

Теория электрохимического перенапряжения, учитывающая структуру двойного электрического слоя
В кинетике протекания стадии переноса заряда особую роль должно играть строение ДЭС на границе раздела двух фаз. Если другие стадии электродного процесса – транспортировка частиц и гомогенное химическое превращение – протекают хотя и вблизи границы раздела электрод – электролит, но далеко за пределами двойного слоя, то собственно электрохимический акт разыгрывается внутри этого слоя. Распределение потенциала в двойном слое и положение реагирующих частиц в нем должны поэтому существенно влиять и на скорость электрохимического акта, и на величину электрохимического перенапряжения. Фрумкин, первым высказавший эту мысль, дал ее количественное оформление на основе некоторых предположений. Первое из них сводится к тому, что реально существующий двойной слой может быть удовлетворительно описан моделью Штерна. Согласно второму предположению в электрохимическом акте участвуют лишь частицы, находящиеся в плотной части ДЭС, то есть непосредственно у поверхности электрода. Как следствие этого, средняя энергия заряженных частиц и их концентрация у поверхности должны быть иными, чем в глубине раствора. Это различие обязано существованию скачка потенциала в диффузной части двойного слоя.
Фрумкин подчеркнул, что потенциал в точке, где происходит перенос заряда, названный им 1-потенциалом, не обязательно совпадает с -потенциалом, он может быть различным для разных ионов. Сейчас обычно принимается, что 1-потенциал приблизительно равен потенциалу внешней плоскости Гельмгольца по модели Грэма. Это, вероятно, справедливо для поверхностно-инактивных частиц. Для специфически адсорбирующихся веществ, находящихся во внутренней плоскости Гельмгольца, более обоснованным кажется отождествление 1-потенциала с потенциалом этой плоскости. В связи с этим представляется целесообразным в дальнейшем изложении сохранить обозначение, предложенное Фрумкиным, то есть 1 .
Для величины катодного перенапряжения в теории Фрумкина получено следующее выражение:
=
![]() ln io
+
ln io
+
![]() –
ln iк
, (1)
–
ln iк
, (1)
где z1 – заряд частиц Ох.
Ток обмена (см. ранее) является функцией концентраций участников электродной реакции:
io
=
![]()
![]()
![]()
и, следовательно,
=
ln
+
![]()
![]() ln cOx
+
ln cRed
+
–
ln iк
(2)
ln cOx
+
ln cRed
+
–
ln iк
(2)
При = 0,5 и n = z1 = 1 уравнение (2) упрощается до
=
![]() ln
+
ln
+
![]() ln cOx
+
ln cRed
– 1
–
ln iк
(3)
ln cOx
+
ln cRed
– 1
–
ln iк
(3)
и отвечает формуле, выведенной Фрумкиным применительно к процессу катодного выделения водорода. Из формулы Фрумкина вытекает, что перенапряжение зависит не только от концентрации непосредственных участников электродной реакции, но и от природы и содержания любых веществ, в первую очередь поверхностно-активных, способных изменить величину 1-потенциала. В частном случае (1 = 0) она превращается в формулу Эрдей-Груза и Фольмера.
Влияние состава раствора на перенапряжение.
Так как состав раствора влияет на величину 1-потенциала, то, соответственно, влияет и на скорость разряда водорода. Рассмотрим разряд ионов водорода в разбавленных растворах кислот с добавкой 1,1-валентного электролита при отсутствии специфической адсорбции ионов раствора. Такие условия реализуются, например, на ртути в растворах HCl + KCl, т.к. области катодного выделения водорода соответствуют большие отрицательные заряды поверхности, при которых ионы Cl– не адсорбируются на ртути, а ионы К+ специфической адсорбционной способностью не обладают. При данных условиях из теории двойного слоя следует, что
1
const
+
![]() ln
c ,
ln
c ,
где с = HCl + KCl – суммарная концентрация 1,1-валентного электролита. Подставив это выражение в уравнение (3), получим приближенное соотношение:
const +
ln
![]() –
ln i
(4)
–
ln i
(4)
При анализе этого соотношения можно выделить три случая:
1. Раствор разбавленной кислоты без добавки постороннего электролита.
с =
![]()
const –
ln i
const –
ln i
Из этого уравнения следует, что для разбавленных растворов (до 0,1 н) результаты укладываются на тафелевскую прямую вне зависимости от концентрации кислоты, т.е. для i = const перенапряжение не должно зависеть от рН. Этот результат связан с взаимной компенсацией двух эффектов: с ростом концентрации кислоты увеличивается концентрация реагирующего вещества и уменьшается перенапряжение, и одновременно сдвигается 1-потенциал в положительную сторону, что приводит, согласно уравнению (3), к росту . На опыте установлено, что на ртути в растворах чистых кислот в области концентраций до 0,1н не является функцией рН; при более высоких концентрациях зависит от рН, уменьшаясь с увеличением концентрации кислоты, причем /рН составляет примерно 60 мВ.
2. Общая концентрация электролита поддерживается постоянной, но изменяется соотношение концентраций соли и кислоты (изменение рН при поддержании постоянной ионной силы раствора). Из уравнения (4) получаем:
const + ln – ln i
Тогда при i = const
const
+
![]() lg
= const – 0,059 рН,
lg
= const – 0,059 рН,
т.е. перенапряжение должно возрастать на 59 мВ (при 25оС) с увеличением рН на единицу. На опыте: при выделении водорода на ртути из растворов кислот с добавкой индифферентного электролита возрастает с ростом рН в широком интервале концентраций кислоты, при этом /рН составляет 58-60 мВ.
3. Концентрация ионов Н3О+ поддерживается постоянной, а концентрация соли меняется:
const – 0,059 lg с – 0,118 lg i ,
т.е. должно расти на 59 мВ при увеличении концентрации 1,1-валентной соли на порядок. Опытные данные: добавки индифферентных солей к разбавленным растворам кислот увеличивают на ртути, причем увеличение концентрации 1-1 зарядного электролита в 10 раз (при постоянном рН) повышает примерно на 55–58 мВ. Первоначальная добавка электролита с поливалентным катионом оказывает большее действие, чем такая же добавка 1-1 зарядного электролита.
Рассмотрим теперь влияние специфической адсорбции ионов на скорость реакции разряда ионов гидроксония.
1. При специфической адсорбции анионов 1-потенциал сдвигается в отрицательную сторону, что, согласно уравнению (3), должно уменьшать перенапряжение. Экспериментальные данные, полученные на ртути (см. рис.), подтверждают вывод об ускорении реакции разряда катионов Н3О+ в присутствии специфически адсорбирующихся анионов, причем ускоряющий эффект проявляется только в области их адсорбции. Действие добавок ослабляется с ростом плотности тока и при высоких ее значениях полностью исчезает, что связано с десорбцией анионов при достижении достаточно отрицательных потенциалов.
|
Поляризационные кривые для реакции выделения водорода на ртути из подкисленных растворов солей с постоянной ионной силой: 1 – Na2SO4 , 1н 2 – KCl , 1н 3 – KBr , 1н 4 – KJ , 1н Постоянная общая концентрация выбирается для того, чтобы исключить эффект увеличения перенапряжения. |
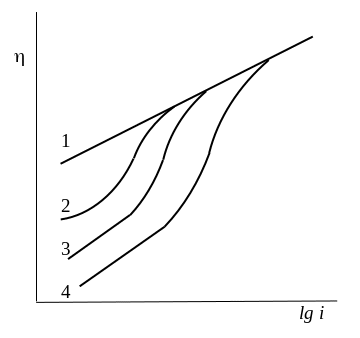
2. Специфически адсорбирующиеся катионы, наоборот, тормозят выделение водорода (повышают перенапряжение), т.к. при их адсорбции 1-потенциал сдвигается в положительную сторону.
3. В присутствии органических веществ перенапряжение водорода изменяется в том интервале потенциалов, где эти вещества адсорбируются на поверхности электрода. В присутствии спиртов или кислот жирного ряда возрастает в области адсорбции этих веществ. После их десорбции , lg i – кривые в растворах кислоты с добавкой органического вещества и без добавки совпадают, причем потенциал десорбции, полученный из поляризационных измерений, хорошо совпадает с данными электрокапиллярных кривых или кривых дифференциальной емкости. Аналогичным образом повышают органические катионы, например, катионы тетрабутиламмония (ТБА). Эти катионы также десорбируются с поверхности, но при более отрицательных потенциалах, вследствие чего их эффект наблюдается в более широком интервале потенциалов.
Действие органических молекул и ионов сказывается на кинетике стадии разряда-ионизации через изменение 1-потенциала и через блокировку поверхности. Последний эффект связан с тем, что разряд реагирующих частиц на заполненной органическими молекулами поверхности происходит, как правило, со значительно меньшей скоростью. При адсорбции молекул алифатических спиртов или кислот, а также при адсорбции органических катионов 1-потенциал сдвигается в положительную сторону, что, согласно уравнению (3), должно приводить к росту . Т.о., в этих условиях сдвиг 1-потенциала и блокировка поверхности действуют в одном направлении, уменьшая скорость стадии разряда ионов Н3О+, причем при малых концентрациях катионов ТБА их влияние на скорость стадии разряда в основном обусловлено сдвигом 1-потенциала.
Кинетика электролитического выделения водорода
В связи с большим практическим значением реакции выделения водорода для ряда технических процессов (электролиз воды, хлорный электролиз, эксплуатация аккумуляторов и гальванических элементов, коррозия) эта электрохимическая реакция изучена наиболее детально.
Электролитическое выделение водорода из кислых и щелочных растворов происходит различными путями. Источником водорода в кислых растворах служат ионы гидроксония:
2Н3О+ + 2е = Н2 + 2Н2О
В щелочных растворах предполагается непосредственное присоединение электронов к молекулам воды:
2Н2О + 2е = Н2 + 2ОН–
Полагают, что последняя реакция может протекать и в кислых растворах, но при высоких плотностях тока. Иногда в кислых средах водород выделяется непосредственно из молекул кислоты (например, при выделении водорода на ртутном катоде из водных растворов угольной кислоты):
2НА + 2е = Н2 + 2А–
Выделение водорода на катоде происходит при потенциале, более отрицательном, чем обратимый потенциал ЕН, отвечающий рН данного раствора:
ЕН
=
![]() ln
ln
![]() = –
рН
= –
рН
В большинстве случаев разность Еi – ЕН отождествляется с активационной поляризацией, т.к. концентрационная поляризация здесь мала (в кислых растворах диффузионные ограничения незначительны вследствие аномально высокой подвижности ионов водорода; в щелочных растворах концентрационная поляризация низка благодаря очень высокой концентрации разряжающихся частиц – молекул воды). Следовательно, величину
Н = Еi – ЕН
которую называют перенапряжением водорода, можно рассматривать как непосредственную меру необратимости собственно электрохимической реакции образования водорода.
Изучение водородного перенапряжения позволяет выяснить механизм этой реакции и представляет большой интерес с теоретической точки зрения, т.к. установленные при этом закономерности можно частично распространить и на другие электрохимические реакции. Изучение водородного перенапряжения имеет также большое практическое значение, потому что современная промышленная электрохимия является преимущественно электрохимией водных растворов, и процессы электролитического разложения воды могут накладываться на любые катодные и анодные реакции. Водородное перенапряжение составляет значительную долю напряжения на ваннах по электролизу воды и растворов хлоридов; знание природы водородного перенапряжения позволяет уменьшить его и тем самым снизить расход электроэнергии и улучшить экономические показатели этих процессов. В других случаях (электролитическое выделение металлов, катодное восстановление неорганических и органических веществ, эксплуатация химических источников тока) знание природы водородного перенапряжения позволяет успешно решать обратную задачу – нахождение путей его повышения. Вот почему изучение процесса катодного выделения водорода и природы водородного перенапряжения всегда находилось и находится в центре внимания электрохимиков.