

Сорбция воды полимером может вызывать его набухание, сопровождающееся увеличением массы, объема, изменением структуры. Предельным случаем набухания является растворение полимера.
Полимеры, применяемые в качестве конструкционных материалов и защитных покрытий, не растворяются в воде, а лишь ограниченно набухают.
Сорбируемая вода может вызывать вымывание водорастворимых ингредиентов – пластификаторов, стабилизаторов, наполнителей, красителей и т.д. По отношению к ряду полимеров вода может быть не только физически, но и химически активной средой.
Под
воздействием воды в полимерах могут
протекать реакции гидролиза. При
гидролизе вода присоединяется по месту
разрыва связей. Гидролизу, как правило,
подвержены гетероцепные полимеры,
имеющие гетероатом в основной цепи.
Например, к гидролизу наиболее
чувствительны соединения, содержащие
ацетальные
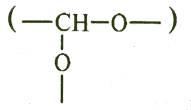 , амидные
, амидные![]() , с
, с










 ложные
эфирные
ложные
эфирные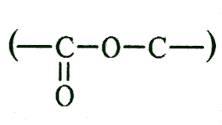 и простые эфирные
и простые эфирные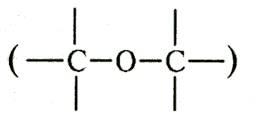
группы. Однако реакции гидролиза в чистой воде для большинства полимеров протекают слишком медленно, и они практически устойчивы к химическому воздействию воды.
Гидролиз катализируется в присутствие кислот и оснований. Особенно сильно катализируют гидролиз HCl,H2SO4,HF.
Введение в полимер алифатических звеньев – (СН2)2– или – (СН2)4– повышает подвижность молекул, следовательно, и проницаемость материала. Водостойкость полимеров увеличивается с введением в полимерную цепь ароматических звеньев. Например, высокую устойчивость к гидролизу проявляют ароматические полиамиды.
Особую роль играет морфология полимера, так как вода проникает только в аморфную часть полимера.
Примером синтетических смол, выдерживающих действие воды даже при температурах ее кипения, являются фенолоформальдегидные смолы.
Если вода не вступает с полимером в химическое взаимодействие и не происходит при этом сколько-нибудь заметных структурных превращений, его прочность не претерпевает существенных изменений при длительном контакте с водой (рис. 17.2).
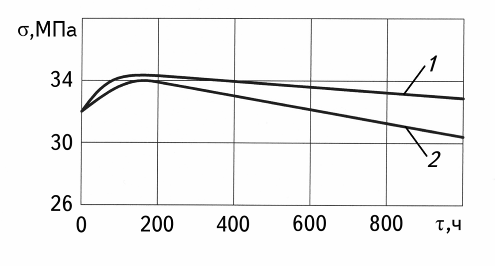
Рис.17.2. Зависимость разрушающего напряжения от времени выдержки полипропилена на воздухе (1) и в воде (2) при температуре 90 0С.
Наличие экстремума при выдержке в течение до 200 – 400 часов связывается с пластифицирующим действием воды, что приводит к увеличению гибкости и облегчению ориентации молекул при растяжении, а также к равномерному распределению напряжений в материале. К этому же эффекту приводит и просто нагревание.
В дальнейшем происходит некоторое ослабление сил взаимодействия между структурными элементами и макромолекулами в материале, вследствие дезориентационных процессов, обусловленных различной степенью набухания материала по толщине, - все это, в конечном итоге, ведет к незначительному снижению прочности.
Однако, при существенном пластифицирующем действии воды, вызывающем сильное набухание, наблюдается и резкое снижение прочностных характеристик полимера.
Водостойкость композиционных материалов обусловлена не только вышеуказанными процессами, протекающими в полимерном связующем (матрице) и армирующем наполнителе, но и нарушением под действием воды адгезионной связи на поверхности их раздела.
Влияние воды на свойства полимерных композиционных материалов можно рассмотреть на примере стеклопластиков. Контакт их с водой приводит к набуханию связующего, проникновению воды к границе раздела стеклянное волокно – полимерное связующее и нарушению адгезионной связи на поверхности раздела.
На поверхности раздела между гидрофильным стеклянным наполнителем и связующим происходит скопление молекул воды в виде капель или пленки. Вода вызывает разрушение стеклянных волокон в результате гидролиза. Интенсивность этого процесса зависит от химического состава стекол (рис. 17.3).Так у стеклопластика, сформированного на стеклянных волокнах щелочного состава (кривая 2, рис. 17.3) снижение модуля упругости идет более резко, чем у стеклопластика на бесщелочном волокне (кривая 1, рис. 17.3).

Рис.17.3. . Изменение модуля упругости стеклопластиков в парах воды при 300С.
эпоксидная смола ЭД – 20, отвержденная на бесщелочном стекле;
эпоксидная смола ЭД – 20, отвержденная на щелочном стекле;
эпоксидная смола ЭД – 20 без стеклонаполнителя.
Накопление воды на гидрофильных центрах поверхности стеклянных волокон может привести к возникновению осмотического давления, достаточного для расслоения композиционного материала. Связь на границе раздела, а вместе с тем механические свойства и водостойкость стеклопластиков, могут быть повышены за счет обработки стеклянных волокон специальными химическими соединениями (гидрофобизаторы, аппреты). Так на рис. 17.4 приведены данные по сорбции воды стеклопластиками, у которых волокна не обрабатывались (кривые 1, 2), и стеклопластиков, у которых волокна обработаны аппретом АМГ – 9 (крива 3) и гидрофобизатором – триметилхлорсиланом (кривая 4).
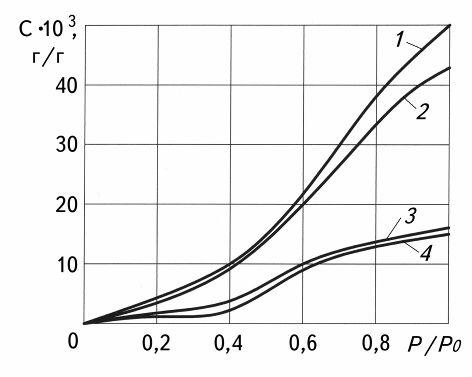
Как видно, при обработке стеклянных волокон водопоглощение стеклопластиков уменьшается более чем в 2 раза, и, главное, оно становится независимым от типа волокон. Это дает возможность использовать для изготовления изделий более дешевые волокна, например, щелочные.
Для повышения водостойкости полимеров используют нанесение водостойких и гидрофобных покрытий, термическую обработку, облучение и др.
Взаимодействие неметаллических материалов с растворами электролитов и другими жидкими средами.
Водные растворы электролитов (кислоты, основания, соли) наиболее широко распространены в различных технологических процессах и являются наиболее агрессивными по отношению к неметаллические материалам. Помимо химического воздействия, растворенные в воде щелочи, органические кислоты и другие соединения по отношению к неметаллам являются сильными поверхностно-активными веществами.
Водные растворы электролитов, являясь многокомпонентными системами, обладают специфическими особенностями сорбции и переноса в неметаллических материалах. В этой связи целесообразно рассмотреть общие закономерности взаимодействия неметаллических материалов с представителями различных электролитов, объединенных в определенные группы по общим характерным признакам.
Прежде всего, поведение электролита в контакте с неметаллическими материалами зависит от силы его взаимодействия с водой.
Сила такого взаимодействия определяется значениями упругости паров электролита и воды над раствором. Растворы с высокой упругостью паров над раствором (например, HCl, HF, HNO3,CH3COOH,NH3и другие) называют летучими электролитами, а с низкой упругостью пара над раствором (H2SO4,H3PO4,NaOH,NaCl,K2Cr2O7и др.) относят к нелетучим электролитам.
Следующий важный фактор – это степень диссоциации электролита, определяющая состав кинетических единиц в растворе и, следовательно, интенсивность их переноса и химического взаимодействия с материалом.
По степени диссоциации электролиты делятся на сильные, полностью диссоциированные в растворе (например, NaCl), и слабые, частично диссоциированные (например, H2SO4, H3PO4).
Кроме того, растворы электролитов оценивают так же по виду взаимодействия с неметаллическими материалами. Для каждой конкретной системы материал – раствор важно знать будет ли преобладать физическое взаимодействие или химическое, т.е. является ли среда по отношению к материалу физически или химически активной.
Если раствор является химически активным, следует оценить характер химического взаимодействия. Например, для полимеров выделяют гидролизующие или окисляющие растворы; для силикатных материалов – выщелачивающие растворы или разрушающие кремнеземный каркас(NaOH и HF).
В любом случае интенсивность взаимодействия неметаллических материалов с растворами электролитов в значительной степени определяется формой существования раствора в материале и механизмом его переноса.
Особенность переноса этих растворов заключается в том, что диффузионный поток зависит не только от взаимодействия его частиц с неметаллическим материалом, но и от их взаимодействия друг с другом в растворе и в материале.
17.5.1 Перенос растворов электролитов в неметаллических материалах.
Как и в случае переноса газов и воды, механизм этого процесса для растворов электролитов определяется соотношением плотной и пористой доли материала.
В пористых материалах, в которых размер пор обуславливает вязкостное течение раствора под гидростатическим напором, поток будет зависеть от вязкости и плотности раствора. Помимо гидростатического напора на интенсивность потока будет оказывать влияние давление насыщенных паров. Поэтому в пористых материалах при наличии фазового потока электролита у летучих электролитов интенсивность потока будет выше, чем для потока нелетучих электролитов в аналогичных условиях. Это отличие особенно ощутимо при малых гидравлических напорах жидкости. Интенсивность переноса растворов электролитов в макропористых неметаллах зависит от уровня заполнения раствором сечения пор. Если жидкость полностью заполняет это сечение, то часть ее, взаимодействуя со стенками пор, ограничена в подвижности, что ослабляет конвективные потоки жидкости.
Если раствор не полностью заполняет макрокапилляры, присутствующая газовая или парогазовая фаза будет способствовать появлению конвективных потоков в пористой среде.
С уменьшением средних размеров пор в неметаллических материалах свободное их заполнение раствором электролита ограничивается.
Когда размер микрокапилляров становится меньше 200 нм, их заполнение уже происходит только за счет адсорбции компонентов раствора на стенках капилляров. В этом случае количество адсорбированных частиц будет целиком определяться парциальным давлением их паров над раствором и уровнем сил адсорбционного взаимодействия с материалом. Величина парциального давления паров над раствором зависит от летучести электролита и концентрации раствора. Уровень адсорбционного взаимодействия определяется гидрофильностью или гидрофобностью материала. Поэтому процессы переноса растворов электролита, связанные так или иначе с адсорбционными явлениями, принято рассматривать отдельно для гидрофобных и гидрофильных неметаллических материалов, подразделяя растворы на летучие и нелетучие.
Над растворами нелетучих электролитов парциальное давление паров воды намного превышает парциальное давление паров электролита, которое практически равно нулю. В этом случае на поверхности материала адсорбируются преимущественно пары воды. При низких значениях парциального давления воды (Р/Р0 <0,25), что наблюдается у концентрированных растворов нелетучих электролитов, вода адсорбируется на поверхности микродефектов в виде монослоя. Для разбавленных растворов (Р/Р0>0,25) при адсорбции образуются полимолекулярные слои воды. При сильном разбавлении (Р/Р0>0,75) становится возможной капиллярная конденсация влаги в микрокапиллярах материала.
Полимерные материалы и композиты на их основе, которые способны сорбировать воду за счет ее молекулярного распределения и переноса в свободном объеме, будут поглощать из нелетучих электролитов в основном только воду. Стало быть, в таком случае речь идет только о водостойкости (водопроницаемости) материала.
Проницаемость нелетучего электролита возможна только тогда, когда в материале имеются сквозные микрокапилляры, заполненные водой, или другие протяженные образования агрегированной воды, по которым возможен перенос электролита. Однако, как правило, при контакте с нелетучими электролитами максимальная сорбция, набухание и проницаемость неметаллов наблюдается в разбавленных растворах, что обусловлено преимущественной ролью воды в этих процессах.
Увеличение сопротивляемости неметаллических материалов действию нелетучих электролитов может быть достигнуто путем их гидрофобизации.
В полярных полимерах и композиционных материалах на основе полиэфирных и эпоксидных смол, фиксируется перенос таких нелетучих электролитов, как серная и фосфорная кислоты, хотя их проницаемость на несколько порядков ниже проницаемости воды и летучих электролитов.
Над растворами летучих электролитов содержатся пары воды и электролита; соотношение между ними зависит от концентрации раствора. Над разбавленными растворами парциальное давление паров воды превышает парциальное давление паров электролита. С повышением концентрации это соотношение меняется в обратную сторону. Поэтому, когда с раствором электролита контактирует микропористый неметаллический материал, то капиллярная конденсация воды в микропорах будет происходить только в случае воздействия разбавленных растворов (Р/Р0>0,75). Летучий электролит при этом может находиться и диффундировать в капиллярах в диссоциированном состоянии. Такой перенос разбавленных летучих электролитов наиболее характерен для полярных неметаллических материалов.
С повышением концентрации раствора летучего электролита на стенках капилляров будут формироваться полимолекулярные, а с дальнейшим ростом концентрации – мономолекулярные слои воды. Капилляры в этих случаях будут не полностью заполнены влагой, и перенос летучего компонента будет осуществляться в газообразном состоянии в свободном пространстве протяженных капилляров. Следует все же учитывать, что часть летучего вещества будет абсорбироваться полимолекулярными слоями воды, находящимися на стенках капилляров. В микропористых материалах поток электролита с ростом концентрации раствора будет увеличиваться. Для значений коэффициентов проницаемости электролита в таких материалах характерен большой разброс, а также их зависимость от толщины образцов.
Таким образом, интенсивность переноса растворов электролитов в капиллярно-пористых неметаллических материалах целиком зависит от степени заполнения микрокапилляров водой. При капиллярной конденсации влаги в микропорах облегчается перенос нелетучих электролитов и затрудняется перенос летучих электролитов.
Летучие электролиты, обладая малой работой выхода из растворов (легко испаряются), адсорбируются на поверхности неметаллического материала в количестве, пропорциональном давлению их паров над раствором. Эти молекулы способны внедряться в свободный объем полимерных материалов и диффундировать аналогично диффузии молекул газа или воды.
Особенностью диффузионного переноса летучих электролитов в полимерных и композиционных материалах на их основе является взаимодействие молекул электролита и воды в материале в процессе их переноса.
В гидрофобных полимерах, сорбирующих ограниченно малое количество воды, такое взаимодействие приводит к связыванию электролита, понижению его подвижности и торможению процесса переноса. В полярных полимерах, особенно при наличии гидрофильных наполнителей, вода ориентируется у полярных групп полимера или на поверхности гидрофильного наполнителя, образуя протяженные области, которые затрудняют свободный перенос молекул летучего электролита. Например, если коэффициенты проницаемости воды в полиэтилене и в эпоксидной смоле являются величинами одного порядка, то значение коэффициента проницаемости HCl в отвержденной эпоксидной смоле на два порядка меньше, чем в полиэтилене. Значит, для защиты от действия водных растворов летучих электролитов более эффективными являются покрытия на основе полярных синтетических смол. Этим, кстати, и объясняется широкое применение систем покрытий на основе эпоксидных смол для защиты металлоконструкций на промышленных объектах.
17.5.2. Стойкость силикатных материалов в кислотах и щелочах.
Взаимодействие силикатных материалов с электролитами, как уже отмечалось, определяется их химическим составом.
Подавляющее большинство современных силикатных материалов, используемых для химзащитных работ, квалифицируется как кислотостойкие материалы. Это обусловлено преимущественным содержанием в них кремнезема SiO2 – кислотного оксида. Кислоты не действуют на него, причем, чем выше концентрация кислоты, тем выше химическое сопротивление материала. Исключение здесь составляют плавиковая кислота (HF) и концентрированная фосфорная кислота при высоких температурах. Растворы плавиковой кислоты любой концентрации действуют как на аморфный кремнезем, так и на кристаллический. Процесс взаимодействия выражается двумя уравнениями:
SiO2 + 4 HF →SiF4 + 2 Н2О (17.19)
SiO2 + 4 HF → Н2SiF6 + 2 Н2О (17.20)
Скорость этих реакций возрастает с повышением концентрации кислоты и температуры.
Горячая фосфорная кислота энергично действует на все разновидности кремнезема с образованием кристаллического фосфата кремнезема SiO2Р2О5.
Как кислотный оксид, SiO2взаимодействует с основаниями. Наибольшей агрессивностью обладают едкие щелочиNaOHи КОН.
Кислотостойкость силикатных материалов возрастает с увеличением содержания SiO2 и падает при уменьшении его количества. Увеличение содержания основных оксидов и солей ведет к снижению кислотостойкости и увеличению стойкости к действию щелочей.
Помимо степени насыщения силикатных материалов металлическими оксидами, их химическая стойкость зависит и от природы силикатообразующего оксида. Чем более химически активен металлический оксид, тем менее химически стойкий силикат он образует. Поэтому химическая стойкость силикатов растет от Ме2ОSiO2через МеОSiO2к Ме2О3SiO2.
Исходя из активности силикатообразующих оксидов, силикаты группы МеОSiO2располагаются по степени возрастания их химической стойкости в следующий ряд:PbOSiO2,BaOSiO2,CaOSiO2,MgOSiO2,ZnOSiO2,
FeOSiO2,MnOSiO2.
Стойкость силикатных материалов к действию кислот или оснований определяется не только химическим составом, но и их структурой, что целесообразно рассмотреть в последующих разделах.
Бетоны на основе портланд цементов (строительные бетоны) не стойки к действию кислот из-за их взаимодействия с Са(ОН)2и другими составляющими цементного камня. Образующиеся продукты реакции вымываются из бетона, если они растворимы, или остаются в его порах в виде рыхлой массы. Скорость коррозии бетонов возрастает с увеличением концентрации кислоты, растворимости получаемых продуктов, скорости движущегося потока.
Строительные бетоны сильно разрушаются под действием соляной, азотной, серной, уксусной кислот. Менее агрессивные слабые растворы фосфорной и кремнефтористоводородной кислот (<1%) из-за малой растворимости кальциевых солей. В этом случае скорость коррозии зависит от толщины и плотности слоя этих солей, образовавшихся на поверхности бетона.
Высокой кислотостойкостью обладают специальные кислотоупорные бетоны на основе жидкого натриевого (Na2SiO3) или калиевого стекла (К2SiO3) с кислотостойкими наполнителями. Об их рецептуре, свойствах и областях применения будет изложено в соответствующем разделе.
17.5.3. Химическая деструкция полимерных материалов под действием растворов электролитов.
По отношению к полимерам из растворов электролитов наиболее агрессивными являются кислоты и основания, менее агрессивными являются соли.
Все окислители (HNO3, конц.H2SO4,H2O2, K2Cr2O7, КМnО4и др.) оказывают сильное разрушающее воздействие на подавляющее большинство полимеров и композиционных материалов на их основе. Исключение здесь составляют фторполимеры, фторопласты.
Химическая деструкция протекает с разрывом химически нестойких связей и сопровождается изменением молекулярной массы полимера.
При химической деструкции в полимере могут происходить следующие превращения макромолекул:
разложение основной цепи макромолекул в полимере, приводящее к уменьшению степени полимеризации;
отщепление молекул мономера от конца макромолекулы – деполимеризация;
превращение группы атомов в составе макромолекулы при сохранении исходной степени полимеризации – полимераналогичные превращения;
образование химических связей между макромолекулами – сшивание, структурирование.
В подавляющем большинстве случаев при контакте полимеров с химическими реагентами эти превращения протекают одновременно с преобладанием того или иного процесса.
Химическая деструкция ухудшает физико-химические и физико-механические характеристики полимеров, приводя к частичной или полной потере их эксплуатационных качеств.
Основными, наиболее распространенными видами химической деструкции полимеров в растворах электролитов являются окислительная и гидролитическая. Окислительная деструкция полимеров является сложным процессом, включающим в себя радикальные, молекулярные, ионно-химические реакции и протекает под действием сред, обладающих окислительными свойствами. Гидролитической деструкции подвергаются гетероцепные полимеры, содержащие гетероатомы ( - N=, - О -, -Si– и др.) в основной или боковых цепях. Кислоты и основания являются сильными катализаторами гидролитических процессов. На протекание гидролитической деструкции большое влияние оказывает строение элементарного звена полимера.
Кроме указанных процессов химической деструкции, различные функциональные группы полимеров могут вступать в реакции сульфирования, нитрования, галогенирования и др.
Способность полимеров вступать во взаимодействие с различными химическими реагентами определяется, прежде всего, наличием функциональных групп, двойных связей и т.п. Поэтому теоретически при соблюдении определенных условий реакционная способность полимеров должна быть такой же, как и у их низкомолекулярных аналогов. Однако на практике это положение не выполняется, и реакционная способность полимеров намного ниже реакционной способности аналогичных низкомолекулярных соединений. Это объясняется рядом особенностей твердых полимеров.
Прежде всего, наличие большого количества функциональных групп в полимере, их близкое расположение друг к другу обуславливают их взаимодействия между собой, что понижает химическую активность материала при контакте с внешней средой. Во-вторых, активные центры в твердом полимере могут быть труднодоступным для молекул агрессивной среды из-за диффузионных ограничений. В-третьих, существенно влияет появление в полимере кристаллических областей. С повышением степени кристалличности возрастает химическая стойкость полимера, т.к. замедляется диффузия агрессивной среды. В-четвертых, при действии химически активных сред на поверхности полимера часто образуются плотные слои из продуктов их взаимодействия, что также снижает диффузию среды к активным центрам. Наконец, пространственные связи в полимере могут также способствовать увеличению его химической стойкости, если сами эти связи не оказываются слабее связей основной цепи. Характер поперечных связей существенно влияет на поведение полимера только в случае проникновения среды в его массу. При поверхностном действии среды, особенно когда наблюдается сильное изменение полимера и образование плотной пленки из продуктов его перерождения, характер поперечных связей на стойкость полимера практически не влияет.
Химическая деструкция полимеров в жидких агрессивных средах является сложным физико-химическим процессом, который протекает в несколько основных стадий:
Адсорбция компонентов агрессивной среды на поверхности полимера;
Диффузия компонентов агрессивной среды в объеме полимера;
Химическая реакция компонентов агрессивной среды с химически нестойкими связями;
Диффузия продуктов химических реакций к поверхности полимера;
Десорбция продуктов с поверхности полимера.
Обычно основными лимитирующими стадиями являются стадии 2 и 3, на которые и обращается внимание исследователей.
В зависимости от соотношения скоростей процессов диффузии и химической реакции деструкция полимеров может протекать в различных областях.
Если скорость диффузии агрессивной среды соизмерима со скоростью химической реакции (Wд.=Wх), процесс деструкции протекает в некоторой реакционной зоне, размер которой увеличивается во времени, достигая в пределе размеров толщины изделия из полимера. Реакция в этом случае протекает во внутренней диффузионно-кинетической области, что характерно при взаимодействии летучих электролитов с гидрофобными и гидрофильными полимерами. Это наблюдается, например, при контакте полиэтилена, полипропилена, поливинилхлорида с концентрированной азотной кислотой.
Если Wд.>Wх, среда быстро проникает в объем полимера, где и происходит химическое взаимодействие – деструкция протекает во внутренней кинетической области. Такое явление характерно для систем гидрофильные полимеры – нелетучие электролиты (например, при взаимодействии поликарбоната с серной кислотой).
Если Wд.<Wх, деструкция протекает во внешней диффузионно-кинетической области, т.е. практически на поверхности полимера, что типично для систем гидрофобный полимер – нелетучий электролит. К примеру, такое происходит с полиэтиленом и полипропиленом в контакте с концентрированной серной кислотой. Этот вариант, с точки зрения прогноза, работоспособности полимера и контроля ситуации наиболее предпочтителен, поскольку изменения эксплуатационных характеристик находятся в соответствие с толщиной «отработанного» слоя полимера.
В общем случае скорость распада химически нестойких связей в полимере под действием растворов электролитов может быть выражена в виде:
W=
![]() = к (
= к (![]() - Сн) Скат.Сраст., (17.21)
- Сн) Скат.Сраст., (17.21)
где
![]() - начальная концентрация химически
нестойких связей в полимере; Сн- концентрация распавшихся связей; Скат.- концентрация катализатора (например,
кислоты или основания) в полимере; Сраст.- концентрация растворителя (например,
воды) в полимере; к - константа скорости
распада химически нестойких связей.
- начальная концентрация химически
нестойких связей в полимере; Сн- концентрация распавшихся связей; Скат.- концентрация катализатора (например,
кислоты или основания) в полимере; Сраст.- концентрация растворителя (например,
воды) в полимере; к - константа скорости
распада химически нестойких связей.
Концентрация катализатора (кислоты или основания) в полимере может быть найдены из уравнения Фика:
![]() = Dкат.
= Dкат.![]() Скат. -
Скат. -![]() Скат.Сi
кi, (17.22)
Скат.Сi
кi, (17.22)
где
![]() - оператор Лапласа; Сi- концентрация функциональных групп в
полимере, способных вступать в реакцию;
Dкат.- коэффициент диффузии
катализатора; кi-
константа скорости реакции катализатора
с функциональными группами полимера.
- оператор Лапласа; Сi- концентрация функциональных групп в
полимере, способных вступать в реакцию;
Dкат.- коэффициент диффузии
катализатора; кi-
константа скорости реакции катализатора
с функциональными группами полимера.
Если растворитель расходуется на распад химически нестойких связей в полимере (например, расходование воды в реакциях гидролиза), то его концентрацию можно определить из уравнения:
![]() = Dраст.
= Dраст.![]() Сраст. – к (
Сраст. – к (![]() - Сн) Скат.Сраст.(17.23)
- Сн) Скат.Сраст.(17.23)
Для каждой области протекания деструкции имеются решения приведенных уравнений, однако определение констант реакций для конкретных полимеров, концентраций катализатора и растворителя затруднено из-за неоднородной структуры полимеров и различной доступности химически нестойких связей.
Полимерным материалам свойственны изменения структуры и свойств по объему, называемыми градиентами структуры.
Для однофазных систем такими градиентами являются наличие стеклообразных и высокоэластических участков, присутствие сильно- и слабосшитых областей, наличие ориентированных и неориентированных областей, наличие зон с различным массмолекулярным распределением (ММР) и различной степенью упорядоченности молекул.
Для гетерогенных систем характерны следующие градиенты их структуры: аморфные и кристаллические области; области с различной степенью микронеоднородности или с различной пространственной и химической структурой. В реальных полимерах, кроме того, наблюдаются разнообразные структурные градиенты, создаваемые при введении стабилизаторов, пластификаторов, красителей, наполнителей и т.д. Перечисленные градиенты порождают анизотропию диффузионных и химических свойств в полимерных материалах, т.е. в них имеются области с различной реакционной способностью, (различные константы к) и различной способностью сорбировать и проводить агрессивную среду (различные значения С0иD). Вследствие этого общая скорость деструкции равна:
WΣ= [к (- Сн) Скат.Сраст.]iVi,(17.24)
Где Vi- относительный объем каждой области.
Изучение кинетики химической деструкции полимерных материалов является чрезвычайно сложной задачей, как в экспериментальном плане, так и при нахождении кинетических параметров для отдельных элементарных актов. Поэтому на практике весьма часто определяют не истинную, а эффективную константу скорости деструкции (кэф.), которая является произведением ряда истинных констант каждой стадии процесса деструкции.
Важной практической задачей является уменьшение скорости химической деструкции, т.е. увеличение химической стойкости (химического сопротивления) полимерных материалов. Пути увеличения химической стойкости направлены на торможение лимитирующих стадии процесса деструкции: адсорбции агрессивных компонентов, их диффузии в материал и распада нестойких химических связей под действием проникающей среды.
Затормозить распад нестойких связей возможно при введении веществ, образующие комплексы с химически нестойкими связями, тем самым, уменьшая их реакционную способность. Однако, этот путь практически малоэффективен.
Более радикальным является уменьшение концентрации доступных химически нестойких связей и концентрации компонентов агрессивной среды в полимере.
Уменьшение концентрации химически нестойких связей достигается за счет:
увеличение степени кристалличности полимера;
введения в полимер наполнителей;
введения в макромолекулу заместителей, стерически затрудняющих подход компонентов агрессивной среды к химически нестойким связям.
Уменьшить концентрацию компонентов агрессивной среды в полимер можно путем:
нанесения на поверхность полимера защитных слоев из веществ, стойких к действию данной среды и растворяющих ее компоненты в весьма малых количествах. К таким веществам относятся кремнийорганические соединения, парафины, металлические и др. покрытия;
введения в полимер инертных наполнителей, способных диффундировать к поверхности полимера и создавать на границе контакта со средой высокую поверхностную концентрацию.
Если процесс деструкции может протекать в объеме полимерного материала, то положительный эффект его торможения достигается за счет:
сшивания макромолекул бифункциональными соединениями, образующими сетчатую структуру, что значительно понижает растворимость компонентов среды в материале;
увеличения степени кристалличности и ориентации макромолекул;
изменения химического строения полимера (например, путем хлорирования);
введения в полимер веществ, связывающих компоненты агрессивной среды.
17.5.4. Взаимодействие неметаллических материалов с органическими растворителями, расплавами металлов и солей.
По отношению к силикатным материалам органические безводные вещества не являются агрессивными средами. Опасность представляют органические вещества, которые содержат примеси, способные разрушать силикатные материалы: сернистые нефти, крекинг бензины, сырые фенолы и др.
Агрессивность сернистых нефтей определяется сернистыми соединениями, из которых наиболее опасными являются меркаптаны и сероводород. В парогазовой фазе из этих веществ образуется активная сера, вызывающая разрушение силикатов. Коррозионно-активными веществами топлив являются также сернистые соединения (сероводород, элементарная сера, меркаптаны) и кислородные соединения, из которых наиболее агрессивными являются органические кислоты.
При действии на полимерные материалы органических растворителей часто наблюдается их неограниченное набухание, переходящее в растворение. Высокая растворяющая способность таких органических веществ обусловлена большим сродством между их молекулами и молекулами полимера. В первом приближении об интенсивность воздействия на полимер органических растворителей можно судить по параметрам растворимости полимера Sпи растворителяSр, которые численно равны корню квадратному из плотности энергии когезии соответственно полимера и растворителя. Под энергией когезии понимают энергию, которую необходимо затратить для удаления молекул друг от друга на расстояние, исключающее межмолекулярное взаимодействие.
Чем ближе значения параметров растворимости полимера и растворителя, тем интенсивнее будет процесс растворения полимера, причем неполярные полимеры хорошо растворяются в неполярных растворителях, полярные – в полярных.
Помимо химической природы полимера и органического растворителя на способность полимеров растворяться влияют и другие факторы. С уменьшением молекулярной массы и увеличением гибкость полимеров их растворимость возрастает. Увеличение плотности упаковки полимера уменьшает его растворимость. Кристаллические полимеры растворяются в органических растворителях только при температурах, близких к температурам плавления. Полимеры с сетчатой пространственной структурой не растворяются в органических растворителях, а лишь могут набухать. Иллюстрацией сказанного является сопоставление растворимости в бензине натурального каучука, который имеет активные двойные углеродные связи, и вулканизованного каучука – резины, когда образуется пространственная сетка за счет насыщения двойных связей вулканизатором – серой. В первом случае имеет место растворение с образованием резинового клея; резина в бензине не растворяется, а лишь может частично набухать.
Увеличение числа поперечных химических связей (степени сшивания макромолекулы) приводит к уменьшению способности полимера набухать в органических растворителях, и при наличии густосетчатой структуры полимер полностью утрачивает способность набухать в этих средах.
В металлургических, химических, нефтехимических и других высокотемпературных технологических процессах, металлические корпуса аппаратов защищают от воздействия расплавов металлов, солей, шлаков и газовых сред покрытиями из огнеупоров (футеровки).
Взаимодействие огнеупоров с такими расплавами является сложным гетерогенным процессом, включающим в себя массоперенос, плавление и химическое взаимодействие, который может протекать с кинетическим или диффузионным контролем.
Когда скорость оплавления (растворения) огнеупора больше скорости миграции расплава и продуктов взаимодействия в огнеупоре, на поверхности последнего образуется насыщенный раствор огнеупора в расплаве, так называемый контактный слой. При этом дальнейшее растворение огнеупора в расплаве прекращается, но может происходить деструкция огнеупора. Контактный слой смывается или стекает, захватывая с собой из поверхностного слоя зерна огнеупора, не успевшие раствориться в расплаве, в результате чего на месте первоначального образуется новый контактный слой и т.д.
Скорость растворения огнеупора Wопределяется следующим уравнением:
W= D S(Снас– С)/x, (17.25)
где D – коэффициент диффузии компонентов расплава в огнеупоре; S– поверхность контакта огнеупора с расплавом; Снас.– концентрация насыщения огнеупора в расплаве; С – начальная концентрация огнеупора в расплаве; х – толщина диффузионного слоя.
При малой скорости оплавления огнеупора расплав по его порам проникает в объем. Здесь идет взаимодействие, результатом чего может быть увеличение жидкой фазы в объеме огнеупора, образование новых фаз, твердых растворов или химических соединений. При повышении температуры скорость диффузионных процессов изменяется мало, а скорость химического взаимодействия – существенно. Химическое взаимодействие огнеупора и расплава зависит от местоположения их катионов в ряду напряжений: любой элемент вытесняет из расплава все другие, следующие за ним в этом ряду, а сам вытесняется теми, что стоят выше.
Коррозия огнеупора будет меньшей, если энергия связи металла огнеупора с кислородом больше энергии связи металла расплава с кислородом или другими элементами. На коррозию огнеупора оказывает влияние смачиваемость его поверхности расплавом и шлаком. Если краевой угол смачивания больше 1800, огнеупор не взаимодействует с расплавом в кинетической области. Смачиваемость, как известно, обусловлена силами физического и химического взаимодействия. Связь огнеупора с оксидными расплавами обусловлена химическими силами и весьма возрастает с повышением температуры.
Глубина проникновения расплава в огнеупор определяется его пористостью, текстурой, объемом открытых и закрытых пор. В процессе пропитки шлаки и расплавы способны разрушать стекловидную связку и превращать закрытую пористость в открытую.
Проникновение расплава в поры огнеупора протекает, в основном, по капиллярному механизму и в общем случае описывается уравнением:
h=
![]() , (17.26)
, (17.26)
где h– высота капиллярного поднятия;![]() -
коэффициент поверхностного натяжения
расплава;
-
коэффициент поверхностного натяжения
расплава;![]()
![]() -
краевой угол смачивания;
-
краевой угол смачивания;![]() -
плотность расплава;g–
ускорение силы тяжести;r– радиус капилляра.
-
плотность расплава;g–
ускорение силы тяжести;r– радиус капилляра.
Зависимость hот времени носит параболический характер.
Все изложенное выше, дает возможность рационально выбирать огнеупоры для конкретных случаев.