

ФИЗическая химия
.docxОБРАТИМЫЕ И НЕОБРАТИМЫЕ ПРОЦЕССЫ , пути изменения состояния термодинамич. системы. Процесс наз. обратимым, если он допускает возвращение рассматриваемой системы из конечного состояния в исходное через ту же последовательность промежут. состояний, что и в прямом процессе, но проходимую в обратном порядке. При этом в исходное состояние возвращается не только система, но и среда. Обратимый процесс возможен, если и в системе, и в окружающей среде он протекает равновесно. При этом предполагается, что равновесие существует между отдельными частями рассматриваемой системы и на границе с окружающей средой. Обратимый процесс - идеализир. случай, достижимый лишь при бесконечно медленном изменении термодинамич. параметров. Скорость установления равновесия должна быть больше, чем скорость рассматриваемого процесса. Если невозможно найти способ вернуть и систему, и тела в окружающей среде в исходное состояние, процесс изменения состояния системы наз. необратимым.
Необратимые
процессы могут протекать самопроизвольно
только в одном направлении; таковы
диффузия, теплопроводность, вязкое
течение и др. Для хим. р-ции применяют
понятия термодинамич. и кинетич.
обратимости, к-рые совпадают только в
непосредств. близости к состоянию
равновесия. Р-ция А + В![]() С
+ D наз. кинетически обратимой или
двусторонней, если в данных условиях
продукты С и D могут реагировать друг
с другом с образованием исходных в-в А
и В. При этом скорости прямой и обратной
р-ций, соотв.
С
+ D наз. кинетически обратимой или
двусторонней, если в данных условиях
продукты С и D могут реагировать друг
с другом с образованием исходных в-в А
и В. При этом скорости прямой и обратной
р-ций, соотв.![]()
![]() , где
, где![]() и
и![]() -константы
скорости, [А], [В], [С], [D]- текущие концентрации
(активности), с течением времени
становятся равными и наступает химическое
равновесие
, в
к-ром
-константы
скорости, [А], [В], [С], [D]- текущие концентрации
(активности), с течением времени
становятся равными и наступает химическое
равновесие
, в
к-ром
![]() -константа
равновесия
., зависящая
от т-ры. Кинетически необратимыми
(односторонними) являются обычно такие
р-ции, в ходе к-рых хотя бы один из
продуктов удаляется из зоны р-ции
(выпадает в осадок, улетучивается или
выделяется в виде малодиссоциированного
соед.), а также р-ции, сопровождающиеся
выделением большого кол-ва тепла.
-константа
равновесия
., зависящая
от т-ры. Кинетически необратимыми
(односторонними) являются обычно такие
р-ции, в ходе к-рых хотя бы один из
продуктов удаляется из зоны р-ции
(выпадает в осадок, улетучивается или
выделяется в виде малодиссоциированного
соед.), а также р-ции, сопровождающиеся
выделением большого кол-ва тепла.
На практике нередко встречаются системы, находящиеся в частичном равновесии, т.е. в равновесии по отношению к определенного рода процессам, тогда как в целом система неравновесна. Напр., образец закаленной стали обладает пространств. неоднородностью и является системой, неравновесной по отношению к диффузионным процессам, однако в этом образце могут происходить равновесные циклы мех. деформации, поскольку времена релаксации диффузии и деформации в твердых телах отличаются на десятки порядков. Следовательно, процессы с относительно большим временем релаксации являются кинетически заторможенными и могут не приниматься во внимание при термодинамич. анализе более быстрых процессов.
Необратимые процессы сопровождаются диссипатив-ными эффектами, сущностью к-рых является производство (генерирование) энтропии в системе в результате протекания рассматриваемого процесса. Простейшее выражение закона диссипации имеет вид:
![]()
где![]() средняя
т-ра, diS-
производство
энтропии,
средняя
т-ра, diS-
производство
энтропии,![]() - т. наз. нескомпенсированная теплота
Клаузиуса (теплота диссипации).
- т. наз. нескомпенсированная теплота
Клаузиуса (теплота диссипации).
Обратимые процессы, будучи идеализированными, не сопровождаются диссипативными эффектами. Микроско-пич. теория О. и н. п. развивается в статистической термодинамике . Системы, в к-рых протекают необратимые процессы, изучает термодинамика необратимых процессов .
12.
Вну́тренняя эне́ргия тела (обозначается как E или U) — это сумма энергий молекулярных взаимодействий и тепловых движений молекулы. Внутренняя энергия является однозначной функцией состояния системы. Это означает, что всякий раз, когда система оказывается в данном состоянии, её внутренняя энергия принимает присущее этому состоянию значение, независимо от предыстории системы. Следовательно, изменение внутренней энергии при переходе из одного состояния в другое будет всегда равно разности между ее значениями в конечном и начальном состояниях, независимо от пути, по которому совершался переход.
Внутреннюю энергию тела нельзя измерить напрямую. Можно определить только изменение внутренней энергии:
![]()
Энтальпи́я, также тепловая функция и теплосодержание — термодинамический потенциал, характеризующий состояние системы в термодинамическом равновесии при выборе в качестве независимых переменных давления, энтропии и числа частиц.
Изменение энтальпии не зависит от пути процесса, определяясь только начальным и конечным состоянием системы. Если система каким-либо путём возвращается в исходное состояние (круговой процесс), то изменение любого её параметра, являющегося функцией состояния, равно нулю, отсюда ΔH = 0, или же
![]()
В
этом случае изменение энтальпии в
изобарическом процессе практически
удобно рассчитывать, зная теплоемкость
при постоянном давлении Cp(T)
(термохимический закон Кирхгофа):

При этом используется эмпирическое разложение теплоёмкости в ряд по степеням Т:
Энтальпия —
величина
аддитивная
(экстенсивная),
т. е. для сложной системы равна сумме
энтальпий её независимых частей
![]() .
Подобно другим термодинамическим
потенциалам, энтальпия определяется
с точностью до постоянного слагаемого,
которому в термодинамике часто придают
произвольные значения (например, при
расчете и построении тепловых диаграмм).
.
Подобно другим термодинамическим
потенциалам, энтальпия определяется
с точностью до постоянного слагаемого,
которому в термодинамике часто придают
произвольные значения (например, при
расчете и построении тепловых диаграмм).
Теплоёмкость тела (обычно обозначается латинской буквой C) — физическая величина, определяющая отношение бесконечно малого количества теплоты δQ, полученного телом, к соответствующему приращению его температуры δT:
![]()
Единица измерения теплоёмкости в системе СИ — Дж/К.
Удельной теплоёмкостью называется количество теплоты, которое необходимо подвести к телу чтобы изменить его температуру на один (1) градус. Количество вещества может быть измерено в килограммах, кубических метрах и молях. В зависимости от того, к какой количественной единице относится теплоёмкость, различают массовую, объёмную и молярную теплоёмкость.
Понятие теплоёмкости определено как для веществ в различных агрегатных состояниях (твёрдых тел, жидкостей, газов), так и для ансамблей частиц и квазичастиц (в физике металлов, например, говорят о теплоёмкости электронного газа). Если речь идёт не о каком-либо теле, а о некотором веществе как таковом, то различают удельную теплоёмкость — теплоёмкость единицы массы этого вещества и молярную — теплоёмкость одного моля его.
Для примера, в молекулярно-кинетической теории газов показывается, что молярная теплоёмкость идеального газа с i степенями свободы при постоянном объеме равна:
![]()
R = 8.31 Дж/(моль К) — универсальная газовая постоянная.
А при постоянном давлении
![]()
13.
Первый закон термодинамики - Изменение внутренней энергии системы при ее переходе из одного состояния в другое равно сумме количества теплоты, подведенного к системе извне, и работы внешних сил, действующих на нее:
![]()
Первый закон термодинамики - количество теплоты, подведенное к системе, идет на изменение ее внутренней энергии и на совершение системой работы над внешними телами:
![]()
При изохорном процессе объем газа остается постоянным, поэтому газ не совершает работу. Изменение внутренней энергии газа происходит благодаря теплообмену с окружающими телами:
![]()
При изотермическом процессе количество теплоты, переданное газу от нагревателя, полностью расходуется на совершение работы:
![]()
При изобарном расширении газа подведенное к нему количество теплоты расходуется как на увеличение его внутренней энергии и на совершение работы газом:
![]()
Адиабатный процесс - термодинамический процесс в теплоизолированной системе.
![]()
Теплоизолированная система - система, не обменивающаяся энергией с окружающими телами.
Закон Гесса — основной закон термохимии, который формулируется следующим образом:
Тепловой эффект химической реакции, проводимой в изобарно-изотермических или изохорно-изотермических условиях, зависит только от вида и состояния исходных веществ и продуктов реакции и не зависит от пути её протекания.
Иными словами, количество теплоты, выделяющееся или поглощающееся при каком-либо процессе, всегда одно и то же, независимо от того, протекает ли данное химическое превращение в одну или в несколько стадий (при условии, что температура, давление и агрегатные состояния веществ одинаковы). Например, окисление глюкозы в организме осуществляется по очень сложному многостадийному механизму, однако суммарный тепловой эффект всех стадий данного процесса равен теплоте сгорания глюкозы.
На рисунке приведено схематическое изображение некоторого обобщенного химического процесса превращения исходных веществ А1, А2… в продукты реакции В1, В2…, который может быть осуществлен различными путями в одну, две или три стадии, каждая из которых сопровождается тепловым эффектом ΔHi. Согласно закону Гесса, тепловые эффекты всех этих реакций связаны следующим соотношением:ΔH1 = ΔH2 + ΔH3 = ΔH4 + ΔH5 + ΔH6
14.
Термохи́мия — раздел химической термодинамики, в задачу которой входит определение и изучение тепловых эффектов реакций, а также установление их взаимосвязей с различными физико-химическими параметрами. Ещё одной из задач термохимии является измерение теплоёмкостей веществ и установление их теплот фазовых переходов.
Тепловой эффект химической реакции или изменение энтальпии системы вследствие протекания химической реакции — отнесенное к изменению химической переменной количество теплоты, полученное системой, в которой прошла химическая реакция и продукты реакции приняли температуру реагентов.
Чтобы тепловой эффект являлся величиной, зависящей только от характера протекающей химической реакции, необходимо соблюдение следующих условий:
Реакция должна протекать либо при постоянном объёме Qv(изохорный процесс), либо при постоянном давлении Qp(изобарный процесс).
В системе не совершается никакой работы, кроме возможной при P = const работы расширения.
15.
Все реакции, протекающие в клетке, можно разделить на Экзергоничесше И Эндергонические. Первые идут с выделением энергии, которая может быть рассеяна в виде тепла в окружающую среду. Вторые требуют энергетических затрат и, как правило, способствуют созданию сложной организации и поддержанию внутреннего порядка. Изменение энергии, которое сопровождает химические реакции, можно проанализировать количественно, используя термодинамические параметры: Т — Абсолютная температура (К); Р — давление; V— объем; Е — Внутренняя полная энергия системы; S — Энтропия; Н— энтальпия, или теплосодержание системы; G — свободная энергия системы (энергия Гиббса).
При постоянных Р, Т и V Изменение общей энергии системы ∆Е в результате химической реакции будет равно изменению теплосодержания системы ∆Н, Т. е. тепловому эффекту реакции. В этих условиях термодинамические параметры связаны соотношением:
∆G = ∆Н - Т∆S.
Для характеристики реакций, которые протекают при постоянных температуре, давлении и объеме, наиболее важным термодинамическим параметром является изменение свободной энергии Гиббса AG. Этот показатель характеризует энергию, которая поглощается или выделяется в ходе реакции или может быть передана другой химической реакции. Для биохимических реакций в растворе при низких концентрациях веществ изменение свободной энергии в результате реакции АА + BВ = сС + DD) Можно определить по уравнению:
∆G = ∆G° + 2,3RTlg([C]c[D]d /[A]a[B]b) [Дж∙моль-1],
Где R — Газовая постоянная (8,31 Дж∙ град-1 моль-1); Т— абсолютная температура (К); [А], [В], [С], [D] — молярные концентрации реагирующих веществ; ∆G° — изменение стандартной свободной энергии данной реакции.
Конста́нта равнове́сия — величина, определяющая для данной химической реакции соотношение между термодинамическими активностями (либо, в зависимости от условий протекания реакции, парциальными давлениями, концентрациями или фугитивностями) исходных веществ и продуктов в состоянии химического равновесия (в соответствии с законом действующих масс). Зная константу равновесия реакции, можно рассчитать равновесный состав реагирующей смеси, предельный выход продуктов, определить направление протекания реакции.
константа равновесия может быть рассчитана по уравнению:
![]()
![]() —
относительные
парциальные давления компонентов,
—
относительные
парциальные давления компонентов,
Зако́н де́йствующих масс устанавливает соотношение между массами реагирующих веществ в химических реакциях при равновесии, а также зависимость скорости химической реакции от концентрации исходных веществ.
В химической термодинамике закон действующих масс связывает между собой равновесные активности исходных веществ и продуктов реакции, согласно соотношению:
ai — активность веществ. Вместо активности могут быть использованы концентрация (для реакции в идеальном растворе), парциальные давления (реакция в смеси идеальных газов), фугитивность (реакция в смеси реальных газов);
νi — стехиометрический коэффициент (для исходных веществ принимается отрицательным, для продуктов — положительным);
Ka — константа химического равновесия. Индекс «a» здесь означает использование величины активности в формуле.
На практике в расчётах, не требующих особой точности, значения активности обычно заменяются на соответствующие значения концентраций (для реакций в растворах) либо парциальных давлений (для реакций между газами). Константу равновесия при этом обозначают Kc или Kp соответственно. Впервые закон действующих масс был выведен из кинетических представлений Гульдбергом и Вааге, а термодинамический вывод его дан Вант-Гоффом в 1885 году[3].
Пример: для стандартной реакции
![]()
константа химического равновесия определяется по формуле
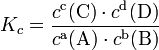
В начале XIX века нормальное распределение затмило собой все остальные, поскольку в работах Гаусса и Лежандра утверждалось о нормальном законе распределения ошибок наблюдений.
Нормальный закон распределения (или распределение Гаусса) задается следующей дифференциальной функцией
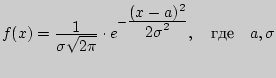 -
параметры.
-
параметры.

16.
Свободная энергия Гиббса (или просто энергия Гиббса, или потенциал Гиббса, или термодинамический потенциал в узком смысле) — это величина, показывающая изменение энергии в ходе химической реакции и дающая таким образом ответ на принципиальную возможность химической реакции; это термодинамический потенциал следующего вида:
![]()
Энергию Гиббса можно понимать как полную химическую энергию системы (кристалла, жидкости и т. д.)
Понятие энергии Гиббса широко используется в термодинамике и химии.
Классическим определением энергии Гиббса является выражение
![]()
где U — внутренняя энергия, P — давление, V — объем, T — абсолютная температура, S — энтропия.
Дифференциал энергии Гиббса для системы с постоянным числом частиц, выраженный в собственных переменных — через давление p и температуру T:
![]()
Для системы с переменным числом частиц этот дифференциал записывается так:
![]()
Здесь μ — химический потенциал, который можно определить как энергию, которую необходимо затратить, чтобы добавить в систему ещё одну частицу.
Свобо́дная эне́ргия Гельмго́льца (или просто свобо́дная эне́ргия) — термодинамический потенциал, убыль которого в квазистатическом изотермическом процессе равна работе, совершённой системой над внешними телами.
Свободная энергия Гельмгольца для системы с постоянным числом частиц определяется так:
![]() ,
где U
— внутренняя
энергия,
T
— абсолютная температура,
S
— энтропия.
,
где U
— внутренняя
энергия,
T
— абсолютная температура,
S
— энтропия.
Отсюда дифференциал свободной энергии равен:
![]() .
.
Видно,
что это выражение является полным
дифференциалом относительно независимых
переменных T
и V.
Поэтому часто свободную энергию
Гельмгольца для равновесного состояния
выражают как функцию
![]() .
.
Для системы с переменным числом частиц дифференциал свободной энергии Гельмгольца записывается так:
![]() ,
,
где
μ
— химический
потенциал,
а N
— число частиц в системе. При этом
свободная энергия Гельмгольца для
равновесного состояния записывается
как функция
![]() .
.
В соответствии с рекомендациями ИЮПАК энергию Гельмгольца в химической термодинамике можно также обозначать как A
Второе начало термодинамики — физический принцип, накладывающий ограничение на направление процессов передачи тепла между телами.
Второе начало термодинамики гласит, что невозможен самопроизвольный переход тепла от тела, менее нагретого, к телу, более нагретому.
Второе начало термодинамики запрещает так называемые вечные двигатели второго рода, показывая что коэффициент полезного действия не может равняться единице, поскольку для кругового процесса температура холодильника не должна равняться 0.
Второе начало термодинамики является постулатом, не доказываемым в рамках термодинамики. Оно было создано на основе обобщения опытных фактов и получило многочисленные экспериментальные подтверждения.
Существуют несколько эквивалентных формулировок второго начала термодинамики:
Постулат Клаузиуса: «Невозможен процесс, единственным результатом которого являлась бы передача тепла от более холодного тела к более горячему»[1] (такой процесс называется процессом Клаузиуса).
Постулат Томсона (Кельвина): «Невозможен круговой процесс, единственным результатом которого было бы производство работы за счет охлаждения теплового резервуара» (такой процесс называется процессом Томсона).
17.
Энтропи́я (от греч. ἐντροπία — поворот, превращение) в естественных науках — мера беспорядка системы, состоящей из многих элементов. В частности, в статистической физике — мера вероятности осуществления какого-либо макроскопического состояния; в теории информации — мера неопределённости какого-либо опыта (испытания), который может иметь разные исходы, а значит и количество информации; в исторической науке, для экспликации феномена альтернативности истории (инвариантности и вариативности исторического процесса).
Энтропия в информатике — степень неполноты, неопределённости знаний.
Понятие энтропии впервые было введено Клаузиусом в термодинамике в 1865 году для определения меры необратимого рассеивания энергии, меры отклонения реального процесса от идеального. Определённая как сумма приведённых теплот, она является функцией состояния и остаётся постоянной при обратимых процессах, тогда как в необратимых — её изменение всегда положительно.
![]() ,
,
где dS — приращение энтропии; δQ — минимальная теплота подведенная к системе; T — абсолютная температура процесса;
Абсолютную энтропию хим. соед. определяют экспериментально, гл. обр. калориметрич. методом, исходя из соотношения:
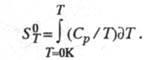
Использование второго начала позволяет определять энтропию хим. р-ций по эксперим. данным (метод электродвижущих сил, метод давления пара и др.). Возможен расчет энтропии хим. соед. методами статистич. термодинамики, исходя из мол. постоянных, мол. массы, геометрии молекулы, частоты нормальных колебаний. Такой подход успешно осуществляется для идеальных газов. Для конденсир. фаз статистич. расчет дает значительно меньшую точность и проводится в ограниченном числе случаев; в последние годы в этой области достигнуты значит. успехи. Энтропии хим. соед. табулированы в справочниках. Как правило, для конденсир. фаз приводят результаты калориметрич. измерений, для газообразных - статистич. расчета.
18.
Поэтому одна из формулировок второго закона термодинамики предупреждает о невозможности построения такого двигателя. В отличие от вечного двигателя первого рода, создающего энергию из ничего, двигатель, действующий при наличии одного источника теплоты, называют вечным двигателем второго рода. [1]
Поэтому одна из формулировок второго закона термодинамики предупреждает о невозможности построения такого двигателя. В отличие от вечного двигателя первого рода, создающего энергию из ничего, двигатель, действующий при наличии одного источника тепла, называют вечным двигателем второго рода. [2]
Существует поэтому несколько формулировок второго закона термодинамики, в которых содержится термин энтропия. Так, например, формулировка этого закона Дифференциал энтропии есть полный дифференциал подчеркивает существование функции энтропии для равновесных систем и ее математические свойства. [3]
Исторически сложилось несколько формулировок второго закона термодинамики. Все они выражают одно и то же содержание, подмечая существование самопроизвольных и несамопроизвольных процессов и различие между ними. [4]
Хими́ческая термодина́мика — раздел физической химии, изучающий процессы взаимодействия веществ методами термодинамики.
Основными направлениями химической термодинамики являются:
Классическая химическая термодинамика, изучающая термодинамическое равновесие вообще.
Термохимия, изучающая тепловые эффекты, сопровождающие химические реакции.
Теория растворов, моделирующую термодинамические свойства вещества исходя из представлений о молекулярном строении и данных о межмолекулярном взаимодействии.
Положение химического равновесия зависит от следующих парамктров реакции: температуры, давления и концентрации. Влияние, которое оказывают эти факторы на химическую реакцию, подчиняются закономерности, которая была высказана в общем виде в 1884 году французским ученым Ле-Шателье. Современная формулировка принципа Ле-Шателье такова:
|
Если на систему,находящуюся в состоянии равновесия, оказать внешнее воздействие, то система перейдет в другое состояние так, чтобы уменьшить эффект внешнего воздействия. |
1. Влияние температуры. В каждой обратимой реакции одно из направлений отвечает экзотермическому процессу, а другое - эндотермическому.
|
N2
+ 3H2 |
Прямая реакция - экзотермическая, а обратная реакция - эндотермическая. Влияние изменения температуры на положение химического равновесия подчиняется следующим правилам:
|
При повышении температуры химическое равновесие смещается в направлении эндотермической реакции, при понижении температуры - в направлении экзотермической реакции. |
2. Влияние давления. Во всех реакциях с участием газообразных веществ, сопровождающихся изменением объема за счет изменения количества вещества при переоходе от исходных веществ к продуктам, на положение равновесия влияет давление в системе. Влияние давления на положение равновесия подчиняется следующим правилам:
|
При повышении давления равновесие сдвигается в направлении образования веществ (исходных или продуктов) с меньшим объемом; при понижении давления равновесие сдвигается в направлении образования веществ с большим объемом |
Таким образом, при переходе от исходных веществ к продуктам объем газов уменьшился вдвое. Значит, при повышении давления равновесие смещается в сторону образования NH3, о чем свидетельствуют следующие данные для реакции синтеза аммиака при 400 0С: