книги студ / Color Atlas of Pathophysiology (S Silbernagl et al, Thieme 2000)
.pdf

|
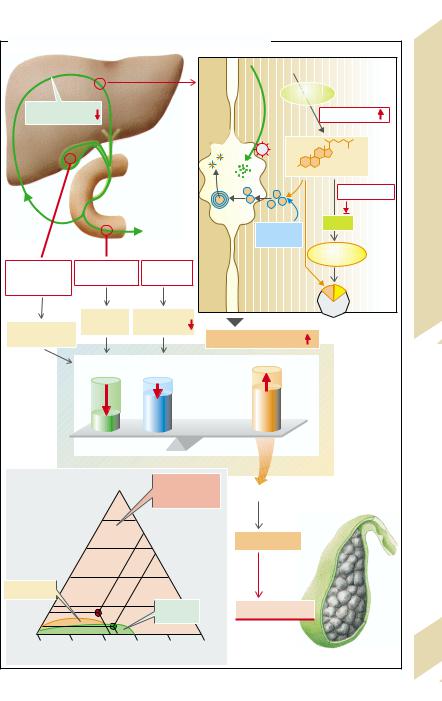
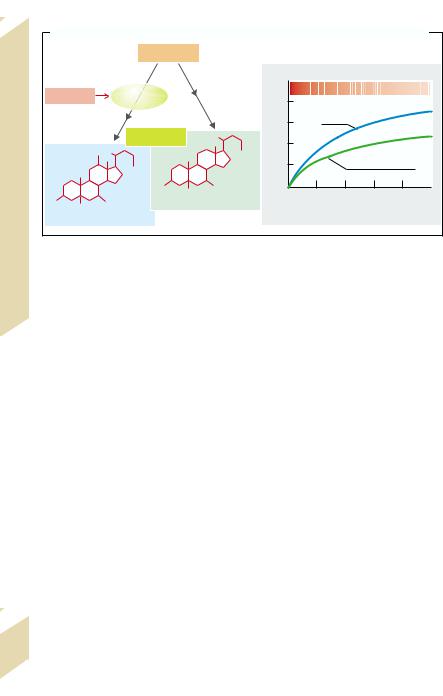
B. Cholesterol/Bile Salts: Dependence on Bile Salt Type and Bile Salt Secretion Rate |
|
||||||||||
|
|
|
Cholesterol |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
–1] |
|
|
|
|
|
|
|
|
|
|
|
|
h |
5 |
|
|
|
|
|
|
|
|
12α- |
|
|
kg–1· |
|
[Cholesterol]/[Bile salts] |
|
|||
|
Estrogens |
|
|
|
|
|
|
|||||
|
|
hydroxylase |
|
|
· |
4 |
|
|
|
|
|
|
|
|
|
|
|
[mmol |
|
|
|
|
|
||
|
|
|
|
|
|
3 |
Cholate |
|
|
|
||
|
|
|
|
|
|
secretion |
|
|
|
|
|
|
|
|
|
Bile salts |
|
|
2 |
|
|
|
|
|
|
|
|
OH |
|
|
|
|
|
|
|
|
||
|
|
|
|
COO– |
1 |
|
Chenodeoxycholate |
|
||||
Intestines, Liver |
|
12 |
COO– |
|
|
Cholesterol |
|
|
||||
|
|
|
|
|
|
|
|
|||||
|
|
HO |
OH |
|
00 |
10 |
20 |
30 |
40 |
50 |
||
HO |
|
OH |
Chenodeoxycholate |
|
Bile salt secretion [mmol · kg–1· h–1] |
|||||||
|
Cholate |
|
|
|
|
(after G. Paumgartner et al.) |
||||||
|
|
|
|
|
|
|
|
|
|
|
||
Stomach, |
! |
|
|
|
|
|
|
|
|
|
|
|
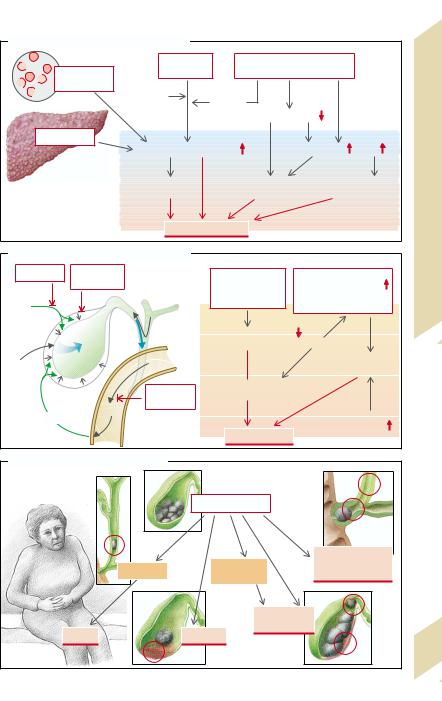
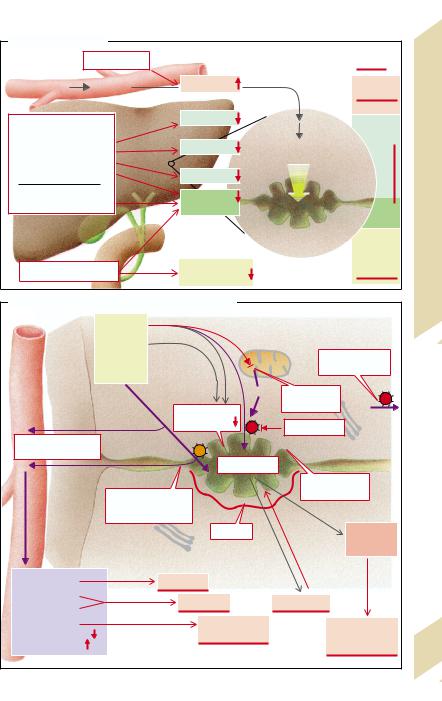
The gallbladder, in which the specific bile |
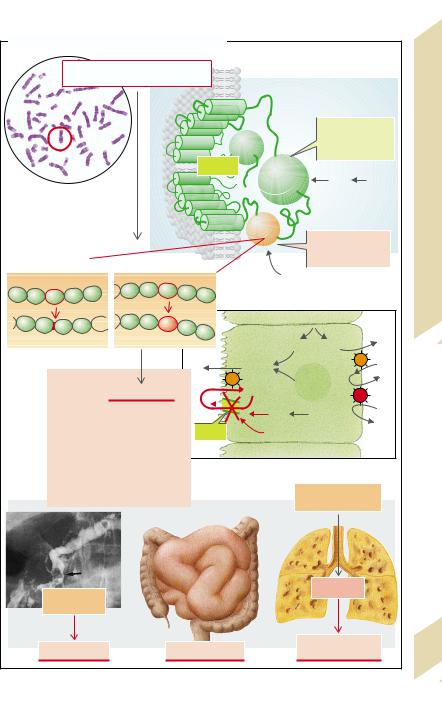
In acute cholecystitis fever and leukocyto- |
|||||||||||
components (Ch, BS, Pch) are concentrated |
sis are added to the symptoms listed above. |
|||||||||||
many times over by withdrawal of water, |
Important causes are trauma to the gallblad- |
|||||||||||
6 |
also plays an important part (→ D) in the for- |
der epithelium caused by stones. Prostaglan- |
||||||||||
|
mation of gallstones (cholelithiasis after cho- |
dins are liberated from the gallbladder epi- |
||||||||||
|
lecystectomy is rare). Disorders of gallblad- |
thelium in addition to phospholipase A2. The |
||||||||||
|
der emptying can be among the causes, ei- |
latter splits phosphatidylcholine to lysoleci- |
||||||||||
|
ther due to insufficient CCK being liberated |
thin (i.e., removal of the fatty acid at C2), |
||||||||||
|
(lack of free fatty acid [FFA] release in the lu- |
which in turn brings about acute cholecysti- |
||||||||||
|
men in pancreatic insufficiency), so that the |
tis. In some circumstances it may lead to |
||||||||||
|
main stimulus for gallbladder contraction is |
gallbladder perforation. |
|
|
|
|||||||
|
weakened, or because after nonselective va- |
Bacterial cholangitis usually occurs when |
||||||||||
|
gotomy the second most important contrac- |
bile flow is stopped because of cholelithiasis. |
||||||||||
|
tion signal, acetylcholine, is absent. Gallblad- |
A rise in pressure with dilation of the bile |
||||||||||
|
der contraction is also weakened in pregnan- |
ducts is the result, and posthepatic cholesta- |
||||||||||
|
cy. This means that not only occasional or ab- |
sis and biliary pancreatitis may also develop. |
||||||||||
|
sent emptying (see above) but also incom- |
In relatively rare cases gallbladder cancer |
||||||||||
|
plete emptying increases the duration for |
develops on the basis of gallstone disease. |
||||||||||
|
which bile remains in the gallbladder. As a re- |
|
|
|
|
|
|
|
|
|||
|
sult, there is enough time for the precipitated |
|
|
|
|
|
|
|
|
|||
|
crystals to form large concrements. A raised |
|
|
|
|
|
|
|
|
|||
|
mucus secretion (stimulated by prostaglan- |
|
|
|
|
|
|
|
|
|||
|
dins) can thus lead to an increased number |
|
|
|
|
|
|
|
|
|||
|
of nuclei of crystallization. |
|
|
|
|
|
|
|
|
|
||
|
Possible |
consequences of |
cholelithiasis |
|
|
|
|
|
|
|
|
|
|
are (→ E): |
|
|
|
|
|
|
|
|
|
|
|
|
Colic. When the cystic duct or the com- |
|
|
|
|
|
|
|
|
|||
|
mon bile duct is transiently blocked by a |
|
|
|
|
|
|
|
|
|||
|
stone, pressure rises in the bile ducts and in- |
|
|
|
|
|
|
|
|
|||
|
creased peristaltic contraction in the region |
|
|
|
|
|
|
|
|
|||
|
of the blockage causes severe visceral pain |
|
|
|
|
|
|
|
|
|||
166in the epigastric area, possibly with radiation into the back, as well as vomiting (→ p.140).
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.