

учебное пособие химия цемента
.pdf- 89 -
вокруг зерна цемента часто обнаруживает слоистость. Вместе с тем в системе вцелом цикличность отвердевания не обнаруживается из-за действия статистического фактора, предполагающего, что в каждом зерне цемента в силу индивидуальной его активности, связанной с размерами, формой, распределением клинкерных минералов и пр., рассматриваемый процесс протекает в собственном временном режиме. В силу же действия законов статистики относительно всей твердеющей системы процесс протекает монотонно.
Таким образом, мы проследили за общими проявлениями процессов образования новых твердых фаз. Теперь нам предстоит разобраться в глубинной сущности этих процессов.
Вопросам образования кристаллической фазы из растворов посвящено множество трудов ученых, в том числе с мировым именем, таких, как Гиббс, Томсон, Кельвин, Фольмер, Ребиндер и др. По вопросам образования кристаллических фаз, сопровождающего твердение вяжущих веществ, большой вклад в мировую науку внесли наши отечественные ученые и, в первую очередь, О.П.Мчедлов-Петросян, В.И.Бабушкин (термодинамика процесса), А.Ф.Полак (кинетическая сторона процесса).
Рассмотрение вопроса начнем с классических представлений. Считается общепризнанным, что основой зарождения частиц новообразований является пересыщенное состояние раствора. Это - промежуточное состояние между ионными растворами и коллоидными системами. При увеличении пересыщения возрастает число ассоциатов в виде квазикристаллов, которые существуют временно и распадаются под действием теплового движения, возникая затем в других местах. Этот процесс носит вероятностно-статистический характер. Субмикрокристаллы находятся в метастабильном равновесии с пересыщенным раствором. Наиболее вероятной причиной образования зародышей кристаллов является механизм столкновения и срастания одноили двухмерных частиц за счет броуновского движения, притяжения и ориентации. Зародыши новой фазы не могут иметь сразу упорядоченную структуру и представляют собой аморфные частицы. Соударяясь в пересыщенном растворе, частицы слипаются, захватывая некоторое количество дисперсионной среды. Но термодинамическая неравновесность системы приводит к формированию кристаллической упорядоченной структуры, так как это сопровождается уменьшением энергии системы и термодинамически выгодно. На длительность существования первичных аморфных частиц большое влияние оказывает температурный фактор: нагрев вызывает относительно быстрое превращение их в кристаллические образования. Если в ходе процесса по каким-либо причинам возникают дислокации (искажения в кристаллической решетке), то это сильно снижает работу по об-разованию кристаллической структуры. В этом плане замечательным является то, что рост кристаллов на «подложке» (границе раздела фаз) вообще не требует образования зародыша,
- 90 -
так как фактически идет «надстройка» кристаллических образований с использованием структурных элементов подложки.
Кристаллизация из пересыщенных растворов возможна лишь при определенных термодинамических условиях, характеризуемых величиной
критического термодинамического потенциала Gкр.. Само появление центров кристаллизации с точки зрения термодинамики является процессом маловероятным, так как их возникновение связано не с убылью, а с ростом свободной энергии, поэтому образовавшиеся зародыши вновь распадаются на составляющие частицы.
Для возникновения устойчивого зародыша необходимо, чтобы его размер (радиус) был равен или превышал размер центра кристаллизации с критическим радиусом rкр. хотя бы на один молекулярный слой. Критическому радиусу соответствует величина Gкр . Дальнейший рост частиц с радиусом r rкр. невозможен, так как это вызвало бы увеличение термодинамического потенциала. Величина Gкр называется также работой образования зародыша. Она затрачивается на то, чтобы создать поверхности в гомогенном пересыщенном растворе.
Значение Gкр связано с rкр зависимостью
G |
4 |
r 2 , |
(28) |
кр. |
3 |
кр. |
|
|
|
|
где - поверхностная энергия на границе раздела фаз «твердое-жидкость». Если в системе имеются «подложки» (rn – радиус подложки), то
величина критического термодинамического потенциала становится равной:
G |
4 (r 2 |
r 2 ), |
(29) |
|
кр. |
3 |
кр |
п |
|
|
|
|
|
|
Появление частиц новообразований на границе раздела фаз |
||||
термодинамически более выгодно, так как |
|
|
|
|
G*кр |
Gкр |
(30) |
||
Величина Gкр уменьшается |
тем |
сильнее, чем ближе по |
своим |
|
кристаллохимическим параметрам материал «подложки» к тем же характеристикам частиц новой фазы (эпитаксия).
Вероятность (W) возникновения частиц новой фазы связана с
величиной |
критического термодинамического |
потенциала |
уравнением |
||
Фольмера: |
|
|
|
|
|
|
W B e |
Gкр. |
|
|
|
|
RT |
, |
(31) |
||
где В – коэффициент;
R - постоянная Больцмана;
Т- температура.
Кристаллизация из пересыщенного раствора идет самопроизвольно,
изменение G в ходе этого процесса можно описать уравнением:
G S n(2 1), |
(32) |
|
|
|
- 91 - |
|
|
|
|
|
|
|
||||
где |
n - число молей вещества, образовавших центр кристаллизации, |
|||||||||||||
|
поверхность которого S; |
|
|
|
|
|
|
|
|
|||||
|
1 и 2 – химические потенциалы растворенного вещества в пересыщен- |
|||||||||||||
|
ном растворе и в твердой фазе соответственно. |
|||||||||||||
|
Для rкр. справедливо выражение |
|
2M |
|
|
|
|
|||||||
|
|
|
rкр. |
|
, |
|
|
(33) |
||||||
|
|
|
|
|
|
|
|
|
||||||
|
|
|
(2 1) |
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
||||
где |
М - молярная масса вещества; |
|
|
|
|
|
|
|
|
|||||
|
- плотность твердого вещества. |
|
|
|
|
|
|
|
||||||
Подставляя значение rкр. |
в уравнение для критического термодинамического |
|||||||||||||
потенциала, имеем: |
|
Gкр. |
|
|
16 3M 2 |
|
. |
(34) |
||||||
|
3 |
2 ( |
|
)2 |
||||||||||
|
|
|
|
|
|
|
|
|
2 |
1 |
|
|
|
|
|
Выразив мольный объем вещества как v |
M |
, |
а разность химических |
||||||||||
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
потенциалов как |
2 |
1 RT ln |
Cп |
, |
|
где |
Сп |
|
и |
Сн – концентрации |
||||
|
|
|
||||||||||||
|
|
|
|
Сн |
|
|
|
|
|
|
|
|
||
пересыщенного и насыщенного растворов, а Сп = Сн + С, |
где С – |
||||||||||
пересыщение, имеем: |
|
|
|
|
|
|
|
|
|
|
|
|
C |
п |
|
|
|
|
|
С |
|
|
|
|
|
|
C |
|
|
|
|||||
ln |
|
|
ln 1 |
|
|
|
|
|
|
. |
(35) |
С |
|
C |
|
С |
|
||||||
|
н |
|
н |
|
н |
|
|
||||
|
|
|
|
|
|
|
|
|
|||
Подставив это значение в выражение для Gкр., получим
|
|
|
16 |
3v2C 2 |
|
||||||
G |
|
|
|
|
|
|
|
н |
, |
(36) |
|
|
|
|
|
|
|
|
|||||
кр. |
|
|
3(RT)2 C 2 |
|
|||||||
|
|
|
|
||||||||
Тогда выражение для вероятности возникновения новой фазы будет |
|||||||||||
иметь вид: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
2 |
2 |
|
|
|
|
|
|
|
|
|
v |
|
Cн |
|
|
||
|
|
|
|
3 ( C)2 , |
|
||||||
W B e 3(RT ) |
(37) |
||||||||||
где - коэффициент.
Скорость возникновения центров кристаллизации в свою очередьпропорциональна вероятности W и, следовательно, для ускорения этого процесса требуется уменьшение величины поверхностного натяжения(например, за счет добавок поверхностно-активных веществ), увеличение степени пересыщения С (например, за счет изменения ионной силы раствора при введении добавок-электролитов) и повышение температуры Т.
Рассмотренные закономерности возникновения первичных частиц новообразований неразрывно связаны с процессом их укрупнения. Этот процесс неизбежен, так как возникающие частицы находятся в коллоидной степени дисперсности и их укрупнение термодинамически выгодно.
- 92 -
Итак, нами рассмотрены классические представления о механизме образования кристаллических фаз, о связи этого процесса с энергетическими, другими параметрами «участников» этого процесса.
Относительно твердения вяжущих веществ представленные закономерности в наибольшей степени подтверждаются исследованиями, выполненными А.Ф.Полаком и его учениками, применительно к гипсовым вяжущим веществам. Именно эти вяжущие в начале главы мы отнесли к группе вяжущих кристаллизационного механизма отвердевания (по ЛеШателье). И действительно, расчеты, выполненные А.Ф.Полаком, относительно вероятности образования зародышей, энергии кристаллообразования, силы внутрикристаллических связей, габитуса кристаллов в основном соответствуют классическим представлениям и, следовательно, принятые в расчетах факторы могут рассматриваться как управляющие воздействия для твердеющей системы.
Что же касается системы «цемент-вода», то здесь процессы структурообразования протекают намного сложнее, и те классические представления, о которых речь шла выше, могут быть приняты лишь в качественном аспекте.
В частности, многие ученые считают, что в зависимости от значений коэффициента физико-химической неоднородности твердеющей системы квазитрехмерные и квазидвухмерные зародыши могут образовываться как на поверхности исходной твердой фазы, так и в растворе; возможен отрыв кристаллов, первоначально образовавшихся на подложке, но преимущественно кристаллизация новообразований будет протекать в соответствии с правилом Гиббса на границе раздела фаз, то есть на поверхности зерен цемента и на поверхности новых фаз.
Вероятность образования зародышей зависит от поверхностной энергии частиц новообразований, химического потенциала участвующих фаз, температуры, значения межфазной энергии, габитуса кристалла.
Критический радиус зародыша может быть определен с помощью формулы Томсона-Кельвина (33), а вероятность существования зародыша – по уравнению Фольмера (37).
Процесс зародышеобразования и первый этап роста кристаллов вначале протекают в кинетической, а затем в диффузионной области.
Структура внутреннего ритма из-за ограниченного объема и большого пересыщения представляет собой плотную рентгеноаморфную метамиктную (состоящую из различных агрегатов) коллоидную фазу цементного камня; структура внешнего ритма может состоять как из зародышей, так и из растущих кристаллов.
Если говорить о конкретных фазах, то, прежде всего, следует учесть комплексообразование и полимеризацию кремнекислородных анионов, в том числе твердофазовый процесс поликонденсации кремнекислородных анионов (SiO4)4- в последовательности: мономер, димер, тример или тетрамер, высшие олигомеры.
- 93 -
Фаза С-S-H, образующаяся при комнатной температуре, содержит, главным образом, димеры (Si2O7)6- и небольшое число линейных тримеров (Si3O10)8-, а также различное количество полимеров SiO4 4- на более поздних стадиях развития.
Фундаментальные исследования вопросов кристаллообразования в силикатных композициях начаты еще в конце 40-х- начале 50-х годов ХХ столетия школой кристаллографов во главе с Н.В.Беловым. Были расшифрованы сложные структуры гидросиликатов кальция. Установлено, что основным кремнекислородным радикалом в гидросиликатах является диортогруппа Si2O7 6-, которая получается из двух тетраэдров, соединенных вершинами через атом кислорода. Анион Si2O7 6- лежит в основе построения новых кремнекислородных цепочек, лент, сеток и колец. Установлено, что в этих структурах есть возможность обратимой замены части ортотетраэдров на равнозарядные и равнообъемные группы гидроксила 4(ОН)-. На рис.18 представлена упрощенная схема структуры самого простого соединения - низкоионного С2S -гидрата. Его кристаллы представляют собой призматические пластинки.
в
|
|
с |
|
|
а |
|
Рис.18. Упрощенная структура С2S -гидрата |
|
Обозначения: |
- Са2+ |
- отдельный ион ОН- |
- Si(O3OH) – тетраэдры
Как следует из схемы, С2S- -гидрат в своем составе содержит изолированные, разобщенные ионами кальция мономерные кремнекислородные анионы с одной группой ОН- вместо О2-.
- 94 -
Кроме того, в структуре этого гидросиликата имеются молекулы воды, тесно связанные с ионами кальция, то есть у ионов кальция здесь проявляется своеобразная роль – комплексообразователя, связывающего воду. Наличие в составе цементного камня С2S -гидрата, имеющего четко выраженную кристаллическую структуру, к сожалению, не придает ему высоких прочностных свойств, поэтому его образования необходимо избегать.
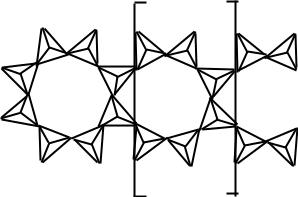
Другие гидросиликаты кальция: ксонотлит, гиллебрандит, тоберморит имеют упорядоченную структуру силоксановой цепи. В ксонотлите и гиллебрандите содержится полимерный радикал с инкрементом (инкремент – форма элементарного звена полимерной цепи) Si6O17 10-. В результате соединения таких групп образуются ленты, получившие название ксонотлитовых. На рис.19 представлена схема ксонотлитовой ленты, на которой квадратными скобками выделен структурный инкремент.
Рис.19. Схема структуры ксонотлитовой ленты
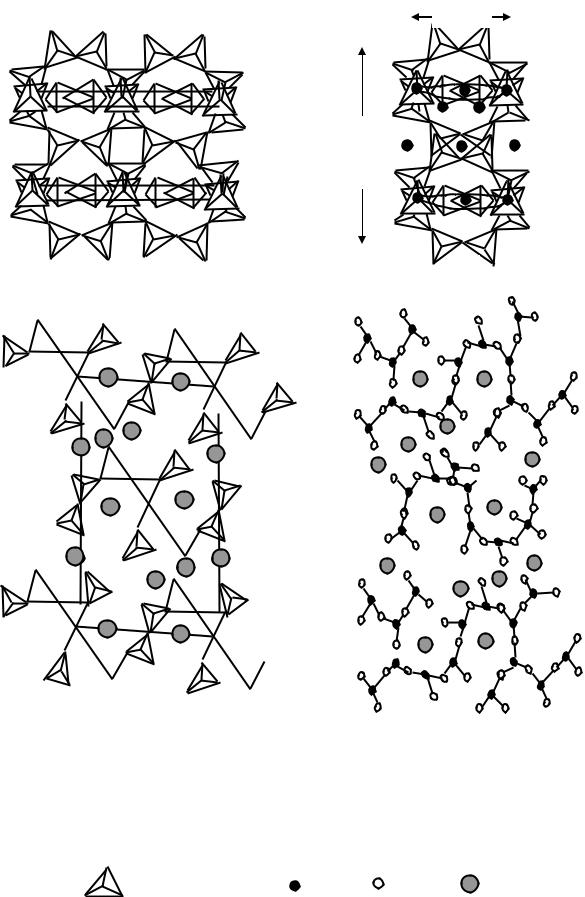
Тоберморит 5СаО . 6SiO2 .5H2O имеет более сложную структуру. Она слагается из сдвоенных ксонотлитовых цепочек, которые в процессе конденсации как бы накладываются друг на друга, образуя гофрированные слои. В структуре тоберморита присутствует более сложный полимерный силоксановый анион Si12O31 14-. Структурная модель тоберморита представлена на рис.20.
Таким образом, гидросиликаты кальция, входящие в состав цементного камня, представляют собой соединения с неупорядоченной, трудноопределяемой рентгенографически структурой. Это, в основном, гидросиликаты группы С-S-H(I) или СSH(B). Такая структура получается в результате определенных пространственных «трудностей» и других факторов, влияющих на строение образующихся соединений как в процессе конденсации кремнекислородных тетраэдров, так и при перестройке одного гидросиликата в другой. Это означает, что гидросиликаты представленных типов являются промежуточными продуктами последовательных превращений таких соединений. В целом же развитие структур с высокой
- 95 -
а) |
б=7,2 |
|
а=11,3
I I
|
|
|
|
|
|
|
|
в) |
|
|
б) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Рис.20. Структура тоберморита
а) схема сцепления и взаимного наложения;
I – ксонтлитовых лент; II – ксонотлитовых колец; |
|||
б) с выделением тетраэдров; в) с выделением силоксановых цепей |
|||
Обозначения: |
|
|
|
- SiO4 4- –тетраэдр |
- Si4+ |
- О |
- Са2+ |
- 96 -
прочностью обеспечивается теми гидратными новообразованиями, частицы которых имеют тенденцию к созданию прочных химических связей и, следовательно, к формированию непрерывного каркаса в твердеющей системе.
И, наконец, школой Н.В.Белова убедительно доказано, что комплексообразование и полимеризация являются основой формирования структур новых гидросиликатных фаз.
Попутно следует заметить, что именно с этими процессами связывают периодические сбросы прочности твердеющим цементным камнем при общей тенденции к ее росту.
6.3.2.3. О скорости гидратационных процессов
Решение практических задач управления процессами твердения цементосодержащих композиционных материалов предполагает, что мы имеем достаточно объективную информацию о скорости этого процесса.
Для самого простого рецептурного управления достаточно иметь четкие представления об общей кинетике процесса о механизме и эффективности действия всевозможных внешних и внутренних факторов. При этом недостающую информацию можно получить путем постановки соответствующих экспериментальных исследований.
Если же ставится задача управления процессом в автоматическом режиме, то для этого необходима математическая модель, отражающая кинетику процесса гидратационного твердения.
Рассмотрим, насколько возможен тот или иной путь. Начнем с элементарных представлений о химической кинетике.
Химическая кинетика изучает общие законы эволюции химических систем. Ее центральным вопросом является учение о скоростях химических реакций. Но вопрос о скоростях реакций самым непосредственным образом
связан с механизмами их протекания.
Интенсивность химических превращений выражают обычно скоростью реакции w, то есть количеством вещества n, реагирующего в единицу времени ( ) в данной системе:
w |
dn |
. |
(38) |
|
|||
|
d |
|
|
Для рассматриваемых процессов более объективным является показатель удельной скорости реакции wуд., характеризуемой количеством вещества, реагирующего в единицу времени в единице реакционного пространства:
w |
уд. |
1 |
dn |
, |
(39) |
|
q |
d |
|
|
где q – размер структурного элемента системы, в котором локализована реакция; для гомогенной реакции, протекающей в объеме V фазы,
- 97 -
q V; для реакции на поверхности S, q S.
Формально скорость химической реакции на основе постулатов, принятых в химической кинетике, может быть представлена как
w |
уд. |
k |
0 |
t E / RT C 1 |
C 2 |
... kПС i |
(40) |
|
|
1 |
2 |
i |
|
||
|
|
|
|
|
|
|
|
где k – константа скорости реакции; |
|
|
|
||||
Сi – концентрация реагента i (i=1,2,…);i – его стехиометрический коэффициент; k0 - предэкспоненциальный множитель;
Е - энергия активации; R - газовая постоянная;
Т - абсолютная температура; П - математический оператор произведения.
Уравнение (40) – простейшее кинетическое уравнение. Существенным его свойством является то, что правая часть представляет собой произведение двух членов, один из которых зависит только от концентрации реагентов, а второй – только от температуры. Заметим также, что в (40) записано по существу утверждение, что скорость элементарной реакции определяется параметрами k0 и Е, а также параметрами i , заданными
стехиометрическим уравнением реакции. Величину i при этом называют порядком реакции по компоненту i , а их сумму (для многостадийных процессов) – наблюдаемым порядком реакции.
Из вышесказанного следует, что уравнение (40) корректно лишь для реакции, протекающей в пределах одного структурного элемента системы. Даже в простейшем случае одной и той же реакции, идущей в объеме одной
из фаз V и на поверхности раздела |
фаз S, |
наблюдаемая скорость реакции |
|||||
выразится уравнением |
|
|
|
|
|
|
|
dn w w |
|
S w |
V , |
(41) |
|||
d |
|
уд(S) |
|
|
уд(V ) |
|
|
или в общем случае |
|
|
|
|
|
|
|
|
w w |
уд( j) |
q |
j |
, |
(42) |
|
|
|
|
|
|
|
||
где w и qj – удельная скорость реакции в пределах структурного элемента j и его размер соответственно.
Существенно, что при этом величины удельной скорости реакции могут иметь различные размерности.
А теперь обратим внимание на то, что уравнения (41,42) справедливы лишь для стационарных процессов, то есть таких, для которых неизменность скорости во времени обеспечивается при условии постоянства во времени константы скорости реакции и концентраций реагентов. В некоторых аппаратах химической технологии эти условия обеспечиваются постоянными подпитками процесса химическими реагентами и отводом продуктов реакции. Такие системы называют открытыми.
- 98 -
Рассматриваемая же нами система твердения, безусловно, относится к классу закрытых систем. Причем, эта система к тому же является гетерогенной, достаточно сложной.
Сложность задач гетерогенной кинетики определяется, по крайней мере, двумя особенностями процессов. Первая из них обусловлена локализацией разных стадий сложного процесса в различных структурных элементах твердеющей системы (фазах и поверхностях раздела фаз), что, в свою очередь, порождает пространственное распределение концентраций и температуры и соответствующие потоки вещества и теплоты.
Вторая особенность связана с тем, что гетерогенная система включает одну или несколько конденсированных фаз. Это обстоятельство приводит к затруднениям обмена веществ как в пределах системы, так и с окружающей средой. И, таким образом, в закрытой системе химический процесс оказывается нестационарным, так как имеют место постоянная убыль концентрации реагирующих веществ, накопление продуктов реакции. А это значит, что при попытке описать химический процесс уравнениями (41,42), мы должны их дополнить параметром времени. Но подобные задачи в химической кинетике решены лишь для реакций первого порядка. Для реакций произвольного порядка получаемые системы дифференциальных уравнений не имеют аналитических решений.
Вообще подобные задачи рассматриваются в относительно «молодом» разделе химической кинетики – макрокинетике. В этой области науки основное внимание уделяется рассмотрению внешней и внутренней задач диффузионной кинетики. Но в целом задача не стала проще и достаточно убедительных решений пока не имеет.
Исследованиям вопросов макрокинетики твердения вяжущих веществ посвящено множество работ ученых. Однако, полученные представления о
кинетике процессов твердения имеют преимущественно качественный характер. Они дают возможность объяснить лишь некоторые стороны этого сложного процесса.
Остановимся на следующих результатах, имеющих, на наш взгляд, как теоретическое, так и практическое значение. Если рассматривать первую стадию твердения – гидратацию, то с наибольшей скоростью она протекает у относительно простых вяжущих веществ – воздушной извести и строительного гипса. Так, например, если взять кусок высокоактивной негидратированной извести и постепенно наносить на него капли воды, то мы будем отмечать интенсивное ее впитывание настолько, что поверхность куска извести определенное время выглядит сухой, но затем (через 5-20 минут) кусок извести начинает увеличиваться в объеме и рассыпаться в белый порошок. Что произошло? Благодаря высокой исходной пористости (вспомним, что при обжиге в результате диссоциации углекислого кальция из куска «ушло» с дымовыми газами 44 % СО2) объем куска извести стал практически равнодоступным для воды и поэтому можно считать, что химическая реакция идет во всем объеме достаточно быстро. При этом