

книги студ / Color Atlas of Pathophysiology (S Silbernagl et al, Thieme 2000)
.pdfA. Causes and After-effects of Bleeding Tendency |
|
|
|
Vascular disorder |
Thrombocyte deficiency |
|
or defect |
|
Lack of |
|
|
plasma factors |
|
|
Mainly joint bleedings |
Bleeding tendency |
|
and blue-black spots |
|
|
|
(hemorrhagic |
Mainly petechial hemorrhages |
|
diathesis) |
|
B. Clotting Tests for Diagnosing Plasmatic Hemorrhagic Diatheses
|
|
|
Common pathway |
|
Endogenous system: |
||
Exogenous system: |
for both systems: |
|
|||||
|
factors VIII, IX, XI, XII |
||||||
|
factor VII |
|
factors II, V, X |
|
|||
|
|
|
as well as HMK |
||||
|
|
|
|
as well as |
|
and prekallikrein |
|
|
|
|
|
Fibrinogen |
|
||
|
|
|
|
|
|
||
|
Quick value |
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Partial thrombo- |
|
|
|
|
Thrombin time |
|
|
plastin time (PTT) |
|
|
|
|
|
|
|
|
Plate 3.17 Hemostasis I
C. Interpretation of Clotting Test Results
Quick |
PTT |
Thrombo- |
Bleeding |
Probable causes |
|
of hemorrhagic diathesis |
|||||
value |
|
cyte count |
time |
||
|
(applied to moderate to severe disorders) |
||||
|
|
|
|
||
normal |
normal |
normal |
normal |
Vascular cause, factor XIII deficiency |
|
|
normal |
normal |
normal |
Factor VII deficiency |
|
normal |
|
normal |
normal |
Heparin administration, deficiency of factor VIII, IX, XI, |
|
|
XII, HMK or prekallikrein |
||||
|
|
|
|
||
normal |
normal |
|
|
Thrombocytopenia |
|
|
|
normal |
normal |
Administration of coumarin derivatives, |
|
|
|
vitamin K deficiency, factors I, II, V, X deficiency |
|||
|
|
|
|
||
normal |
|
normal |
|
v. Willebrand’s disease |
|
|
|
|
|
Liver damage, consumption coagulopathy, sepsis |
|
|
|
|
(after E. Lechler) |
|
|
reduced |
|
prolonged |
61 |
|
|
|
||||
|
|||||
|
|
|
|
|
|
|
|
|
|
Photo : Siegenthaler W. Differentialdiagnose innerer Krankheiten. |
|
|
|
|
|
17th ed. Stuttgart: Thieme; 1993. |
|
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

3 Blood
62
!
A bleeding tendency (hemorrhagic diathesis [HD]) may be caused by disorders of the coagulation or fibrinolysis system (plasma HD) as well as disorders of TC (thrombocytic HD), or of vascular defects (vascular HD). While in plasma disorders minimal mechanical injury result in hematomas (bruises) and bleeding into joints, thrombocytic and vascular HDs are characterized by punctuate, tiny insect bite–like cutaneous bleedings (petechiae; → A, photo).
The probable cause of a more serious HD can be elucidated with a few simple clotting tests (→ B). In the Quick test, plasma is transiently made incoagulable with substances that form complexes with Ca2+ (citrate, oxalate, or EDTA); an excessive amount of Ca2+ and tissue thrombokinase are then added and the resulting clotting time is compared with serial dilutions of normal plasma. If, for example, the test plasma coagulates after the same time interval as 1:1 diluted normal plasma, the Quick value is 50% (normal range: 70– 125%). Low Quick values denote that either factor VII (exogenous system) or the cascade initiated by factor X is abnormal or is influenced by vitamin K antagonists (→ B,C,D). To measure partial thromboplastin time (PTT), kephalin, kaolin (substitute for contact activation), and Ca2+ are added to the citrated plasma and the time until clotting (= PTT) is measured (normal: 25 – 38 s). If it is increased, the disorder lies either in the endogenous activation or in the common final pathway from factor X onward (→ B, C). To measure (plasma) thrombin time, thrombin is added to the citrated plasma and the clotting time determined (normal: 18 – 22 s); the result can reveal fibrinogen deficiency (→ B), or can be used to monitor treatment with heparin, which enhances the inhibitory action of antithrombin III on thrombin (→ E). An abnormality in the platelets usually goes hand in hand with prolonged bleeding time (bleeding > 5 min after, e.g., a prick into the ear lobe). A TC deficiency (thrombocytopathy; < 50 × 103/µL blood) can be distinguished from an abnormality of platelet function (thrombocytopathy) by means of a platelet count (→ C).
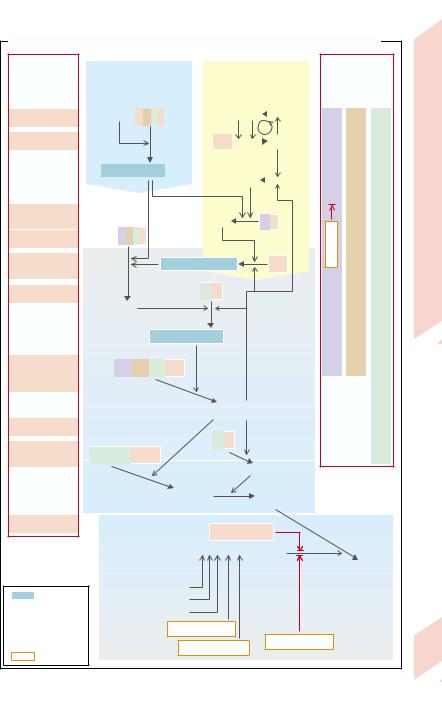
Plasma hemorrhagic diathesis (coagulopathies) are caused by congenital or acquired clotting factor deficiency. The hereditary co-
agulopathies (→ D1) can affect practically each of the plasma factors, but deficiency of some of the factors may produce relatively few symptoms (e.g., factors of the contact phase, factor XI). The most common (one of 10 000 newborn boys) of the the X-chromo- somal recessive forms is classical hemophilia (Type A). This was, for example, inherited from Queen Victoria by numerous male descendants of European royal houses (women are carriers). The most common bleeding sites are the muscles and the large joints of the leg, the latter becoming markedly deformed with time (hemophilic arthropathy). Hemophilia A is due to the absence, reduced formation, or defect of factor VIII. The fivefold rarer hemophilia B (factor IX deficiency) is similar in its mode of inheritance and symptoms to hemophilia A. The rare homozygous hereditary deficiency of factor I (afibrinogenemia), of factor II (hypoprothrombinaemia), of factors V, VII, and X leads especially to marked bleeding after severe injury or operations. Homozygous deficiency of α2-antiplasmin, an important inhibitor of fibrinolysis (→ D3) also results in a he- mophilia-like bleeding tendency. Factor XIII deficiency is characterized by fibrin instability so that bleedings occur only after a long interval (up to 11⁄2 days). The routine clotting tests are usually normal in factor XIII deficiency, because actual clotting is unchanged.
Acquired coagulopathies (→ D2) occur when formation of the various factors is reduced, when they are inhibited (e.g., by administration of heparin [→ E] or by immune coagulopathies, e.g., factor VIII antibodies), or if their consumption is high (consumption coagulopathy). As most of the clotting factors are formed in the liver, liver damage (in particular liver cirrhosis; → p.172ff.) results in clotting disorders. Simultaneously occurring portal hypertension further increases the risk of hemorrhages (mainly from esophageal varices; → p.170ff.) because platelets are sequestered in the enlarged spleen, resulting in thrombocytopenia (see below). As several clotting factors are vitamin K–dependent (see above), a coagulopathy can also be caused by deficiency or inhibition of vitamin K (→ D2). Causes of vitamin K deficiency are:
–obstructive jaundice, in which fat-soluble vitamins (e.g., vitamin K1 from green plants
!
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
D. Angeborener und erworbener Faktormangel als Ursache einer Blutungsneigung
1
angeborener Faktormangel
Faktor VII
Faktor XII
Faktor IX:
Hämophilie B
Faktor X
Faktor VIII:
Hämophilie A
Faktor V
Faktor II:
Hypoprothrombinämie
Faktor XIII
Faktor I:
Afibrinogenämie
a2-Antiplasmin
Komplex
PL Phospholipide  bewirkt
bewirkt  wird zu
wird zu  hemmt
hemmt
Medikament
exogene Aktivierung |
endogene Aktivierung |
2 |
||||||||||||
erworbener |
||||||||||||||
(Gewebsverletzung) |
|
(Kollagenkontakt) |
||||||||||||
|
Faktormangel |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
Kontaktphase |
durch: |
|||||||
|
|
|
|
|
|
|
||||||||
|
Ca2+, III |
|
|
|
|
HMK KK |
|
|
|
PKK |
|
|||
|
VII |
|
|
|
|
|
Wirkung-K- |
|||||||
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
XIIa |
||||
|
PL– Ca2+– VIIa |
|
|
|
|
|
||||||||
|
|
XII |
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
Vitamin |
|||||
|
|
|
|
|
|
|
XIa |
|
|
|
|
XI |
||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
||||
IXa |
IX |
Mangel-K-Vitamin Cumarine |
|
|
X |
|
|
|
|
PL– Ca2+– IXa – VIIIa |
VIII |
Leberschaden |
Verbrauchskoagulopathie |
|
V |
|
|||
|
|
|
|
|
Xa |
|
|
|
|
PL– Ca2+–Xa –Va |
|
|
|
|
Prothrombin (II) |
|
|
|
|
Fibrinbildung |
Thrombin (IIa) |
||
|
|||
|
|
XIII |
|
Fibrinogen (I) |
|
|
|
|
|
XIIIa |
|
Fibrinmonomer |
vernetztes |
||
Fibrin |
|||
|
|
||
3 |
|
a2-Antiplasmin |
|
Fibrinolyse |
|
||
Plasminogen  Plasmin
Plasmin
lösliche
GewebeplasminogenFibrinopeptide
aktivator
Plasmakallikrein
Urokinase
Streptokinase
Staphylokinase Tranexamsäure
Plate 3.18 Hemostasis II
63
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

3 Blood
!
or synthetic vitamin K3) fail to be absorbed due to the lack of bile salts (→ p.168);
–generalized malabsorption (→ p.152ff.);
–destruction by antibiotics of the intestinal flora, which through its synthesizing vita-
min K2 contributes significantly to supplying the body with this substance.
The inhibition of the vitamin K effect by coumarin derivatives (phenprocoumon, warfarin, acenocumarol) is used for oral prophylaxis of thrombosis (anticoagulant treatment).
Consumption coagulopathy (= disseminated intravascular coagulation; → D2) is a coagulation disorder caused by acute or chronic activation of thrombin with clot formation and platelet activation that secondarily results in hyperfibrinolysis. It is caused by large amounts of tissue thromboplastin entering the bloodstream, for example, in amniotic fluid embolism, extensive brain injury, malignant disease (e.g., leukemia), or sepsis (e.g., petechiae in meningococcal septicemia [Wa- terhouse–Friedrichsen syndrome]). Vascular causes are seen, for example, in aortic aneurysm (→ p. 236ff.), or in vascular malformations as well as in ABO blood group mismatches, and due to enzyme action with certain snake poisons.
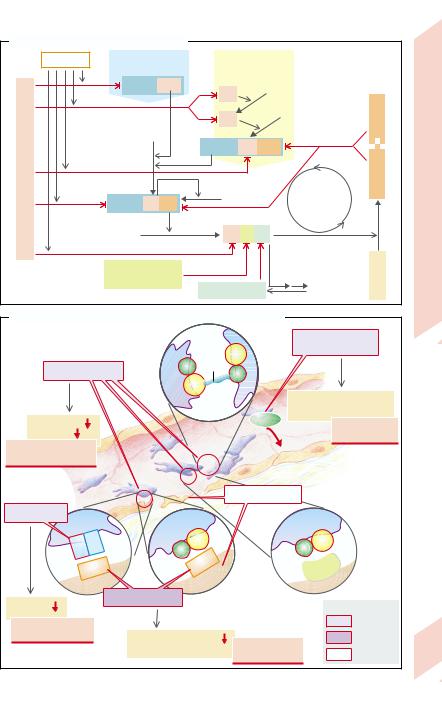
The two groups of hemorrhagic diathesis caused by platelet abnormalities are thrombocytopenias and thrombocytopathies. Acquired thrombocytopenias (TCPs) are the most common HD. TCP is due to diminished platelet formation (aplastic TCP, e.g., in bone marrow tumors, radiation damage, or cobalamine or folate deficiency), to increased platelet destruction (thrombocytoclastic TCP), or platelet sequestration in an enlarged spleen. Markedly increased bleeding tendency occurs when the number of platelets falls below 20 × 103/µL. Idiopathic TCP (Werlhof’s disease) is relatively frequent, its acute form developing one to three weeks after a viral infection (shortened
platelet survival time due to immune complexes). The chronic form occurs as an autoimmune disease. Drug allergy can produce TCP through the action of drugs (e.g., quinine or sulfonamides) as haptens (→ p. 52). Acquired thrombocytopathies occur in uremia and dysproteinemia (platelet coating). They can also be caused by such drugs as acetylsalicylic acid via their inhibitory effect on cyclo-oxygenase, an effect that is used in thrombosis prophylaxis.
Congenital thrombocytic HDs are the auto- somal-dominant and autosomal-recessive hereditary thrombocytopenias (abnormal platelet production) with the following functional disorders:
–Membrane defects such as 1) deficiency of platelet glycoprotein Ib (→ F1) that disturb adhesion (Bernard–Soulier syndrome); 2) deficiency of glycoprotein complex IIa/IIIb (→ F2), which inhibits aggregation and adhesion (Glanzmann–Naegeli thrombasthenia);
–Diverse defects of storage or secretion, for example, deficiency of cyclo-oxygenase and thromboxane synthetase, in which ADP release is reduced (storage pool deficiency);
(→ F3).
Among the forms of HD of vascular cause are the different kinds of hereditary von Willebrand’s (vW) disease, a defect of vascular endothelium in which the vW factor is reduced or defective (→ F4). This weakens platelet adhesion and secondarily leads to factor VIII deficiency, because the vW factor acts as a kind of carrier for this factor (complex formation). Finally, there are a number of functional disorders and tissue changes in the vascular wall and connective tisue that are either congenital (purpura simplex; Osler–Weber–Rendu disease; Schönlein–Henoch disease), or acquired (scurvy in vitamin C deficiency; drug-medi- ated immune reactions).
64
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
E. Inhibition of Blood Clotting System |
|
|
|
|
|
Heparin |
Exogenous |
Endogenous |
|
|
|
|
activation |
activation |
|
|
|
|
PL– Ca2+– VIIa |
XIIa |
XI |
|
|
|
|
|
|
||
|
|
|
|
S |
|
|
|
|
|
|
|
|
|
XIa |
|
IX |
Protein |
|
|
|
|
||
|
|
|
|
|
|
III |
X |
|
|
|
|
|
PL– Ca2+– IXa – VIIIa |
|
|
||
Antithrombin |
PL– Ca2+– Xa –Va |
V |
|
feed-back |
Protein Ca |
|
|
|
|
Negative |
|
Prothrombin |
Thrombin |
Thrombomodulin |
|
||
|
|
|
|
||
α2 |
macroglobulin |
|
|
C |
|
Fibrinopeptides |
Fibrin |
Protein |
|||
α1 |
antitrypsin |
||||
F. Causes of Thrombocytic and Vascular Bleeding Tendency
|
|
|
Fibrinogen |
3 |
Defects of storage |
|
|
|
|
IIIa |
and secretion |
|
|
|
|
|
|
2 |
IIb/IIIa reduced |
IIb |
|
|
|
|
|
|
|
|
|
|
|
Thromboxan synthetase and |
||
|
|
|
cyclooxygenase deficiency |
|
|
Aggregation |
|
|
ADP |
Storage pool |
|
|
|
|
|
||
Adhesion |
|
|
|
deficiency |
|
Glanzmann-Naegeli |
|
Endothelium |
|
|
|
thrombasthenia |
|
|
|
|
|
|
|
Extracellular |
|
||
|
|
Thrombo- |
|
||
|
|
matrix |
|
||
|
|
cytes |
|
|
|
1 |
|
|
Endothelial defect |
|
et al.) |
Ib reduced |
|
|
|
|
|
Ib |
IX |
IIIa |
IIIa |
|
Heimpel |
IIb |
IIb |
|
after |
||
|
|
|
|||
|
|
vWF |
|
|
|
|
vWF |
Fibronectin |
(mod. |
||
|
4 |
||||
|
|
||||
|
|
|
|
|
|
Adhesion |
|
vWF reduced |
|
|
|
|
|
|
Causes: |
|
|
Bernard-Soulier |
|
|
|
thrombocytic |
|
|
|
|
|
|
|
syndrome |
|
Thrombocyte adhesion |
|
vascular |
|
|
|
v. Willebrand’s |
|
||
|
|
F VIII deficiency |
various |
|
|
|
|
disease |
|
||
|
|
|
|
||
Plate 3.19 Hemostasis III
65
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
4 |
Respiration, Acid–Base Balance |
F. Lang |
 Overview
Overview
Breathing through the lungs has two functions: firstly, to supply O2 to the blood and, secondly, to regulate the acid–base balance via the CO2 concentration in the blood. The mechanics of breathing serve to ventilate the alveoli, through whose walls O2 can diffuse into the blood and CO2 can diffuse out. Respiratory gases in the blood are largely transported in bound form. The amount transported depends, among other factors, on the concentration in blood and on pulmonary blood flow (perfusion). It is the task of respiratory regulation to adapt ventilation to the specific requirements.
A number of disorders can affect breathing in such a manner that ultimately sufficient O2 uptake and CO2 release can no longer be guaranteed.
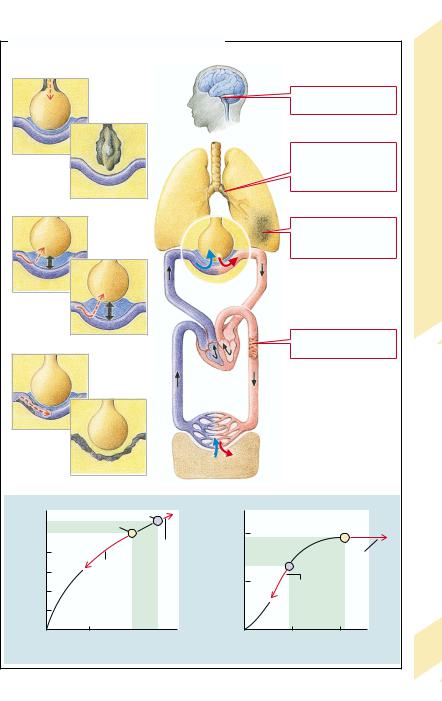
In obstructive lung disease (→p. 76) flow resistance in the respiratory tract is raised and ventilation of the alveoli is thus impaired (→A1). The primary consequence is hypoventilation in some alveoli (abnormal distribution; →p. 72) or of all alveoli (global hypoventilation). If alveolar ventilation ceases completely, a functional arteriovenous shunt occurs. However, hypoxia leads to constriction of the supplying vessels, thus diminishing blood flow to the underventilated alveoli.
In restrictive lung disease (→p. 74) the loss of functioning lung tissue reduces the area of diffusion and in this way impairs gaseous exchange. There is also a reduced area of diffusion in emphysema (→p. 78), a condition characterized by alveoli that have a large lumen but are also diminished in number. Disorders
of diffusion can also be caused by an increased distance between alveoli and blood capillaries (→A2; →p. 70,80). If alveoli and capillaries are completely separated from one another, this results in both a functional dead space (nonperfused alveoli) and an arteriovenous shunt.
Restrictive and obstructive lung disease as well as cardiovascular disease may affect lung perfusion (→A3; →p. 80). Decreased perfusion results in a reduced amount of gases being transported in blood, despite adequate O2 saturation and CO2 removal in the alveoli. If flow resistance is increased, severe consequences for the circulation are possible, because the entire cardiac output (CO) must pass through the lungs (→p. 80).
Breathing is also impaired in dysfunction of the respiratory neurons (→p. 82) as well as of the motoneurons, nerves, and muscles that are controlled by them (→p. 68). The changes in breathing movement that occur when the breathing regulation is abnormal (→Table 1) do not, however, necessarily lead to corresponding changes of alveolar ventilation.
Consequences of inadequate breathing can be hypoxemia (→A5; →p. 84), hypercapnia or hypocapnia (increased or decreased CO2 content, respectively; →A4; →p. 86ff.) in arterialized blood. The supply of O2 to the cells as well as the removal of CO2 from the periphery do not only depend on adequate respiration but also on unimpaired oxygen transport in the blood (→chap. 3) and on intact circulation (→chap. 7).
Table 2 Definition of Some Parameters
of Ventilation
|
Table 1 Terms for Various Breathing Activities |
||
|
|
|
|
|
|
Hyperpnea |
increased breathing movement |
|
|
Eupnea |
normal breathing movements |
|
|
Hypopnea |
decreased breathing movements |
|
|
Apnea |
arrested breathing |
|
|
Bradypnea |
decreased rate of breathing |
|
Tachypnea |
increased rate of breathing |
|
|
|
Dyspnea |
labored breathing |
|
|
|
(subjective feeling) |
|
|
Asphyxia |
inability to breathe |
66 |
Orthopnea |
labored breathing, except in |
|
|
|
the sitting or upright position |
|
|
|
|
|
|
|
|
|
Tidal volume (VT) |
volume of normal inspira- |
|
tion and expiration |
Vital capacity (VC) |
volume of maximal |
|
expiration after maximal |
|
inspiration |
Maximal breathing |
maximal ventilation (L/min) |
˙ |
achieved in a short period |
capacity (Vmax) |
|
|
of time (usually 10 s) |
Compliance (C) |
lung distensibility |
Forced expiration volume |
maximal volume expired |
(FEV1) |
in 1 second |
Functional residual |
total residual volume |
capacity (FRC) |
after normal expiration |
|
|
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
A. Pathophysiology of Respiration (Overview) |
|
|
|
|
|||||
|
|
|
|
|
|
|
Abnormal |
|
|
|
|
|
|
|
|
|
regulation of breathing |
|
|
|
|
1 |
|
|
|
|
|
Obstructive |
|
|
|
|
|
|
|
|
|
|
|
Abnormal |
|
|
|
|
|
|
lung diseases, |
|
|
ventilation |
|
|
|
|
|
|
increased |
|
|
|
|
|
|
|
|
|
|
flow resistance |
|
|
|
|
|
|
|
|
|
Restrictive |
Overview |
|
|
|
|
|
CO2O2 |
|
|
lung diseases, |
|
|
|
|
|
|
|
|
loss of tissue |
||
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
4.1 |
|
|
|
|
|
|
|
|
|
|
Abnormal |
|
|
|
|
|
|
|
Plate |
|
diffusion |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
Abnormal |
|
|
|
|
|
|
|
|
|
oxygen transport |
|
|
|
|
|
|
Heart |
|
|
|
|
|
|
3 |
|
|
Capillaries |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Abnormal |
|
|
|
|
|
|
|
|
|
perfusion |
|
|
|
|
O2 |
|
|
|
|
|
|
|
|
|
CO2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Tissue |
|
|
|
|
4 |
|
|
Venous |
|
5 |
|
|
|
|
in blood |
|
|
|
|
|
|
|
|
|
25 |
Arterial |
|
|
blood |
200 |
|
|
|
|
|
|
|
|
|
|
||||
20 |
|
|
Hypercapnia |
|
|
|
|
||
] (mmol/L) |
|
|
](mL/L) in |
|
|
Hyperoxemia (at >50 kPa) |
|
||
15 |
Hypocapnia |
|
100 |
|
|
||||
10 |
|
Hypoxemia |
|
||||||
|
|
|
|||||||
– 3 |
|
2 |
|
|
|||||
[HCO |
5 |
|
[O |
|
|
||||
|
|
|
|
||||||
|
0 |
2.5 |
5 |
7.5 |
|
0 |
5 |
10 |
|
|
|
PCO2 (kPa) in blood |
|
|
PO2 (kPa) in blood |
67 |
|||
|
|
|
|
|
|
|
|
1 kPa = 7.5mmHg |
|
|
|
|
|
|
|
|
|
|
|
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

Ventilation, Perfusion
|
To reach the alveoli, inspired air must pass |
|
|
through those respiratory pathways in which |
|
|
no gaseous exchange takes place (dead space), |
|
|
i.e., normally the mouth, pharynx and larynx, |
|
|
trachea, bronchi and bronchioles. On its way |
|
|
the air will be warmed, saturated with water |
|
|
vapor, and cleansed. |
|
Balance |
The tidal volume (VT) contains, in addition |
|
to the volume of air that reaches the alveoli |
||
|
||
|
(VA), the volume of air that remains in the |
|
Acid–Base |
dead space (VD). If tidal volume is less than VD |
|
veolar ventilation rises with increasing VT. Al- |
||
|
(normally ca. 150 ml), the alveoli are not venti- |
|
|
lated with fresh air (→A, right). When tidal |
|
|
volume is greater than VD, the proportion of al- |
|
Respiration, |
veolar ventilation may even be reduced during |
|
either physiologically (e.g., during work) or |
||
|
hyperpnea, if the depth of each breath, i.e., VT, |
|
|
is low and mainly fills the dead space. |
|
|
Increased ventilation can occur as a result of |
|
4 |
pathophysiologically (e.g., in metabolic acido- |
|
|
sis; →p. 88) increased demand, or due to an |
|
|
inappropriate hyperactivity of the respiratory |
|
|
neurons (→p. 82). |
|
|
Decreased ventilation can occur not only |
|
|
when the demand is reduced, but also when |
|
|
the respiratory cells are damaged, or when |
|
|
neural or neuromuscular transmission is ab- |
|
|
normal. Further causes include diseases of the |
|
|
respiratory muscles, decreased thoracic mobi- |
|
|
lity (e.g., deformity, inflammation of the |
|
|
joints), enlargement of the pleural space by |
|
|
pleural effusion or pneumothorax (→p. 74) as |
|
|
well as restrictive or obstructive lung disease |
|
|
(→p. 74ff.). |
|
|
Changes in alveolar ventilation do not have |
|
|
the same effect on O2 uptake into the blood |
|
|
and CO2 release into the alveoli. Because of |
|
|
the sigmoid shape of the O2 dissociation curve, |
|
|
O2 uptake in the lungs is largely independent |
|
|
of alveolar partial pressure (PAO2). If there is |
|
|
only minor hypoventilation, the partial pres- |
|
|
sure of O2 in the alveoli and thus in blood is re- |
|
|
duced, but the O2 dissociation is at the flat part |
|
|
of the curve, so that the degree of hemoglobin |
|
|
saturation and thus O2 uptake in blood is prac- |
|
|
tically unchanged (→B, right). On the other |
|
68 |
hand, the simultaneous increase in CO2 partial |
|
pressure in the alveoli and blood leads to a no- |
ticeable impairment of CO2 release (→B, left).
Massive hypoventilation lowers the O2 partial pressure in the alveoli and blood, so that oxygen is at the steep part of the O2 binding curve of hemoglobin and O2 uptake is therefore impaired much more than CO2 release is. Hyperventilation increases the O2 partial pressure in the alveoli and blood, but cannot significantly raise the level of O2 uptake into the blood because the hemoglobin is already saturated. However, hyperventilation boosts CO2 release.
Lung perfusion is increased, for example, during physical work. It can be reduced by heart or circulatory failure (→p. 224), or by constriction or occlusion of pulmonary vessels (→p. 80).
A moderate increase in lung perfusion while ventilation remains unchanged increases O2 uptake virtually in proportion to the amount of blood flow (→C, right). Even though the alveolar O2 partial pressure falls slightly because of the increased O2 uptake from the alveoli into the blood, this has little influence on O2 saturation in the blood (see above). It is only when the alveolar partial pressure of O2 falls into the steep part of the O2 dissociation curve that a decrease of alveolar O2 partial pressure significantly affects O2 uptake into blood. At those O2 partial pressures a further increase in lung perfusion only slightly increases O2 uptake. Furthermore, at very high lung perfusion flow, the contact time in the alveoli is not sufficient to guarantee that partial O2 pressure in blood approaches that in the alveoli (→p. 70). If lung perfusion is reduced, O2 uptake is proportionally decreased.
CO2 removal from blood is dependent on lung perfusion (→C, left) to a lesser extent than O2 uptake. In case of reduced lung perfusion (but constant ventilation and venous CO2 concentration) the CO2 partial pressure in the alveoli falls and thus favors the removal of CO2 from the blood. This, in turn, attenuates the effect of the reduction in perfusion. At raised lung perfusion an increase of alveolar CO2 concentration prevents a proportional rise in CO2 release.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.
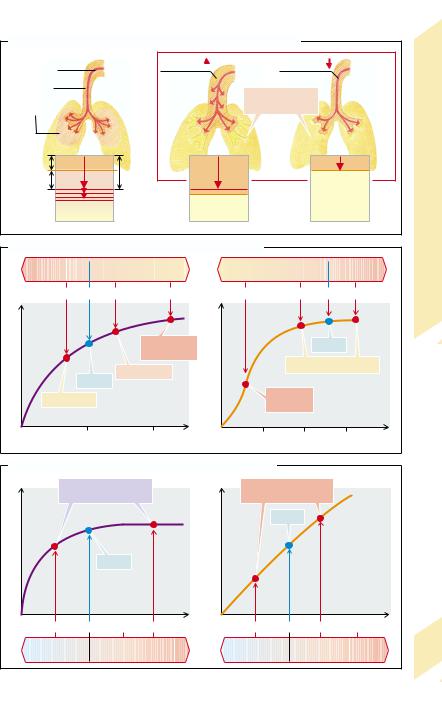
A. Dead Space (VD), Alveolar Volume (VA) and Tidal Volume (VT)
Tidal |
Dead space |
|
Tidal volume |
volume (VT) |
|
||
|
|
|
|
Dead |
|
|
|
space (VD) |
|
|
No alveolar |
Alveolar proportion |
|
|
|
of VT (VA) |
|
|
ventilation |
VD |
|
|
|
|
|
|
|
|
|
|
Perfusion |
|
VA |
|
VT |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
||
VA = VT – VD |
|
|
|
VD > VT |
|
|
|
VT = VD |
Ventilation, |
|||
B. O2 and CO2 of Arterial Blood in Abnormal Ventilation |
|
|
|
|
|
|||||||
Hyper- |
|
|
|
Hypo- |
Hypo- |
|
|
|
|
Hyper- |
||
|
|
|
|
|
|
|
|
|||||
ventilation |
|
|
ventilation |
ventilation |
|
|
ventilation |
4.2 |
||||
15 |
10 |
7 |
(L/min) |
4.5 |
4.5 |
(L/min) |
7 |
10 |
15 |
|||
Plate |
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
||
]blood |
|
|
Severe |
] blood |
|
|
Normal |
|
|
|||
|
|
hypercapnia |
|
|
HbO2 |
|
|
|||||
|
|
|
|
|
|
|
|
|||||
2 |
|
Hypercapnia |
2 |
|
|
practically unchanged |
|
|||||
[CO |
Normal |
[HbO |
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|||
Hypocapnia |
|
|
|
|
|
Severe |
|
|
|
|||
|
|
|
|
|
hypoxia |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|||
|
5 |
|
10 |
|
|
5 |
|
10 |
15 |
|
||
|
PCO2 blood (kPa) |
|
|
|
PO2 blood (kPa) |
|
|
|||||
C. CO2 Release and O2 Uptake at Different Perfusion Levels |
|
|
|
|
|
|||||||
Minor effect |
|
|
|
|
Marked effect |
|
|
|
||||
on CO2 output |
|
|
|
on O2 uptake |
|
|
|
|||||
|
|
|
|
|
|
|
Normal |
|
|
|
||
output |
Normal |
|
uptake |
|
|
|
|
|
|
|||
2 |
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|||
CO |
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
||
3 |
6 L/min 9 |
12 |
|
3 |
|
6 L/min 9 |
|
12 |
|
|||
Reduced |
|
|
Increased |
Reduced |
|
|
|
Increased |
69 |
|||
perfusion |
|
|
perfusion |
perfusion |
|
|
|
perfusion |
||||
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.

4 Respiration, Acid–Base Balance
70
Diffusion Abnormalities
O2 has to diffuse from the alveoli to hemoglobin in the erythrocytes, and CO2 from the erythrocytes into the alveoli. The amount of gas (M˙ ) that diffuses across the diffusion barrier between alveoli and blood per unit time is proportional to the diffusion area (F) and the difference in partial pressure between alveolar gas (PA) and blood (Pblood), and inversely proportional to the length of the diffusion pathway (d):
M˙ = K × F (PA – Pblood)/d.
Krogh’s diffusion coefficient K is about 20 times greater for CO2 than for O2. The diffusion capacity D (= K × F/d) is about 230 mL × min–1 × kPa–1 (1.75 L × min–1 × mmHg–1) in a healthy person.
A diffusion abnormality exists when the ratio of diffusion capacity to lung perfusion (or cardiac output) is reduced.
The diffusion capacity may be reduced by increased distance (→A). When a pulmonary edema occurs (→p. 80), raised intravascular pressure means plasma water is exuded into the interstitial pulmonary tissue or into the alveoli, and thus increases the diffusion distance. Inflammation causes a widening of the space between alveoli and blood capillaries as a result of edema and the formation of connective tissue. In interstitial lung fibrosis (→ p. 74), the connective tissue forces alveoli and blood capillaries apart. It is the distance between hemoglobin and alveolar gas which matters. Thus, the distance can also be slightly increased by vessel dilation (inflammation) or anemia.
A diminished diffusion capacity may also be caused by a reduction of the diffusion area
(→A), as after unilateral lung resection, loss of alveolar septa (pulmonary emphysema; →p. 78), or in loss of alveoli in pneumonia, pulmonary tuberculosis, or pulmonary fibrosis (see above). The diffusion area can also be reduced by alveolar collapse (atelectasis; →p. 72), pulmonary edema, or pulmonary infarction (→p. 80).
Diffusion abnormalities become obvious when cardiac output is large (→A), blood flows rapidly through the lungs, and the contact time of blood in the alveoli is thus quite
brief. In effect, diminution of the diffusion area (e.g., after unilateral lung resection) also means a shorter contact time in the remaining lung tissue, because the same amount of blood will now pass through a reduced amount of lung tissue per unit of time. An increased O2 demand during physical exercise forces an increase in cardiac output and can thus reveal a diffusion abnormality.
Abnormal diffusion primarily affects O2 transport. In order for the same amount of gas to diffuse per time, the O2 gradient must be twenty times greater than the CO2 gradient. Should the diffusion capacity in an alveolus be diminished while ventilation remains constant, O2 partial pressure will fall in the blood leaving the alveolus. If all alveoli are similarly affected, O2 partial pressure will fall in the pulmonary venous (and thus systemic arterial) blood. If O2 consumption remains constant, O2 partial pressure will necessarily be lower also in deoxygenated (systemic venous) blood (→B2). For this reason patients with a diffusion abnormality get blue lips on physical exertion (central cyanosis; →p. 84). The primary effects of abnormal diffusion on CO2 transport and acid–base metabolism are much less marked. Hypoxia stimulates the respiratory neurons, and the resulting increase in ventilation can produce hypocapnia. However, the hypoxemia due to abnormal diffusion can only be slightly improved by hyperventilation. In the example given (→B3), doubling of the alveolar ventilation at unchanged O2 consumption increases alveolar O2 partial pressure by only 4 kPa to 17 kPa (30 mmHg to 129 mmHg), but the increased O2 gradient does not normalize the O2 saturation of the blood. At the same time, respiratory alkalosis develops, despite the abnormal diffusion, because of the increased CO2 removal (→p. 86). Hypoxemia due to abnormal diffusion can be neutralized with O2-enriched inspiratory air (→B4). The degree of hypoxemia can be lessened by decreasing O2 consumption.
Silbernagl/Lang, Color Atlas of Pathophysiology © 2000 Thieme
All rights reserved. Usage subject to terms and conditions of license.