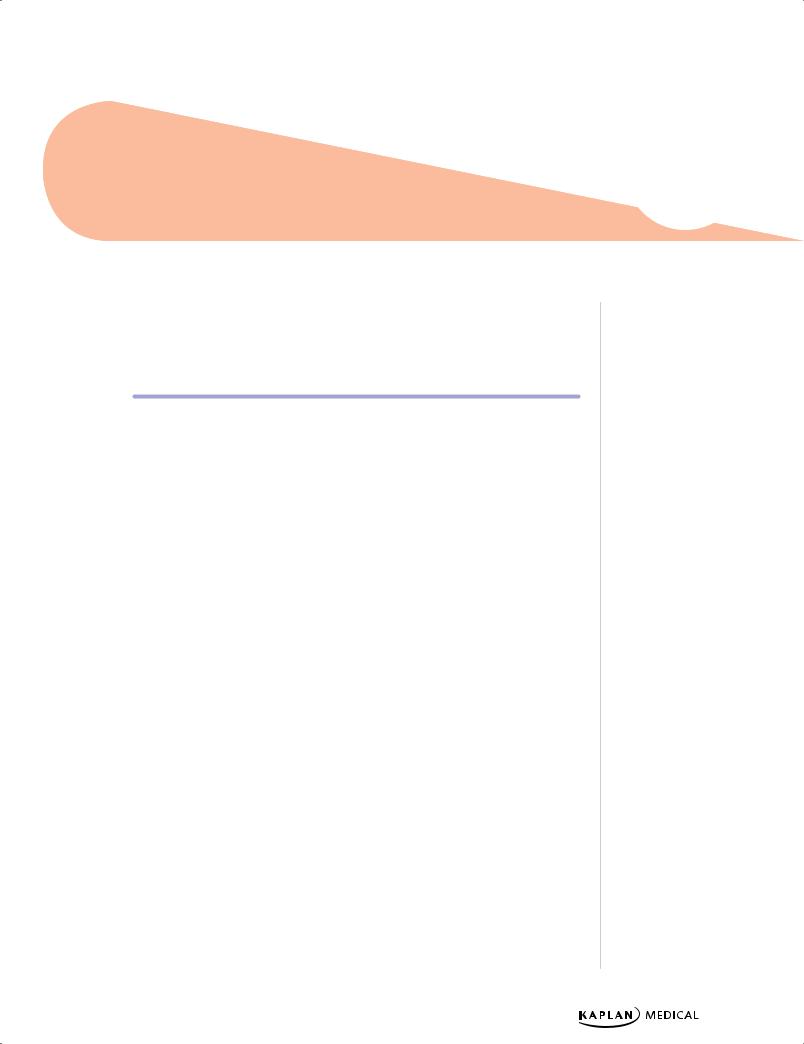
Figure 19-1. Skull X-ray Demonstrating “Hair on End” Appearance of Thalassemia
Published by dr-notes.com |
207 |
|
|
|
USMLE Step 2 CK λ Pediatrics
Note
Minor bleeds = von Willebrand
Deep bleeds = hemophilia
Beta Thalassemia Major (Cooley Anemia)
A 9-year-old has a greenish-brown complexion, maxillary hyperplasia, splenomegaly, and gallstones. Her Hb level is 5 g/dL and MCV is 65 mL.
•Mutations in the beta gene result from point mutations (>200 known mutations), which are collected into the following clinical groupings: beta thalassemia trait, minima, minor, intermedia, and major (no beta chains, Cooley anemia).
•Excess alpha globin chains → alpha tetramers form; increase in HbF (no problem with gamma-chain production)
•Presents in second month of life with progressive anemia, hypersplenism, and cardiac decompensation (Hb <4 mg/dL)
•Expanded medullary space with increased expansion of face and skull (hair-on- end); extramedullary hematopoiesis, hepatosplenomegaly
•Labs
−Infants born with HbF only (seen on Hgb electrophoresis)
−Severe anemia, low reticulocytes, increased nucleated RBCs, hyperbilirubinemia microcytosis
−No normal cells seen on smear
−Bone-marrow hyperplasia; iron accumulates → increased serum ferritin and transferrin saturation
•Treatment
−Transfusions
−Deferoxamine (assess iron overload with liver biopsy)
−May need splenectomy
−Bone-marrow transplant curative
HEMORRHAGIC DISORDERS
Evaluation of Bleeding Disorders
History provides the most useful information for bleeding disorders.
•von Willebrand disease (vWD) or platelet dysfunction → mucous membrane bleeding, petechiae, small ecchymoses
•Clotting factors—deep bleeding with more extensive ecchymoses and hematoma
•Laboratory studies
−Obtain platelets, bleeding time, PT, PTT
°If normal, von Willebrand factor (vWF) testing and thrombin time
°If abnormal, further clotting factor workup
−Bleeding time—platelet function and interaction with vessel walls; qualitative platelet defects or vWD (platelet function analyzer)
–Platelet count—thrombocytopenia is the most common acquired cause of bleeding disorders in children
−PTT—intrinsic pathway: from initiation of clotting at level of factor XII through the final clot (prolonged with factor VIII, IX, XI, XII deficiency)

−PT—measures extrinsic pathway after activation of clotting by thromboplastin in the presence of Ca2+; prolonged by deficiency of factors VII, XIII or anticoagulants; standardized values using the International Normalized Ratio (INR)
−Thrombin time—measures the final step: fibrinogen → fibrin; if prolonged: decreased fibrin or abnormal fibrin or substances that interfere with fibrin polymerization (heparin or fibrin split products)
−Mixing studies: if there is a prolongation of PT, PTT, or thrombin time, then add normal plasma to the patient’s and repeat labs
°Correction of lab prolongation suggests deficiency of clotting factor.
°If not or only partially corrected, then it is due to an inhibitor (most common on inpatient basis is heparin).
°If it becomes more prolonged with clinical bleeding, there is an antibody directed against a clotting factor (mostly factors VIII, IX, or XI).
°If there is no clinical bleeding but both the PTT and mixing study are prolonged, consider lupus anticoagulant (predisposition to excessive clotting).
−Clotting factor assays—each can be measured; severe deficiency of factors VIII or IX = <1% of normal; moderate = 1–5%; mild = >5%
−Platelet aggregation studies—if suspect a qualitative platelet dysfunction, ristocetin
Table 19-3. Clinical Findings in Coagulopathies
|
|
Factor VIII |
|
|
Factor IX |
|
|
vWF |
|
|
|
|
|
|
|
|
|
|
|
Platelet |
Normal |
Normal |
|
Normal |
|
|
|
|
|
|
|
|
|
|
PT |
Normal |
Normal |
|
Normal |
|
|
|
|
|
|
|
|
|
|
PTT |
↑ |
↑ |
|
↑ |
|
|
|
|
|
|
|
|
|
|
Bleeding time |
Normal |
Normal |
|
↑ |
|
|
|
|
|
|
|
|
|
|
Factor VIII |
↓ |
Normal |
|
Normal |
|
|
|
|
|
|
|
|
|
|
Factor IX |
Normal |
↓ |
|
Normal |
|
|
|
|
|
|
|
|
|
|
vWF |
Normal |
Normal |
|
↓ |
|
|
|
|
|
|
|
|
|
|
Sex |
Male |
Male |
|
Male/female |
|
|
|
|
|
|
|
|
|
|
Treatment |
Factor VIII, |
Factor IX |
|
Fresh frozen plasma, |
|
desmopressin |
|
|
|
|
cryotherapy, DDAVP |
|
|
|
|
|
|
|
|
|
|
Hemophilia A (VIII) and B (IX)
•85% are A and 15% B; no racial or ethnic predisposition
•X-linked
•Clot formation is delayed and not robust → slowing of rate of clot formation
−With crawling and walking—easy bruising
−Hallmark is hemarthroses—earliest in ankles; in older child, knees and elbows
−Large-volume blood loss into iliopsoas muscle (inability to extend hip)—vague groin pain and hypovolemic shock
−Vital structure bleeding—life-threatening
Published by dr-notes.com |
209 |
|
|
|
USMLE Step 2 CK λ Pediatrics
Note
There is no way to clinically differentiate factors VIII and IX deficiencies. You must get specific factor levels.
210
•Labs
−2× to 3× increase in PTT (all others normal)
−Correction with mixing studies
−Specific assay confirms:
°Ratio of VIII:vWF sometimes used to diagnose carrier state
°Normal platelets, PT, bleeding time, and vW Factor
•Treatment
−Replace specific factor
−Prophylaxis now recommended for young children with severe bleeding (intravenous via a central line every 2–3 days); prevents chronic joint disease
−For mild bleed—patient’s endogenous factor can be released with desmopressin (may use intranasal form)
−Avoid antiplatelet and aspirin medications
−DDAVP increases factor VIII levels in mild disease
von Willebrand Disease (vWD)
•Most common hereditary bleeding disorder; autosomal dominant, but more females affected
•Normal situation—vWF adheres to subendothelial matrix, and platelets then adhere to this and become activated; also serves as carrier protein for factor VIII
•Clinical presentation—mucocutaneous bleeding (excessive bruising, epistaxis, menorrhagia, postoperative bleeding)
•Labs—increased bleeding time and PTT
•Quantitative assay for vWFAg, vWF activity (ristocetin cofactor activity), plasma factor VIII, determination of vWF structure and platelet count
•Treatment—need to increase the level of vWF and factor VIII
−Most with type 1 DDAVP induces release of vWF
−For types 2 or 3 need replacement → plasma-derived vWF-containing concentrates with factor VIII
Other Bleeding Disorders
Vitamin K deficiency
•Newborn needs intramuscular administration of vitamin K or develops bleeding diathesis
•Postnatal deficiency—lack of oral intake, alteration in gut flora (long-term antibiotic use), malabsorption
•Vitamin K is fat soluble so deficiency associated with a decrease in factors II, VII,
IX, and X and proteins C and S
•Increased PT and PTT with normal platelet count and bleeding time
Liver disease
•All clotting factors produced exclusively in the liver, except for factor VIII
•Decreases proportional to extent of hepatocellular damage
•Treatment—fresh frozen plasma (supplies all clotting factors) and/or cryoprecipitate (supplies fibrinogen)