

I. Зменшення об'єму циркулюючої крові:
1) крововтрата (геморагічний шок);
2) втрата плазми крові при великому ексудативному запаленні (опіковий шок);
3) вихід рідини з кровоносних судин у тканини при генералізованому підвищенні проникності судин (анафілактичний шок);
4) зневоднення (ангІдремічний шок);
5) перерозподіл крові в судинному руслі (тромбоз і емболія магістральних вен).
II. Зменшення хвилинного об'єму серця:
1) порушення скоротливої функції серця (інфаркт міокарда);
2) тампонада серця (розрив серця, ексудативний перикардит);
3) аритмії (фібриляція шлуночків).
III. Зменшення загального периферичного опору - генерал із оване розширення судин:
1) падіння нейрогенного тонусу артеріол (больові форми шоку);
2) зменшення базального тонусу судин під дією біологічно активних речовин (анафілактичний, панкреатичний шок) або токсичних продуктів (травматичний, турнікетний, інфекційно-токсичний шок).
IV. Порушення реологічних властивостей крові:
1) синдром внутрішньо судинного дисемінованого зсідання крові (панкреатичний шок);
2) агрегація формених елементів крові (септичний, інфекційно-токсичний шок);
3) згущення крові - гемоконцентрація (ангІдремічний шок).
12.5. Чим визначаються важкі наслідки шоку?
Розлади загальної гемодинаміки і мікроциркуляції, що виникають за умов шоку, небезпечні насамперед порушеннями кровообігу: а) мозкового; б) вінцевого; в) ниркового. Результатом зазначених розладів є прогресуюче порушення центральної регуляції життєво важливих функцій, аж до розвитку коми; виникнення явищ гострої серцево-судинної і ниркової недостатності. Патогенні фактори, що виникають при цьому, — гіпоксія, ацидоз та інтоксикація - ведуть до генералізованого і незворотного ушкодження клітин.
12.6. Поясніть патогенез травматичного шоку.
Травматичний шок розвивається внаслідок великих за обсягом ушкоджень тканин. У його клініці розрізняють дві стадії: збудження (еректильну) і гальмування (торпідну).
Стадія збудження короткотривала, для неї характерний стан збудження центральної нервової системи, що виникає в результаті надходження великої кількості больових імпульсів з ушкоджених тканин. Розвивається больовий стрес, що виявляє себе посиленням функцій системи кровообігу, дихання, деяких ендокринних залоз
(аденогіпофіза, мозкової і кіркової речовини надниркових залоз, нейросекреторних ядер гіпоталамуса) з вивільненням у кров великої кількості кортикотропіну, адреналіну, норадреналіну, вазопресину (див. розд. 33).
Стадія гальмування більш тривала (від кількох годин до доби) і характеризується розвитком у центральній нервовій системі гальмівних процесів. Генера-лізоване гальмування захоплює й центри життєво важливих функцій (кровообігу, дихання), вони порушуються, унаслідок чого розвивається кисневе голодування. Гіпоксія, у свою чергу, збільшує порушення в серцево-судинному й дихальному центрах. Розлади гемодинаміки і зовнішнього дихання прогресують - "зачароване коло "замикається.
Крім нервово-рефлекторних механізмів у виникненні і розвитку травматичного шоку певну роль відіграє токсемія, обумовлена всмоктуванням у кров продуктів розпаду нежиттєздатних тканин. Особливе значення надають так званому ішемічному токсину.
Участь токсичних продуктів у патогенезі травматичного шоку доводиться дослідами з "перехресним кровообігом", коли після відтворення шоку в однієї тварини явища цього патологічного процесу виникають у другої, інтактної, пов'язаної з першою загальним кровообігом.
12.7. Що таке колапс?
Колапс — це судинна недостатність, що швидко розвивається і характеризується в першу чергу падінням судинного тонусу, а також гострим зменшенням об'єму циркулюючої крові. При цьому відбувається зменшення припливу венозної крові до серця, зниження серцевого виштовху, падіння артеріального й венозного тиску, порушуються перфузія тканин і обмін речовин, настає гіпоксія головного мозку, пригнічуються життєво важливі функції організму.
12.8. Що таке краш-синдром?
Краш-синдром (синдром тривалого роздавлювання) - це патологічний процес, який розвивається у потерпілих у результаті тривалого (4-8 г і більше) роздавлювання м'яких тканин кінцівок уламками зруйнованих будинків, споруд, брилами ґрунту при обвалах у шахтах та ін.
У перебігу краш-синдрому розрізняють три періоди: 1) ранній (до 3-х діб) з переважанням явищ шоку; [ 2) проміжний (з 3-ї до 12-ї доби) з переважанням гострої ниркової недостатності; 3) пізній (з 8-12-ї доби до 1-2 міс), або період видужання, з переважанням місцевих симптомів.
У розвитку краш-синдрому велике значення мають три патогенетичних фактори:
а) больове подразнення;
б) травматична токсемія, обумовлена всмоктуванням токсичних продуктів аутолі-зу тканин з вогнища ураження;
в) плазмо- і крововтрата, пов'язані з набряком і крововиливами в зоні роздавлених або довготривало ішемізованих тканин.
12.9. Що таке кома?
Кома— це патологічний стан, що характеризується глибоким пригніченням функцій центральної нервової системи і виявляє себе втратою свідомості, відсутністю рефлексів на зовнішні подразники і розладами регуляції життєво важливих функцій організму.
12.10. Як класифікують коматозні стани?
Залежно від етіології розрізняють екзогенні й ендогенні коми.
1. Екзогенні коми виникають унаслідок дії патогенних факторів зовнішнього середовища або в результаті дефіциту факторів, необхідних для існування організму. Прикладами екзогенних ком є травматична (виникає при ушкодженні головного мозку), гіпоксична, гІпер- і гіпотермічна, екзотоксична, аліментарно-дистрофічна (виникає при важкому голодуванні).
2. Ендогенні коми розвиваються внаслідок порушень діяльності функціональних систем організму. Так, при розладах функції системи кровообігу може виникати апоплексична кома, системи крові — гемолітична, видільної системи - уремічна, печінки — печінкова, ендокринної системи — діабетична, гіпоглікемічна, гіпокор-тикоїдна, тиреотоксична та інші види коми.
12.11. Які механізми лежать в основі патогенезу коматозних станів?
1. Енергодефіцитний механізм. Порушення синтезу АТФ у нейронах центральної нервової системи призводить до генералізованого порушення їхніх функцій. Зазначений механізм є провідним у розвитку гіпоксичної, апоплексичної, респіраторної, гемолітичної, гіпоглікемічної, аліментарно-дистрофічної, екзотоксичної ком.
2. Порушення синаптичної передачі в центральній нервовій системі. Вони можуть бути пов'язані з: а) порушенням синтезу, транспорту, депонування і секреції нейромедіаторів; б) витісненням нейромедіаторів так званими псевдомедіаторами (хибними медіаторами); в) надмірною активацією гальмівних постсинаптичних рецепторів; г) блокадою збудливих постсинаптичних рецепторів. Зазначений механізм відіграє велику роль у розвитку печінкової, уремічної і екзотоксичної ком.
3. Загальні водно-електролітні порушення. Мають значення зміни осмотичного тиску крові й порушення балансу електролітів в організмі. Цей механізм є провідним у розвитку гіперосмолярної діабетичної коми, гіпертермічної, гіпокортикоїд-ної та гіпопаратиреоїдної ком.
4. Порушення кислотно-основного стану. Лежать в основі виникнення ацидотич-ної діабетичної, лактацидемічної, хлоргідропенічної та деяких інших ком.
5. Підвищення внутрішньочерепного тиску. Цей механізм діє при первинних ураженнях головного мозку. Він є основним у патогенезі коми при менінгітах та енцефалітах, а також травматичної коми при черепно-мозкових травмах.
13. Місцеві розлади кровообігу Порушення мікроциркуляції
13.1. Назвіть основні форми місцевих розладів кровообігу.
Артеріальна гіперемія, венозна гіперемія, ішемія, стаз, тромбоз, емболія.
13.2. Що таке артеріальна гіперемія?
Артеріальна гіперемія - це збільшення кровонаповнення органа або тканини за рахунок надмірного надходження крові по артеріальних судинах.
13.3. Які функціональні зміни й клінічні ознаки характеризують артеріальну гіперемію?
При артеріальній гіперемії спостерігають розширення дрібних артерій, артеріол, вен і капілярів; прискорення течії крові в них, пульсацію дрібних артерій і капілярів, збільшення числа видимих оком судин, збільшення тиску в артеріолах, капілярах і венах. У результаті зазначених змін виникає почервоніння, підвищується місцева температура, збільшується об'єм гіперемованої ділянки, підвищується тургор тканини, посилюються обмін речовин і функція органа.
73.4. Які фактори можуть бути причиною артеріально)' гіперемії? Що мають на увазі, коли кажуть про фізіологічну й патологічну артеріальну гіперемію?
Причиною артеріальної гіперемії може бути вплив фізичних, хімічних і біологічних факторів зовнішнього середовища; збільшення навантаження на орган або ділянку тканини; психогенні впливи.
ФЬіологічною називають артеріальну гіперемію, що виникає під дією звичайних фізіологічних подразників (збільшення навантаження на орган, психогенні впливи). Основними її різновидами є робоча й реактивна гіперемія.
Робоча гіперемія - це збільшення течії крові в органі під час посилення його функції (збільшення вінцевого кровообігу при посиленні роботи серця, гіперемія слинних залоз під час приймання їжі та ін.).
Реактивна гіперемія являє собою збільшення течії крові після його короткочасного обмеження. Розвивається зазвичай у нирках, головному мозку, шкірі, кишках, м'язах.
Патологічна артеріальна гіперемія виникає під дією незвичайних (патологічних) подразників або в результаті підвищення чутливості судин до звичайних впливів. Вона супроводжує розвиток таких патологічних процесів, як запалення, алергія, опіки, гарячка, її клінічними прикладами можуть бути інфекційний або алергічний висип, почервоніння обличчя при багатьох інфекційних хворобах (кір, скарлатина, висипний тиф), почервоніння половини обличчя при невралгії трійчастого нерва і т. д.
13.5. Назвіть основні механізми розвитку патологічної артеріальної гіперемії.
Розрізняють два механізми: нейрогенний і пов'язаний з дією місцевих хімічних (метаболічних) факторів.
Нейрогенна артеріальна гіперемія залежно від конкретних механізмів її розвитку може бути нейротонічного і нейропаралітичного типів.
Гіперемію, пов'язану з дією місцевих гуморальних факторів, іноді називаютьмі-опаралітичною, підкреслюючи тим самим первинність порушень скоротливих властивостей гладких м'язів судин під впливом хімічних агентів.
13.6. У чому сутність нейротонічного механізму розвитку артеріальної гіперемії?
Нейротонічний тип артеріальної гіперемії розвивається при збільшенні імпуль-сації по судинорозширювальних нервах (вазодилататорах). До останніх належать парасимпатичні нерви і симпатичні холінергічні нерви, медіатором яких є ацетилхолін.
В експерименті на тваринах цей тип артеріальної гіперемії відтворюють шляхом подразнення судинорозширювальних нервів. Так, подразнення chorda tympani (гілка n. facialis) викликає артеріальну гіперемію і посилення секреції піднижньощелепної слинної залози (дослід К. Бернара).
У клініці нейротонічна гіперемія виникає найчастіше рефлекторно у зв'язку з подразненням екстеро- та інтерорецепторів, а також при подразненні судинорозширювальних нервів та їх центрів. Цим, зокрема, пояснюється почервоніння обличчя і шиї при патологічних процесах у внутрішніх органах (яєчниках, серці, печінці).
13.7. Поясніть нейропаралітичний механізм розвитку артеріальної гіперемії.
Нейропаролітична артеріальна гіперемія розвивається при припиненні імпуль-сації по симпатичних адренергічних нервах, що мають судинозвужувальну дію. В експерименті на тваринах її моделюють за допомогою хірургічних втручань і фармакологічних способів. Часто застосовують перетинання симпатичних адренергічних волокон і нервів. Так, при перетинанні симпатичних волокон сідничного нерва спостерігають розширення судин лапки жаби (дослід А. Вальтера), а при видаленні шийного вузла симпатичного стовбура- почервоніння й підвищення температури вуха кроля збоку операційного втручання (дослід К. Бернара).
Серед фармакологічних способів відтворення артеріальної гіперемії нейропаралітичного типу — блокада передачі нервових імпульсів у симпатичних вузлах за допомогою гангліоблокаторів, порушення утворення, депонування й виділення катехоламінів закінченнями симпатичних нервів (застосування симпатолітиків); блокада а-адрено-рецепторів судинних гладких м'язів за допомогою відповідних адреноблокаторів.
13.8. Які гуморальні фактори можуть викликати розвиток артеріальної гіперемії?
Артеріальна гіперемія міопаралітичного типу може розвиватися за умов дії трьох груп гуморальних факторів:
1) неорганічних іонів (калію, водню) і дефіциту кисню;
2) метаболітів (молочної кислоти, органічних кислот циклу Кребса, АДФ, АМФ, аденозину);
3) біологічно активних речовин (гістаміну, серотоніну, кінінів, простагландинів).
Зазначені фактори спричинюють розширення судин, діючи або безпосередньо на гладкі м'язи судинної стінки, або опосередковано, через вплив на ендотелій судин.
13.9. Яка роль ендотелію кровоносних судин у розвитку артеріальної гіперемії?
Під впливом цілого ряду місцевих гуморальних факторів, зокрема, біологічно активних речовин, ендотеліальні клітини судин виділяють речовину, що отримала була назву фактора релаксації ендотеліального походження. Нині відомо, що цією речовиною є оксид азоту (II), який утворюється з амінокислоти аргініну під впливом ферменту NO-синтази. Оксид азоту діє на гладком'язові клітини судинної стінки й викликає їхню гіперполяризацію. Результатом цього є зменшення базального тонусу кровоносних судин та їх розширення під дією тиску крові.
13.10. Назвіть можливі наслідки артеріальної гіперемії.
У більшості випадків артеріальна гіперемія супроводжується збільшенням інтенсивності обміну речовин і посиленням діяльності органа, що є пристосуванням до дії підвищеного функціонального навантаження.
Однак можливі й несприятливі наслідки. При артеріосклерозі, наприклад, різке розширення судини може супроводжуватися розривом її стінки і крововиливом у тканину. Особливо часто подібне явище спостерігається в головному мозку.
13.11. Що таке венозна гіперемія?
Венозна гіперемія - це збільшення кровонаповнення органа або ділянки тканини в результаті утрудненого відтоку крові по венах.
13.12. Які фактори можуть бути причиною венозної гіперемії?
Порушення відтоку крові по венах може бути пов'язане з такими чинниками:
1) внутрішньосудинними (закупорка вен тромбом або емболом);
2) позасудинними (здавлювання вен пухлиною, рубцем, збільшеною маткою, набряковою рідиною);
3) факторами самої судинної стінки (конституціональна слабкість еластичного апарату вен, недостатній розвиток і знижений тонус гладком'язових елементів їхніх стінок);
4) порушеннями загальної гемодинаміки (ослаблення функції правого шлуночка серця, зменшення присмоктувальної дії грудної клітки, утруднення течії крові в малому колі кровообігу).
13.13. Якими ознаками виявляє себе венозна гіперемія?
Для венозної гіперемії характерне збільшення об'єму органа або ділянки тканини, ціаноз, місцеве зниження температури, набряк, підвищення тиску у венах і капіля-
pax застійної ділянки, уповільнення течії крові, вихід еритроцитів за межі судинного русла (діапедез). На завершальному етапі гіперемії можливий маятникоподібний рух крові і стаз. Тривале розширення вен призводить до розтягнення їхньої стінки, що може супроводжуватися гіпертрофією м'язової оболонки, явищами флебосклерозу і варикозного розширення вен.
13.14. Які місцеві й загальні порушення можуть бути наслідком венозної гіперемії?
Місцеві зміни при венозній гіперемії пов'язані в основному з кисневим голодуванням (гіпоксією) тканини. Гіпоксія при цьому спочатку обумовлена обмеженням припливу артеріальної крові, а потім дією на тканинні ферментні системи продуктів порушеного обміну, наслідком чого є порушення утилізації кисню.
Кисневе голодування при венозній гіперемії обумовлює розлади тканинного метаболізму, викликає атрофічні і дистрофічні зміни, надмірне розростання сполучної тканини (наприклад, цироз печінки при венозному застої, викликаному недостатністю функції серця).
Якщо венозна гіперемія має генералізований характер, то можливим є ряд загальних гемодинамічних порушень з важкими наслідками. Найчастіше вони виникають при закупорці великих венозних судин - ворітної, нижньої порожнистої вен. Скупчення крові в зазначених судинних резервуарах (до 90 % всієї крові) супроводжується різким зниженням артеріального тиску, порушенням живлення життєво важливих органів (серця, мозку), що може призвести до смерті.
13.15. Що таке ішемія?
Ішемія - це зменшення кровонаповнення органа або ділянки тканини в результаті обмеження або повного припинення припливу артеріальної крові. Ішемію називають ще місцевим недокрів'ям.
13.16. Які ознаки характерні для ішемії?
Ішемія характеризується зблідненням ділянки органа, зниженням його температури, порушенням чутливості (відчуття оніміння, поколювання, "повзання мурашок"), больовим синдромом, зменшенням швидкості течії крові і об'єму органа, зниженням артеріального тиску на ділянці артерії, розташованій нижче перешкоди; зниженням напруги кисню в ішемізованій ділянці органа або тканини, зменшенням утворення тканинної рідини і зниженням тургору тканини, порушенням функції органа або тканини, дистрофічними змінами.
13.17. Назвіть основні типи ішемії залежно від причини й механізмів її виникнення.
Основними типами ішемії є компресійна, обтураційна і ангіоспастична.
Компресійна ішемія виникає в результаті здавлювання артерій ззовні лігатурою, рубцем, пухлиною, стороннім предметом та ін.
Обтураційна ішемія є наслідком часткового звуження або повного закриття просвіту артерій атеросклеротичною бляшкою, тромбом або емболом.
Ангіоспастична ішемія виникає внаслідок спазму артерій, викликаного емоційним впливом (страх, хвилювання, гнів), фізичними факторами (холод, травма, механічне подразнення), хімічними агентами, біологічними подразниками (токсини бактерій) і т. д. В основі спазму можуть лежати нервові рефлекторні механізми або безпосередня дія подразників на гладкі м'язи судин (вплив вазопресину, ангіотензи-ну II, ендотеліну).
13.18. Чим визначається характер обмінних, функціональних і структурних порушень у тканині при її ішемії?
Характер таких порушень визначається ступенем кисневого голодування. Важкість гіпоксії, у свою чергу, залежить від швидкості розвитку й типу ішемії, її тривалості, локалізації, характеру колатерального кровообігу, функціонального стану органа або тканини.
Ішемія, що виникає на ділянці повної обтурації або компресії артерій, за інших рівних умов викликає важчі зміни, ніж при спазмі. Ішемія, що швидко розвивається, як і тривала, протікає важче порівняно з ішемією, що розвивається повільно, або нетривалою.
Ішемія життєво важливих органів (мозок, серце) має важчі наслідки, ніж ішемія нирок, легень, селезінки, а ішемія останніх - важчі, якщо порівнювати з ішемією скелетної, м'язової, кісткової або хрящової тканини. Мозок і серце характеризуються високим рівнем енергетичного обміну, але, незважаючи на це, їх колатеральні судини функціонально не здатні компенсувати порушення кровообігу. Навпаки, скелетні м'язи й особливо сполучна тканина, завдяки низькому рівню енергетичного обміну в них, більш стійкі в умовах ішемії.
Утруднення припливу артеріальної крові при підвищеній функціональній активності органа або тканини небезпечніше, ніж у стані спокою.
13.19. Якими послідовними стадіями характеризується патогенез порушень в ішемізованій тканині?
Виділяють такі стадії патогенезу
I. Порушення енергетичного обміну. Вони виявляють себе зниженням ефективності циклу Кребса і тканинного дихання, активацією гліколізу, а в кінцевому підсумку - зменшенням вмісту в клітинах макроергічних сполук — креатинфосфату і АТФ. Порушення утворення енергії на ділянці ішемії патогенетично пов'язане з недостатньою доставкою кисню й необхідних для окиснення субстратів, зниженням активності й синтезу ферментів, виходом ферментів з ушкоджених клітин, роз'єднанням окиснення й фосфорування.
II. Порушення енергозалежних процесів у клітинах, викликане зменшенням утворення енергії. При цьому порушуються специфічні функції клітин (скоротлива, секреторна та ін.), механізми активного транспорту речовин, зокрема, робота іонних насосів; знижується біосинтез білків неколагенового типу, що беруть участь у структурній організації клітин і тканин. В остаточному підсумку розвиваються ушкодження клітин і некробіотичні зміни, які в найважчих випадках закінчуються утворенням осередку некрозу - інфаркту.
III. Посилення біосинтезу компонентів сполучної тканини - колагену, глікозамі-ногліканів, глікопротеїнів, що є основою для наступного склерозування ішемізо-ваної ділянки тканини або органа.
13.20. Що таке стаз?
Стаз - це уповільнення й зупинка течії крові в капілярах, дрібних артеріях і венах.
13.21. Назвіть основні варіанти стазу та їхні причини.
Розрізняють ішемічний, венозний та істинний (капілярний) стаз.
Ішемічний і венозний стаз розвивається як наслідок ішемії та венозної гіперемії, а тому має ті ж причини, що й зазначені місцеві розлади кровообігу.
Причиною істинного стазу можуть бути фізичні (холод, тепло), хімічні (отрути, концентрований розчин натрію хлориду й інших солей, скипидар) і біологічні фактори (токсини мікроорганізмів).
У патогенезі істинного стазу велике значення надають двом факторам:
1) внутрішньокапілярній агрегації еритроцитів, тобто їх склеюванню й утворенню конгломератів, що утрудняють течію крові;
2) уповільненню течії крові в капілярах унаслідок згущення крові. В останньому випадку провідна роль належить підвищенню проникності стінок судин, розташованих у зоні стазу.
13.22. Що таке тромбоз?
Тромбоз -це процес прижиттєвого утворення на внутрішній поверхні стінки судин згустків крові, що складаються з її елементів. Ці згустки отримали назву тромбів.
Тромби бувають пристінковими (частково зменшують просвіт судини) і закупорювальними.
Залежно від будови розрізняють білі, червоні й змішані тромби. У першому випадку тромб утворюють тромбоцити, лейкоцити, а також невелика кількість білків плазми; у другому - еритроцити, скріплені нитками фібрину; змішані тромби складаються з білих та червоних шарів, що чергуються між собою.
13.23. Назвіть три основні фактори, що сприяють тромбоутворенню (тріада Вірхова).
1. Ушкодження судинної стінки. Воно може виникати під дією фізичних факторів (механічна травма, електричний струм), хімічних, біологічних (ендотоксини мікроорганізмів), а також у результаті порушення живлення й метаболізму судинної стінки.
2. Порушення активності систем зсідання і протизсідання у крові та судинній стінці. Для утворення тромбів велике значення має як підвищення активності системи зсідання крові за рахунок збільшення в ній концентрації прокоагулянтів, так і зниження активності антикоагулянтної і фібринолітичної систем.
3. Уповільнення течії крові. Цей чинник дає можливість пояснити, чому у венах тромби утворюються частіше, ніж в артеріях, у венах нижніх кінцівок - частіше, ніж у венах верхніх кінцівок; а також високу частоту тромбоутворення при декомпенсації кровообігу, тривалому перебуванні хворої людини в ліжку.
13.24. З яких фаз складається процес утворення тромбу? У чому їх сутність?
Процес тромбоутворення має дві фази: клітинну і плазматичну.
Сутність клітинної фази полягає в адгезії, агрегації й аглютинації тромбоцитів (докладно див. розд. 26).
Плазматична фаза (фаза коагуляції крові) являє собою ланцюг послідовних біохімічних реакцій зсідання крові, кінцевим результатом яких є утворення фібрину -важливого компонента тромбу.
13.25. Які негативні наслідки може мати тромбоутворення в умовах патології?
"При різних захворюваннях утворення тромбів може супроводжуватися важкими наслідками, зумовленими гострим порушенням кровообігу в зоні судини, в якій виник тромб (ішемія при тромбозі артерій і застій крові при тромбозі вен). Кінцевим етапом може бути розвиток некрозу (інфаркту) у басейні тромбованої, позбавленої колатералей судини. Особливо велика роль тромбозу вінцевих артерій у розвитку інфаркту міокарда. Тромбоз артерій може призводити до трофічних порушень із наступним розвитком гангрени кінцівок при атеросклерозі, облітеративному ендартеріїті, цукровому діабеті.
13.26. Що таке емболія?
Емболія — це закупорка судин тілами, принесеними течією крові або лімфи. Ці тіла називаються емболами.
13.27. Які виділяють види емболії?
Залежно від характеру емболів та їхнього походження розрізняють емболію екзогенну й ендогенну.
За локалізацією виділяють емболію великого і малого кола кровообігу, а також системи ворітної вени.
Дуже рідко буває так звана ретроградна емболія, коли рух ембола відбувається не за гемодинамічними законами, а відповідно до сили тяжіння самого ембола, і парадоксальна емболія, що спостерігається при незарощенні міжпередсердної або між-шлуночкової перегородки, у результаті чого емболи з вен великого кола кровообігу й правої половини серця переходять у ліву, обминаючи мале коло.
13.28. Назвіть основні причини емболії екзогенного походження.
Залежно від природи й характеру емболів, що надходять ззовні, розрізняють такі види екзогенної емболії: повітряну, газову, бактеріальну, паразитарну, емболію твердими сторонніми тілами.
Повітряна емболія виникає при пораненні великих вен голови й шиї, які слабко спадаються й тиск у яких близький до нуля або негативний. У результаті в ушкоджені вени засмоктується повітря, особливо на висоті вдиху, з наступною емболією судин малого кола кровообігу.
Газова емболія розвивається при різкому перепаді атмосферного тиску від підвищеного до нормального (у робочих кесонів і водолазів) або від нормального до зниженого (при швидкому піднятті на висоту або під час розгерметизації кабіни висотного літального апарата). При цьому зменшується розчинність газів у тканинах і крові, відбувається десатурація, тобто перехід газів з розчиненого стану в газоподібний, і закупорка бульбашками цих газів (у першу чергу азоту) капілярів, розташованих головним чином у системі великого кола кровообігу.
13.29. Назвіть основні причини емболії ендогенного походження.
До ендогенної емболії відносять емболію тромбом (тромбоемболію), жирову емболію, тканинну емболію, емболію навколоплідними водами.
Джерелом тромбоемболії є тромб, що відірвався, найчастіше при асептичному або гнійному його розплавленні.
Жирова емболія виникає при потраплянні в кровоносні судини крапель жиру. Причиною цього найчастіше є ушкодження (роздроблення, сильний струс) кісткового мозку, підшкірної або тазової клітковини.
Тканинна емболія може бути обумовлена занесенням у кровоносне русло частинок різних тканин організму при їх ушкодженні. Особливе значення має емболія судин клітинами злоякісних пухлин, оскільки вона є основним механізмом утворення метастазів.
Емболія навколоплідними водами виникає в результаті потрапляння навколоплідних вод під час пологів в ушкоджені судини матки на ділянці плаценти, що відокремилася.
13.30. Що розуміють під мікроциркуляцією? Назвіть основні типи порушень мікроциркуляції.
Мікроциркуляція — це рух крові й лімфи по мікроциркуляторних кровоносному і лімфоносному руслах.
Мікроциркуляторне кровоносне русло складається з судин, діаметр яких не перевищує 100 мкм, тобто артеріол, метартеріол, капілярних судин, венул і артеріоло-ве-нулярних анастомозів (рис. 38).
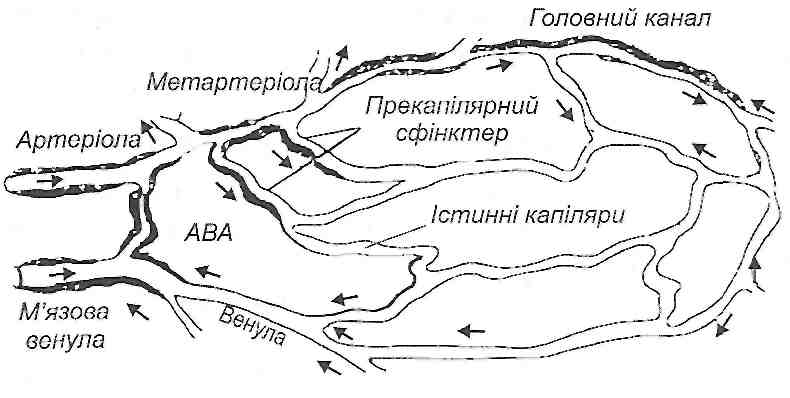
Рис. 38. Схема будови мікроциркупяторного русла: ABA - артеріовенозний анастомоз
Мікроциркуляторне лімфоносне русло представлене початковим відділом лімфатичної системи, у якому відбувається утворення лімфи і надходження її в лімфатичні капіляри.
Порушення мікроциркуляції поділяють на три типи: внутрішньосудиннї, порушення, пов 'язані зі змінами самих судин; позасудинні.
- Великі успіхи у вивченні закономірностей мікроциркуляції в нормі і при патології пов'язані з іменем О. М. Чернуха.
13.31. У чому сутність феномена під назвою "сладж"?
Сладж є внутрішньо судинним порушенням мікроциркуляції, пов'язаним зі зміною реологічних властивостей крові. Основні фактори таких змін — це порушення суспензійної стабільності крові, а також підвищення її в'язкості.
Головними особливостями крові при сладжі є прилипання один до одного еритроцитів, лейкоцитів і тромбоцитів, збільшення в'язкості крові, що утруднює її рух по мі-кросуцинах. При цьому течія крові різко уповільнюється й нагадує переміщення мулу по дну ріки (назва "сладж" походить від англ. sludge - густа грязь, баговиння, мул).
Залежно від розмірів агрегатів клітин крові, характеру їхніх контурів і щільності впакування еритроцитів розрізняють такі типи сладжу: класичний (великі розміри агрегатів, нерівні обриси контурів і щільне впакування еритроцитів), декстриновий (різна величина агрегатів, округлі обриси, щільне впакування еритроцитів) і аморфний гранулоподібний (величезна кількість дрібних агрегатів у вигляді гранул, що складаються всього з декількох еритроцитів).
13.32. Як здійснюється обмін води між плазмою крові й інтерстиціальною рідиною?
В основі обміну води між плазмою крові й інтерстиціальною рідиною лежать три механізми (рис. 39).
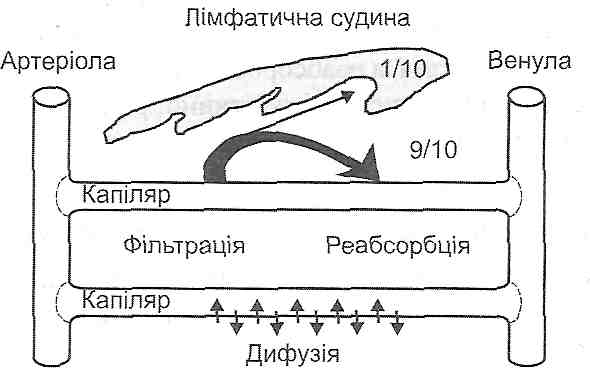
Рис. 39. Обмін рідини між кровоносними капілярами і міоісклітинним простором
1. Двостороння дифузія. Має найбільше значення. її швидкість настільки велика, що при проходженні крові через капіляри рідина плазми встигає 40 разів повністю обмінятися з рідиною міжклітинного простору.
Швидкість дифузії води через загальну обмінну поверхню капілярів становить 60 л за 1 хв, або 85 000 л за добу.
2. Фільтрація-реабсорбція. Це механізм перерозподілу води, що визначається двома факторами: а) гідростатичним тиском крові і міжклітинної рідини; б) онкотичним тиском плазми крові і рідини інтерстиціального простору. Співвідношення між цими факторами таке, що в артеріальній частині капіляра відбувається фільтрація води, а у венозній частині ~Ji реабсорбція.
Швидкість фільтрації через загальну обмінну поверхню капілярів організму становить 14 мл/хв., або 20 л за добу, а швидкість реабсорбції- 12,5 мл/хв., або 18 л за добу.
3. Мікр о везикулярний транспорт. Це механізм активного транспорту через ендотелій капілярів. Його основу становить явище ендоцитозу (піноцитоз, мікропіноцитоз).
13.33. Як впливають зміни гідростатичного і онкотичного тиску крові та міжклітинної рідини на інтенсивність процесів фільтрації-реабсорбції води в капілярах?
Напрямок руху води й інтенсивність процесів фільтрації-реабсорбції визначаються величиною ефективного фільтраційного тиску (Реф).
![]()
де Рг й Рок - гідростатичний і онкотичний тиск крові; Р^ і Рот - гідростатичний і онкотичний тиск тканинної рідини.
За умов норми в артеріальному кінці капілярів Р = 9 мм рт. ст. і рідина переходить із просвіту судин у тканину (фільтрація), а на венозній ділянці Р = -6 мм рт. ст, тому рідина переходить у зворотному напрямку — з тканин у капіляри (реабсорбція).
Фільтрація води збільшується, а реабсорбція зменшується при збільшенні гідростатичного тиску крові й онкотичного тиску інтерстиціальної рідини, а також при зменшенні онкотичного тиску плазми крові й тканинного гідростатичного тиску.
Фільтрація води зменшується, а реабсорбція зростає при зменшенні гідростатичного тиску крові й онкотичного тиску міжклітинної рідини, а також при збільшенні онкотичного тиску плазми крові й гідростатичного тиску рідини інтерстиціального простору.
13.34. Що таке недостатність лімфообігу? Назвіть основні її форми.
Недостатність лімфообігу - це стан, при якому лімфатичні судини не виконують свою основну функцію - здійснення постійного й ефективного дренажу інтерстиціального простору.
Розрізняють такі форми недостатності лімфообігу. 1. Механічна недостатність. Виявляє себе утрудненням відтоку лімфи у зв'язку з наявністю органічних (здавлювання пухлиною, рубцем, облітерація лімфатичних
судин при їх запаленні, тромбозі та ін.) або функціональних причин (підвищення тиску в магістральних венозних судинах, спазм лімфатичних судин, припинення м'язових скорочень та ін.).
2. Динамічна недостатність. Виникає тоді, коли об'єм транссудації міжклітинної рідини перевищує можливості лімфатичної системи забезпечувати ефективний дренаж інтерстиціальної тканини,
3. Резорбційна недостатність. Обумовлена структурними змінами інтерстиціальної тканини, накопиченням білків і осадженням їхніх патологічних видів в інтерстиції.
Основними проявами недостатності лімфообігу в гострій стадії є набряк, накопичення білків і продуктів їхнього розпаду в інтерстиціальній тканині, а в хронічній стадії - розвиток фіброзу й склерозу.
14. Запалення
74.7. Дайте визначення поняття "запалення".
Запалення - це типовий патологічний процес, що виникає в результаті ушкодження тканини й виявляє себе комплексом структурних, функціональних і метаболічних порушень, а також розладами мікроциркуляції.
74.2. Чому запалення називають типовим патологічним процесом?
Загальні закономірності розвитку запалення виявляють себе завжди, незалежно від його причини, локалізації, виду організму та його індивідуальних особливостей.
Запалення може виникати в різних органах і тканинах (ангіна, пневмонія, апендицит), його причиною можуть бути механічна травма, вплив температури, віруси, бактерії та ін.; воно може розвиватися у тварин і в людини. У кожному конкретному випадку запалення має свої особливості. Однак є щось загальне, яке виявляє себе завжди. Це загальне й становить сутність запалення як типового патологічного процесу.
14.3. Назвіть зовнішні ознаки запалення.
Припухлість (tumor), почервоніння (rubor), жар (color), біль (dolor) і порушення функції (functio laesd). Ці ознаки відомі як пеншада Цельса—Голена.
14.4. Що може бути причиною запалення?
Фактори, здатні викликати розвиток запалення, називають флогогенними. Та-кими є:
1) фактори фізичного походження (механічні, термічні, іонізуюча радіація, ультрафі- \ олетове випромінювання та ін.);
2) хімічні фактори (кислоти, луги, солі важких металів, феноли, альдегіди та ін.);
3) біологічні агенти (віруси, бактерії, найпростіші).
Стосовно організму флогогенні агенти можуть бути екзогенними й ендогенними. Екзогенні надходять в організм або діють із зовнішнього середовища. Ендогенні утворюються в самому організмі (токсичні продукти обміну, жовчні кислоти та ін.).
74.5. Які методи використовують при вивченні запалення?
Конгейм уперше запропонував вивчати зміни місцевого кровообігу при запаленні на брижі жаби (дослід Конгейма).
Подружжя Кларків розробило таку методику. На двох протилежних ділянках шкіри вуха кроля видаляли епідерміс і на його місце вставляли диски зі слюди. У такому прозорому віконці можна було безперервно спостерігати кровообіг при запаленні в тонкому шарі тканини між дисками.
Пізніше Сельє запропонував вивчати кровообіг при запаленні в судинах защічних мішків хом'яка, роздутих повітрям.
Широко використовують також біохімічні методи дослідження (Менкін), що дозволяють вивчати біологічно активні речовини і порушення метаболізму у вогнищі запалення.
Порівняльно-еволюційний підхід до вивчення запалення був запропонований /. /. Мечтіковим.
14.6. З яких компонентів складається патогенез запалення?
У патогенезі запалення розрізняють:
1) альтерацію;
2) порушення мікроциркуляції'з явищами ексудації і еміграції;
3) проліферацію.
Ці компоненти іноді називають стадіями запалення (рис. 40). Однак варто пам'ятати, що зазначені процеси не є строго послідовними, оскільки вони перекриваються в часі.
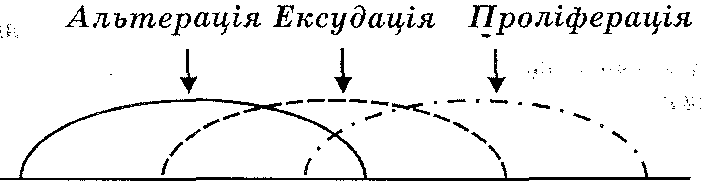
Рис. 40. Стадії патогенезу запалення
14.7. У чому сутність стадії альтерації?
В основі альтерації лежать дві групи явищ:
1) ушкодження клітин і позаклітинних структур;
2) утворення медіаторів запалення.
14.8. Що таке первинна і вторинна альтерація?
Первинна альтерація — це ушкодження тканини, яке виникає внаслідок безпосередньої дії флогогенних агентів.
Вторинна альтерація — це ушкодження тканини, що виникає в результаті дії факторів, які утворилися внаслідок первинної альтерації.
14.9. Які фактори викликають розвиток вторинної альтерації у вогнищі запалення?
1. Медіатори запалення (лізосомні фактори, активований комплемент, лімфокіни-лімфотоксини).
2. Вільні радикали й пероксиди.
3. Гіпоксія, що виникає в результаті місцевих розладів кровообігу.
4. Місцевий ацидоз.
5. Підвищення осмотичного і онкотичного тиску у вогнищі запалення.
14.10. Назвіть причини розвитку місцевого ацидозу у вогнищі запалення.
Розрізняють первинний і вторинний ацидоз. Первинний ацидоз виникає в перші ЗО хв. унаслідок деполімеризації основної інтерстиціальної речовини і вивільнення карбоксильних і сульфатних груп.
Вторинний ацидоз розвивається пізніше й обумовлений порушеннями обміну речовин у вогнищі запалення. До його виникнення причетні накопичення молочної кислоти (активація гліколізу), вихід з ушкоджених клітин недоокиснених продуктів циклу Кребса (три- і дикарбонових кислот), вивільнення вільних жирових кислот, амінокислот і фосфорної кислоти в результаті гідролітичного розщеплення тригліце-ридів, фосфоліпідів, білків, АТФ.
14.11. Чому в осередку запалення розвиваються гіперосмія й гіперон кія?
Збільшення осмотичного тиску у вогнищі запалення (гіперосмія) зумовлене насамперед виходом іонів калію з ушкоджених клітин, а також вивільненням калію зі зв'язаного з внутрішньоклітинними білками стану. Останнє є результатом протеолізу, що відбувається в клітинах за умов їхнього ушкодження.
Збільшення онкотичного тиску (гіперонкія) обумовлене:
1) надходженням білків у тканину із крові в процесі ексудації (плазмове джерело);
2) розщепленням великих білкових молекул на дрібніші під дією лізосомних ферментів (тканинне джерело).
14.12. Які порушення обміну речовин закономірно виникають у вогнищі запалення?
Запалення завжди починається з посилення обміну речовин. У гострому періоді запалення переважають процеси розпаду, катаболізму. Відбувається збільшення
інтенсивності споживання кисню й активація процесів гліколізу. Під дією лізосомних гідролаз великі молекули розщеплюються на дрібні. Це все характеризують терміном "пожежа обміну". Аналогія полягає не тільки в тому, що обмін речовин у вогнищі запалення різко підвищений, але й у тому, що "горіння" іде не до кінця, а з утворенням недоокиснених продуктів.
Пізніше відбувається активація анаболічних процесів, що забезпечують явища відновлення (репарації). Збільшується синтез нуклеїнових кислот, різко зростає утворення глікозаміногліканів, глікопротеїнів, колагену й інших компонентів сполучної тканини.
Велике значення у вивченні біохімічних змін у вогнищі запалення мали роботи Менкіна і Альперна.
14.13. Що таке медіатори запалення? Назвіть основні їх класи.
Медіатори запалення — це біологічно активні сполуки, які утворюються у вогнищі запалення і визначають його патогенез.
Розрізняють медіатори клітинного (утворюються в клітинах) і плазмового (утворюються й надходять із плазми крові) походження.
До медіаторів клітинного походження відносять лізосомні фактори, продукти вільнорадикального окиснення, продукти тканинних базофілів, похідні арахідонової кислоти, цитокіни, фактори росту.
Медіаторами плазмового походження є кініни, продукти активації комплементу, продукти активації системи зсідання крові і фібринолітичної системи.
14.14. Яку роль відіграють лізосомні фактори в патогенезі запалення?
До лізосомних факторів відносять: лізосомні ферменти (кислі й нейтральні гід-ролази) і неферментні катіонні білки.
Роль лізосомних ферментів, основним джерелом яких є лейкоцити, полягає в ініціюванні таких змін.
1. Вони викликають вторинну альтерацію.
2. Беруть участь в утворенні і активації інших медіаторів запалення: стимулюють дегрануляцію тканинних базофілів, активують калікреїн-кінінову систему, систему комплементу; вивільнюють арахідонову кислоту з фосфоліпідів клітинних мембран.
3. Безпосередньо підвищують проникність капілярів завдяки дії еластази, колагена-зи і гіалуронідази на компоненти базальної мембрани судинної стінки.
4. Викликають розвиток фізико-хімічних і метаболічних змін у вогнищі запалення: активують гідролітичне розщеплення речовин, чим сприяють розвитку місцевого ацидозу й гіперонкії.
Неферментні катіонні білки лізосом викликають вторинну альтерацію, підвищують проникність судин, активують хемотаксис лейкоцитів.
14.15. Які фактори можуть викликати дегрануляцію тканинних базофілів у вогнищі запалення?
1. Безпосередня дія флогогенного агента на тканинні базофіли (механічне ушкодження, температура, продукти бактерій, хімічні речовини - лібератори гістаміну).
2. Комплекси антиген-антитіло.
3. Активні протеази, зокрема, лізосомні.
4. Побічні продукти активації комплементу - СЗа, С5а.
14.16. Які медіатори запалення вивільняються при дегрануляції тканинних базофілів ? Яку вони мають дію у вогнищі запалення?
1. Біогенні аміни — гістамін і в деяких видів тварин (зокрема щурів) - серотонін. Основні ефекти гістаміну, що мають важливе значення в патогенезі запалення: а) розширення артеріол, що веде до розвитку артеріальної гіперемії в осередку запалення;
б) підвищення проникності мікросуцин (венул) - одна з причин запального набряку;
в) подразнення нервових закінчень, що зумовлює розвиток болю;
г) спазм гладких м'язів бронхів, матки, кишок. Цим, зокрема, пояснюють порушення функції зазначених органів при їхньому запаленні.
2. Гепарин. Другий основний компонент гранул тканинних базофілів є глікозаміно-гліканом. Його вважають протизапальним медіатором, оскільки він (1) має антикоагулянту дію, (2) гальмує адгезію і агрегацію тромбоцитів, (3) зв'язує біогенні аміни, (4) пригнічує активацію комплементу та калікреїн-кінінової системи.
3. Фактори, що впливають на клітини крові. До них можна віднести поліпептиди: (1) фактор еміграції еозинофілів, (2) фактор еміграції нейтрофілів, а також сполуку фосфоліпідного походження — (3) фактор*агрегації тромбоцитів (ФАТ).
Останній відіграє особливо важливу роль у патогенезі запалення. Утворюючись одразу після стимуляції тканинних базофілів, ФАТ зумовлює такі ефекти у вогнищі запалення:
а) активує процеси агрегації тромбоцитів та вивільнення їхніх гранул. Як наслідок, з тромбоцитів в осередок запалення виходять серотонін адреналін, аденінові нуклеотиди (АТФ, АДФ, АМФ), арахідонова кислота та тромбок-сани, тромбоцитарний фактор росту та інші;
б) навіть у дуже низьких концентраціях зумовлює розширення артеріол (артеріальну гіперемію) і збільшення проникності венул. Кількісно ці ефекти ФАТ відповідно в 100 і 10000 разів сильніші за дію гістаміну;
в) значно посилює адгезію лейкоцитів до ендотелію судин (крайове стояння) і стимулює хемотаксис нейтрофілів та макрофагів у вогнищі запалення.
Крім наведених вище медіаторів запалення під час дегрануляції тканинних базофілів вивільнюються лейкотрієни (повільно реагуюча субстанція анафілаксії), гідролітичні ферменти, катіонні білки.
14.17. Як відбувається активація калікреїн-кінінової системи? Назвіть основні функціональні ефекти кінінів.
У плазмі крові є неактивний протеолітичний фермент колікреїноген. З появою в крові активних протеаз (лізосомні ферменти, фактор Хагемана, трипсин, тромбін, плазмін та ін.) відбувається відщеплення ділянки молекули калікреїногену, в результаті чого він перетворюється на активний фермент — калікреїн.
. Під дією калікреїну відбувається відщеплення від а2-глобуліну плазми крові (кі-ніногену) пептидів, які отримали назву кініни. Найважливішими кінінами є калідин і брадикінін, що складаються відповідно з 9 і 10 амінокислотних залишків (рис. 41).
У вогнищі запалення кініни викликають: 1) розширення артеріол (артеріальну гіперемію); 2) підвищення проникності судинної стінки; 3) подразнення нервових закінчень (біль).
14.18. Які медіатори запалення є похідними арахідонової кислоти? Як вони утворюються і як діють ?
Похідними арахідонової кислоти є простагландини, тромбоксани, простациклі-ни, лейкотрієни (рис. 42).
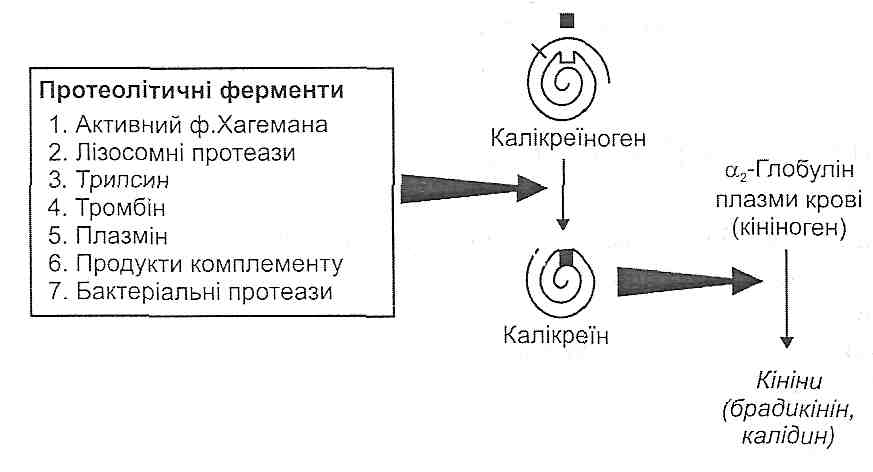
Рис. 41. Механізми активації калікреїн-кінінової системи
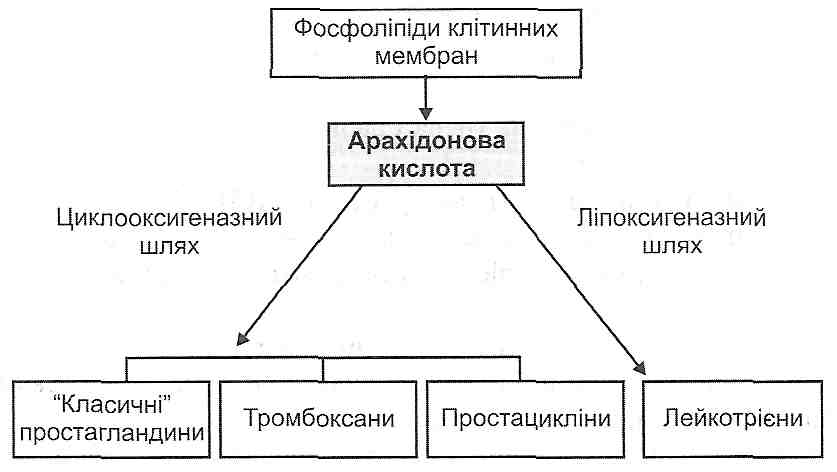
Рис. 42. Похідні арахідонової кислоти - медіатори запалення
Під дією ферменту фосфоліпази А2 (активується іонами кальцію) відбувається вивільнення арахідонової кислоти з фосфоліпідів клітинних мембран. Далі можливі два шляхи її перетворення: циклооксигеназний і ліпоксигеназний. У результаті активації першого утворюються "класичні" простагландини Е2, D2, F2 , тромбоксани й простацикліни, при активації другого - лейкотрієни С4, D4, E4.
Простагландини утворюються практично у всіх клітинах. Вони мають властивість розширювати артеріоли, звужувати венули, підвищувати проникність судинної стінки, зменшувати поріг больової чутливості нервових закінчень.
Тромбоксани утворюються в тромбоцитах. Вони викликають звуження артеріол і агрегацію тромбоцитів.
Простацикліни переважно вивільняються ендотеліальними клітинами судин і є антагоністами тромбоксанів. З їх дією пов'язано розширення артеріол і пригнічення агрегації тромбоцитів.
Місцем утворення лейкотрієнів є лейкоцити і тканинні базофіли. У вогнищі запалення вони стимулюють хемотаксис лейкоцитів і підвищують проникність кровоносних судин.
14.19. Що таке цитокіни? Яку роль вони відіграють у патогенезі запалення?
Цитокіни - це збірне поняття для позначення великої групи біологічно активних речовин білково-пептидної природи, що регулюють взаємодію між різними типами клітин.
Цитокіни синтезуються (1) активованими лімфоцитами (лшфокіни);2) моноцитами і макрофагами (монокіни), а також (3) багатьма іншими клітинами (нейтрофілами, фібробластами, ендотеліальними клітинами, тканинними базофілами, клітинами нейроглії та ін.).
На сьогодні описано понад 50 різних цитокінів. Залежно від функціональних ефектів їх поділяють на чотири групи.
I. Інтерлейкіни (ІЛ). Це сполуки, що регулюють взаємодію між різними видами лейкоцитів. Відомі нині 18 видів інтерлейкінів беруть участь у здійсненні імунних реакцій, у патогенезі алергії.
II. Інтерферони (ІНФ). Зазначена група білків здійснює природний неспецифічний противірусний захист.
III. Гемопоетичні колонієстимулятивні фактори (КСФ). Ці сполуки (гемопоети-ни) здійснюють регуляцію кровотворення в червоному кістковому мозку.
IV. Фактори, що пригнічують ріст пухлин, зокрема, фактор некрозу пухлин (ФНП)
За участю в патогенезі запалення цитокіни поділяють на (1) прозапсиїьні і (2) протизапальні. Спричинювані ними ефекти виявляють себе на місцевому рівні (в осередку запалення) і на рівні організму (системна дія).
Цитокіни є причетними до розвитку основних подій, що складають суть запального процесу, а саме:
а) вторинної альтерації. З-поміж інших цитокінів прямий стосунок до ушкодження клітин і позаклітинних компонентів має ФНП-р (лімфотоксин) — продукт активації макрофагів і Т-лімфоцитів. Високі його концентрації (1) спричинюють цитоліз (т.зв. кілінг-ефект), (2) посилюють генерацію вільних радикалів у вогнищі запалення; (3) індукують синтез колагеназ і, як наслідок, сприяють деградації колагену;
б) еміграції лейкоцитів. Ряд цитокінів (ІЛ-1, ФНП, ІНФ-у) індукують синтез адге-зивних білків в ендотеліальних клітинах, що сприяє розвиткові крайового стояння (прилипання до поверхні ендотелію) нейтрофілів, моноцитів і лімфоцитів. Крім того, деякі інтерлейкіни (ІЛ-6, ІЛ-8) значно посилюють хемотаксис лейкоцитів у вогнищі запалення;
в) проліферації. Однією з властивостей багатьох цитокінів є їхня мітогенна активність, що виявляє себе посиленням процесів проліферації в осередку запалення. Водночас, деякі цитокіни (ІЛ-1, ФНП) стимулюють синтез колагену і новоутворення кровоносних судин (ангіогенез);
г) загальних проявів запалення (див. запит. 14.37). У розвитку таких змін особливо велике значення має інтерлейкін-1 (ІЛ-1) (рис. 43).
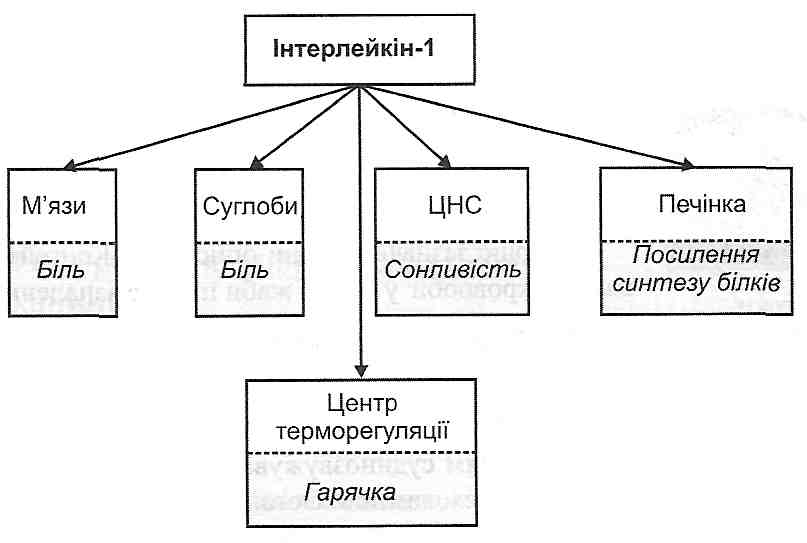
Рис. 43. Деякі системні ефекти інтерлейкіну-1
Серед відомих сьогодні цитокінів є й протизапальні медіатори, зокрема ІЛ-10, який пригнічує синтез лейкоцитами багатьох біологічно активних сполук — активних учасників запального процесу.
14.20. Яке значення має комплемент і продукти його активації в патогенезі запалення?
Активований комплемент здатний викликати вторинну альтерацію у вогнищі імунного запалення.
Побічні продукти його активації- СЗа й С5а- викликають дегрануляцію тканинних базофілів і є сильними хемотаксинами для нейтрофілів.
Проміжні продукти, зокрема СЗЬ, мають властивості опсонінів, тобто полегшують фагоцитоз бактерій. Крім того, проміжні продукти, що виявляють протеазну активність, можуть активувати калікреїн-кінінову систему і систему зсідання крові.
14.21. Які продукти активації системи зсідання крові і фібринолітичної системи можуть впливати на патогенез запалення?
У розвитку запалення можуть мати значення:
1) фібршопептиди (відщеплюються від фібриногену при перетворенні його у фібрин) - збільшують проникність судин і активують хемотаксис лейкоцитів;
2) продукти деградації фібрину — збільшують проникність судин;
3) активні протеази (тромбін, плазмін) — активують калікреїн-кінінову систему й систему комплементу.
14.22. Назвіть стадії порушень місцевого кровообігу у вогнищі запалення. Хто їх уперше описав?
I. Короткочасна ішемія (тривалість від 10—20 с до кількох хвилин).
II. Артеріальна гіперемія (триває 20—30 хв, максимум до 1 год).
III. Венозна гіперемія.
IV. Стаз. Уперше зазначені зміни описав Ю. Конгейм (1867), вивчаючи кровообіг у брижі жаби під час запалення.
14.23. Який механізм лежить в основі короткочасної ішемії на початку запалення?
Короткочасну ішемію на початку запалення обумовлює рефлекторний спазм артеріол. Він пов'язаний зі збудженням судинозвужувальних адренергічних нервів і виділенням їхніми закінченнями катехоламінів. Останні, діючи на а-адренорецепто-ри, викликають скорочення гладких м'язів судинної стінки.
Ішемія, що виникає, є короткочасна, тому що швидко настає виснаження катехо-ламінових депо в нервових закінченнях і відбувається руйнування вивільнених медіаторів відповідними ферментами, зокрема, моноаміноксидазою. Крім того, вазокон-стрикція в деяких тканинах може перекриватися судинорозширювальним впливом холінергічних нервів, що реалізується за типом аксон-рефлексу.
14.24. Назвіть механізми розвитку артеріальної гіперемії у вогнищі запалення.
1. Нейрогенні механізми (нейротонічний і нейропаралітичний) (див. розд. 13). Вони мають значення в перші хвилини розвитку артеріальної гіперемії.
2. Вплив фізично-хімічних факторів: ацидозу, збільшення вмісту іонів калію в тканині, гіпоксії та ін.
3. Вплив продуктів метаболізму: молочної кислоти, АДФ, АМФ, аденозину.
4. Дія медіаторів запалення:
а) гістаміну і серотоніну;
б) кінінів (брадикініну і калідину);
в) простагландинів і простациклінів.
14.25. Які фактори викликають перехід артеріальної гіперемії у венозну в процесі розвитку запалення?
Можна виділити дві групи таких факторів.
І. Внутрішньосудинні фактори:
1) збільшення в'язкості крові;
2) мікротромбоутворення;
3) зсідання крові;
4) крайове стояння лейкоцитів;
5) агрегація еритроцитів;
6) набрякання ендотеліальних клітин. II. Позасудинні фактори:
1) здавлювання венозних судин набряковою рідиною;
2) втрата венулами еластичних властивостей внаслідок розщеплення колагену і еластину лізосомними ферментами (В. В. Воронін).
14.26. Що таке ексудація? Які механізми лежать в основі виходу рідкої частини крові з судин у запалену тканину?
Ексудація - це вихід рідини й розчинених у ній компонентів плазми крові із кровоносних судин у тканину. У широкому значенні слова це поняття включає й еміграцію лейкоцитів.
В основі ексудації лежать такі механізми:
1) підвищення проникності судинної стінки;
2) збільшення гідростатичного тиску в судинах;
3) збільшення осмотичного і онкотичного тиску в тканині.
14.27. Назвіть механізми підвищення проникності судинної стінки при запаленні.
1. Активація мікровезикулярного транспорту через ендотеліальні клітини.
2. Утворення наскрізних трансклітшних каналів в ендотеліоцитах (є наслідком значного посилення мікровезикулярного транспорту).
3. Збільшення просвіту міжендотеліальних щілин (відбувається в результаті скорочення й округлення ендотеліоцитів).
4. Десквамація (злущування) ендотелію. Є проявом первинної й вторинної альтерації.
5. Деполімеризація речовин, що з'єднують ендотеліальні клітини і є компонентами базальної мембрани судинної стінки.
14.28. Що є причиною підвищення проникності судинної стінки у вогнищі запалення?
До основних причин підвищення проникності судин відносять:
а) продукти дегрануляції тканинних базофілів (гістамін і серотонін);
б) кініни (брадикінін і калідин);
в) простагландини й деякі лейкотрієни;
г) лізосомні ферменти (еластаза, колагеназа, гіалуронідаза) і неферментні катіонні білки;
ґ) фібринопептиди й продукти деградації фібрину;
д) ацидоз.
14.29. Яка динаміка підвищення проникності судин при запаленні?
Виділяють дві фази підвищення проникності судин. Пік першої (ранньої) фази припадає на 10-15 хв від початку запалення. Основною причиною її розвитку є гістамін.
Друга (пізня) фаза починається через 1 год від початку запалення й триває кілька діб. До її розвитку причетні всі інші медіатори запалення, що підвищують проникність судин.
14.30. Що таке еміграція лейкоцитів? Які лейкоцити і в якій послідовності емігрують у вогнище запалення?
Еміграція — це перехід лейкоцитів крові із кровоносних судин у тканину.
Уперше послідовність еміграції лейкоцитів описав І. Мечніков. Спочатку в запалену тканину виходять полінуклеарні фагоцити, зокрема нейтрофіли. Вони знищують мікробів, що стали причиною ушкодження тканини. Потім у вогнище виходять мононуклеарні фагоцити - моноцити. Вони фагоцитують загиблі клітини, тканинний детрит, розчищаючи тим самим "поле бою". На кінцевих етапах, особливо при імунному запаленні, у тканину надходять лімфоцити.
14.31. Що таке крайове стояння лейкоцитів? Які його механізми?
Крайове стояння лейкоцитів (маргінація) — це перехід лейкоцитів із циркулюючого пулу в пристінковий (маргінальний). Воно триває від кількох хвилин до 1 години. В основі цього явища лежать такі механізми.
1. При уповільненні течії крові (венозна гіперемія) лейкоцити як найлегші формені елементи відкидаються за законами фізики на периферію.
2. Відбувається випадання ниток фібрину на поверхні ендотелію. Гладка в нормі поверхня ендотелію стає шорсткою. "Бахрома", що утворилася, затримує лейкоцити.
3. Має місце електростатична взаємодія лейкоцитів з ендотеліальними клітинами, її пояснюють втратою лейкоцитами поверхневого негативного заряду і утворенням "кальцієвих містків" між лейкоцитами й ендотеліоцитами.
4. На поверхні лейкоцитів і ендотеліальних клітин з'являються так звані "адгезивні білки ", які специфічно взаємодіють один з одним.
14.32. Що таке адгезивні білки лейкоцитів і ендотеліальних клітин? Що викликає їхню появу?
Адгезивні білки синтезуються в лейкоцитах та ендотеліальних клітинах і зберігаються у внутрішньоклітинних везикулах. При активації клітин відбувається злиття везикул з плазматичною мембраною, у результаті чого зазначені білки виявляються вмонтованими в цю мембрану. Специфічні ділянки молекул розташовуються зовні і можуть взаємодіяти з відповідними структурами адгезивних білків інших клітин.
Активаторами утворення і включення адгезивних білків у мембрану лейкоцитів є:
а) побічний продукт активації комплементу С5а;
б) лейкотрієни В4;
в) фактор активації тромбоцитів (ФАТ).
Аналогічні процеси в ендотеліальних клітинах викликають: а) ендотоксини бактерій;
б) інтерлейкін-1;
в) фактор некрозу пухлин.
14.33. Яким чином лейкоцити долають судинну стінку, емігруючи в запалену тканину?
Емігруючи в тканину, лейкоцити долають два бар'єри капілярної стінки: ендотелій і базальну мембрану. Нейтрофіли і макрофаги проходять крізь ендотелій через міжендотеліальні щілини. Вони випускають свої псевдоподії в простір між ендотелі-оцитами і "розсовують" клітини.
Подолання базальної мембрани може бути обумовлено двома механізмами. Перший з них полягає в явищі тиксотропії-при контакті нейтрофіла з базальною мембраною її колоїди переходять зі стану гелю в стан золю (відбувається розрідження мембрани). Нейтрофіл легко проходить через золь, після чого золь знову перетворюється на щільний гель. Другий механізм полягає у виділенні нейтрофілами нейтральних протеаз (еластази, колагенази), які розщеплюють волокнисті компоненти базальної мембрани.
14.34. У чому полягає сутність стадії проліферації в патогенезі запалення?
Стадія проліферації охоплює:
1) розмноження клітин, тобто власне проліферацію;
2) синтез позаклітинних компонентів сполучної тканини - колагену, еластину, про-теогліканів, глікопротеїнів. Ці події супроводжуються значним посиленням ана-болічних процесів.
14.35. Які фактори викликають активацію розмноження клітин у вогнищі запалення?
I. Зменшення концентрації в тканині кейлонів. Кейлоїш — це речовини білкової природи, які утворюються зрілими клітинами. Вони є інгібіторами клітинного поділу. При ушкодженні і загибелі клітин у вогнищі запалення концентрація кейлонів у тканині зменшується, а отже, знімається гальмівний вплив кейлонів на малодиференційовані (камбіальні) клітини. Вони починають ділитися. Поділ триває доти, доки концентрація кейлонів не збільшиться до рівня, що існував у неушкодженій тканині.
II. Збільшення концентрації в тканині стимуляторів проліферації— факторів росту. Фактори росту надходять у тканину з плазми крові або є продуктами клітин, що перебувають в осередку запалення. Прикладами можуть бути фактор росту епідермісу, фактор росту фібробластів, фактор росту тромбоцитарного походження, фактор росту нервів, фактор некрозу пухлин, інсуліноподібні фактори росту (соматомедини), лімфокіни (мітогенні фактори). Дія зазначених регуляторів здійснюється через активацію внутрішньоклітинних протеїнкіназ (протеїнкі-нази С і тирозинових протеїнкіназ).
14.36. Які механізми лежать в основі розвитку місцевих клінічних ознак запалення?
1. Почервоніння (rubor) пов'язано з розвитком артеріальної, а потім і венозної гіперемії.
2. Підвищення місцевої температури (color) обумовлено підвищенням інтенсивності катаболічних процесів у вогнищі запалення, а також артеріальною гіперемією, під час якої в тканину надходить багато теплої артеріальної крові.
3. Основу набряку (tumor) становлять механізми ексудації (див. запит. 14.26).
4. Біль (dolor) виникає в результаті подразнення рецепторів медіаторами запалення (гістамін, кініни), а також механічним тиском ексудату, ацидозом і гіперосмією.
5. Порушення функції (functio leasa) є наслідком ушкодження й загибелі клітин.
14.37. Які загальні прояви характерні для запалення?
1. Гарячка. Розвивається внаслідок виділення нейтрофілами і макрофагами так званих лейкоцитарних пірогенів (інтерлейкіну-1).
2. Лейкоцитоз. При гострому запаленні, спричиненому гноєтворними мікробами, він характеризується абсолютним збільшенням кількості нейтрофілів у периферичній крові (нейтрофільоз) і зміщенням лейкоцитарної формули вліво. В основі цієї реакції лежать вихід лейкоцитів з резервного пулу червоного кісткового мозку в кров (дія інтерлейкіну-1 і фактора некрозу пухлин), а також стимуляція лейкопо-езу колонієстимулятивним фактором.
3. Підвищення вмісту в крові "білків гострої фази запалення".
4. Збільшення швидкості осідання еритроцитів (ШОЕ). Пов'язано зі збільшенням вмісту в плазмі крові грубодисперсних білків (глобулінів, фібриногену), у результаті чого зменшується поверхневий негативний заряд еритроцитів і вони легко склеюються.
5. Інтоксикація. Обумовлена надходженням у кров продуктів альтерації із запаленої тканини.
14.38. Що таке "білки гострої фази запалення"? Що є причиною підвищення їхньої кількості при запаленні?
"Білки гострої фази запалення" ("реактанти гострої фази") - це білки, концентрація яких у плазмі крові при гострому запаленні збільшується більш як на 50 %. Вони утворюються в основному в печінці під впливом:
а) продуктів, що надходять у кров з вогнища запалення (продуктів альтерації);
б) інтерлейкіну-1 макрофагального походження.
Нині відомо більше 10 "білків гострої фази запалення". Всі вони в основному мають захисне значення. До них відносять:.
1) інгібітори протеаз (орозомукоїд, а-антитрипсин);
2) антиоксиданти (гаптоглобін, церулоплазмін);
3) імуноглобуліни й антитілоподібніречовини (антитіла, С-реактивний білок).
14.39. Як впливають гормони на перебіг запалення? Який механізм протизапальної дії глюкокортикоїдів ?
За механізмом впливу на запалення гормони поділяють на прозапальні й протизапальні. Прикладом прозапальних гормонів, тобто гормонів, що посилюють запалення, можуть бути мінералокортикоїдщ зокрема альдостерон- Під його впливом істотно підвищується проникність стінок судин, у результаті чого збільшується ексудація.
Найважливішими протизапальними гормонами є глюкокортикоїди. У високих (лікувальних) дозах вони викликають дерепресію гена, що кодує структуру особливого білка - ліпокортину. Ліпокортин є природним внутрішньоклітинним інгібітором фосфоліпази Аг Пригнічення цього ферменту ліпокортином має два важливих наслідки: 1) з одного боку, зменшується утворення лізофосфоліпідів, у результаті чого зменшуються явища альтерації (стабілізація мембрани) (див. розд. 11); 2) з другого боку, зменшується утворення арахідонової кислоти та її похідних (простагландинів, тромбоксанів, простациклінів, лейкотрієнів). Ця обставина пояснює інші протизапальні ефекти глюкокортикоїдів — зменшення ексудації і еміграції лейкоцитів, ослаблення порушень мікроциркуляції. У результаті пригнічення процесів клітинного поділу й біосинтезу білка зменшуються явища проліферації у вогнищі запалення.
14.40. Чому запалення слід вважати патологічним процесом, а не захисною компенсаторною реакцією організму?
Запалення, як і будь-який патологічний процес, являє собою єдність і боротьбу двох начал: власне патологічного і захисно-компенсаторного. У запаленні завжди наявні руйнівний і захисний компоненти.
Власне патологічними явищами при запаленні слід вважати первинну і вторинну альтерацію, набряк, фізично-хімічні зміни й порушення обміну речовин у вогнищі запалення, біль і порушення функції.
Захисне значення мають еміграція лейкоцитів, знищення флогогенних агентів, активація процесів репарації ушкодженої тканини, загальні реакції організму, спрямовані на збільшення його резистентності (гарячка, лейкоцитоз, збільшення вмісту "білків гострої фази запалення").
Іноді одна й та сама реакція має одночасно риси патологічної і захисної. Наприклад, розлади місцевого кровообігу, з одного боку, викликають ексудацію, призводять до кисневого голодування тканини (патологічний бік), з другого — сприяють еміграції лейкоцитів, перешкоджають поширенню збудника в організмі, локалізуючи його в запаленій тканині (захисний бік).
З огляду на такий характер запалення, лікар повинен спрямовувати свої зусилля на боротьбу з власне патологічними явищами і, в міру своїх можливостей, посилювати захисно-компенсаторні.
15. Гарячка
15.1. Що таке гарячка?
Гарячка - це типовий патологічний процес, що розвивається у вищих гомойотермних організмів у відповідь на пірогенні подразники і виявляє себе перебудовою терморегуляції, спрямованою на активне підвищення температури тіла.
75.2. Які речовини називають пірогенними (пірогенами)? Як їх класифікують ?
Шрогени — це речовини, які є причиною розвитку гарячки. їх поділяють на:
а) інфекційні й неінфекційні;
б) природні й штучні;
в) екзогенні й ендогенні;
г) первинні й вторинні.
15.3. Наведіть приклади інфекційних і неінфекційних пірогенів.
До інфекційних пірогенів відносять:
1) ендотоксини грамнегативних бактерій (пірогенну дію має фрагмент токсину — ліпоїд А);
2) екзотоксини грампозитивних бактерій (дифтерійний, правцевий);
3) продукти діяльності патогенних грибків;
4) рикетсії;
5) віруси.
Неінфекційними пірогенами є:
1) компоненти несумісної за групами перелитої крові (трансфузійна гарячка);
2) екзогенні білки (білки молока при парентеральному його введенні);
3) продукти розпаду тканин.
15.4. Що таке природні і штучні пірогени?
Природними називають пірогени, які існують у природі або утворюються природно з непірогенних речовин.
Штучні пірогени отримують шляхом обробки нативних бактеріальних токсинів і використовують з лікувальною метою (піротерапія). Найвідомішими є пірогенал, отриманий із Pseudomonas aeruginosa, і пірексаль, виділений із Salmonella abortus equi.
15.5. Що називають екзогенними й ендогенними пірогенами?
Екзогенні пірогени надходять або їх вводять ззовні. При введенні екзогенного пірогену парентерально гарячка виникає через 45-90 хв.
Ендогенні пірогени утворюються в самому організмі. До них відносять:
1) продукти первинної і вторинної альтерації, що утворилися у вогнищі запалення;
2) продукти, які надходять у кров з осередків некрозу (наприклад, при інфаркті міокарда);
3) метаболіти стероїдних гормонів;
4) комплекси антиген-антитіло;
5) лейкоцитарні пірогени - продукти діяльності нейтрофілів і макрофагів.
15.6. Що таке первинні і вторинні пірогени?
Первинними називають пірогени, які безпосередньо не впливають на центр терморегуляції, їх пірогенна дія опосередковується утворенням і вивільненням так званих лейкоцитарних пірогенів.
Вторинними називають лейкоцитарні пірогени, які утворюються і вивільнюються лейкоцитами під впливом первинних пірогенів. Вторинні пірогени мають здатність безпосередньо впливати на центр терморегуляції й викликати розвиток гарячки.
Сьогодні відомо, що одним із вторинних лейкоцитарних пірогенів є інтерлейкін-1. Ця речовина утворюється і вивільнюється лейкоцитами (нейтрофілами й макрофагами) у процесі активації фагоцитозу. Поряд з дією на центр терморегуляції інтерлейкін-1 виявляє цілу низку інших ефектів, що визначають клінічні прояви гарячки (див. розд. 14).
15.7. Які існують докази того, що дія первинних пірогенів пов'язана з утворенням вторинних?
1. Первинні пірогени мають досить високий латентний період дії (у людини 45 хв. і більше), у той час як латентний період дії вторинних пірогенів — 10 хв. і менше.
2. До дії первинних пірогенів розвивається толерантність (звикання), тобто для отримання гарячки треба збільшувати дозу пірогену з кожним наступним введенням. У той же час при повторних введеннях вторинних пірогенів толерантність не виникає.
3. Якщо фагоцитарна функція нейтрофілів і макрофагів порушена (наприклад, при лейкопенії, наповненні фагоцитів тушшю), то при введенні первинних пірогенів гарячка не виникає. ;
4. При безпосередньому введенні первинних пірогенів у терморегуляторні центри гіпоталамуса гарячка не розвивається, у той час як введення в ці центри вторинних лейкоцитарних пірогенів закономірно супроводжується підвищенням температури тіла.
15.8. Які послідовні процеси становлять сутність патогенезу гарячки?
У патогенезі гарячки можна виділити кілька етапів.
I. Індукція утворення й вивільнення вторинного лейкоцитарного пірогену (інтер-лейкіну-1) первинними пірогенами.
II. Вплив інтерлейкіну-1 на центр терморегуляції й перебудова його роботи.
III. Етап клінічних проявів гарячки (зміна температури тіла).
15.9. Де розташований і що являє собою з функціональної точки зору центр терморегуляції?
Нейрони центру терморегуляції містяться в передньому (преоптична ділянка) і задньому (дорсо- і вентромедіальні ядра) гіпоталамусі. Беручи до уваги функціональні характеристики, виділяють чотири групи нейронів (рис. 44).
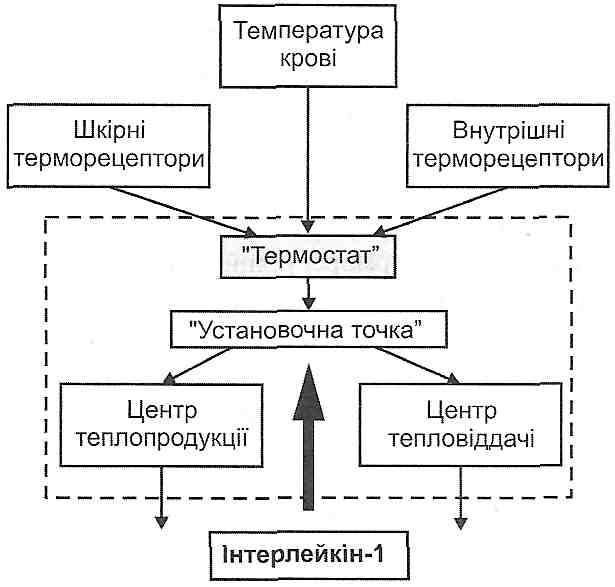
1. Нейрони "термостата" - це група термочутливих нейронів, що безпосередньо сприймають температуру крові, яка протікає через гіпоталамус. Сюди ж надходить інформація від шкірних і внутрішніх периферичних терморецепторів. "Термостат" виводить середню температуру ядра тіла.
2. Нейрони "установочної точки" - це група термонечутливих нейронів, які за-1 дають рівень температури ядра тіла. Інформація від "термостата" надходить до нейронів "установочної точки", де відбувається порівняння наявної температури ядра із запрограмованим рівнем.
Якщо середня температура ядра виявиться вищою за температуру "установочну точки", то це породжує сигнали, які гальмують центр теплопродукції і збуджують центр тепловіддачі. І навпаки, зменшення температури ядра нижче температури "установочної точки" викликає активацію центру теплопродукції і гальмування центру тепловіддачі.
3. Центр теплопродукції. Його нейрони знаходяться у задньому гіпоталамусі. їх збудження викликає збільшення утворення теплоти.
4. Центр тепловіддачі міститься в преоптичній зоні переднього гіпоталамуса. При його збудженні зростає виділення теплоти організмом.
15.10. Який механізм дії інтерлейкіну-1 на центр терморегуляції?
Інтерлейкін-1 взаємодіє зі специфічними рецепторами на мембрані нейронів, які входять у групу клітин "установочної точки". Унаслідок активації рецепторів збільшується активність зв'язаного з ними ферменту - фосфоліпази А,. Цей фермент вивільнює з фосфоліпідів плазматичної мембрани арахідонову кислоту, з якої утворюються простагландини групи Е (рис. 45).
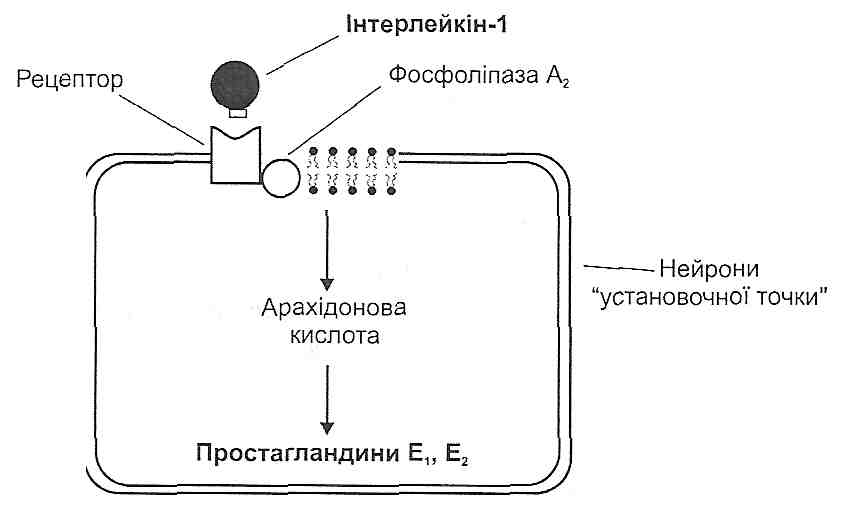
Рис. 45. Вплив інтерлейкіну-1 на центр терморегуляції
Простагландини зменшують чутливість нейронів "установочної точки" до імпульсів, що надходять від нейронів "термостата". У результаті цього звичайні сигнали про нормальну температуру ядра починають сприйматися нейронами "установочної точки" як інформація про зменшення температури. Це, у свою чергу, викликає стандартну реакцію, спрямовану на підвищення температури, - активацію центру теплоутворення й гальмування центру тепловіддачі (рис. 46).
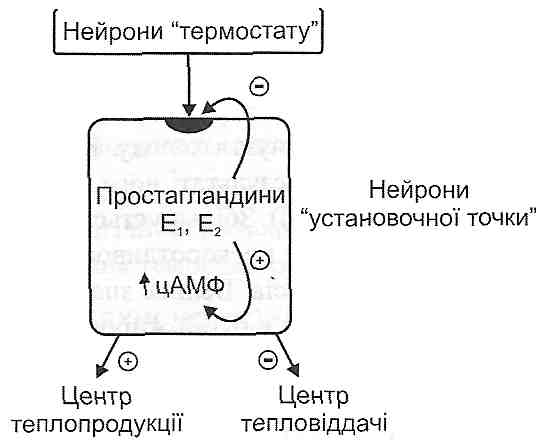
Рис. 46. Роль простагландинів у патогенезі гарячки: "+ " — активація; "—" — гальмування
15.11. Які існують докази ролі простагландинів Ев патогенезі гарячки?
1. Введення простагландинів Е мікроін'єкторами в центр терморегуляції (у ділянку "установочної точки" гіпоталамуса) викликає підвищення температури тіла.
2. Фармакологічні препарати, які інгібують циклооксигеназу (фермент синтезу простагландинів), мають жарознижувальний ефект при гарячці. До таких препаратів, зокрема, належать ацетилсаліцилова кислота, індометацин.
3. Ці ж самі препарати не впливають на рівень нормальної температури тіла. Звідси висновок, що простагландини Е не беруть участі в регуляції температурного гомеостазу в нормі, а утворюються тільки при гарячці.
4. Глюкокортикоїди, що пригнічують активність фосфоліпази А2 через ліпокортино-вий механізм (див. розд. 33), зменшують утворення простагландинів Е, завдяки чому гальмують розвиток гарячки.
15.12. Назвіть стадії гарячки.
Клінічно виділяють три стадії гарячки (рис. 47):
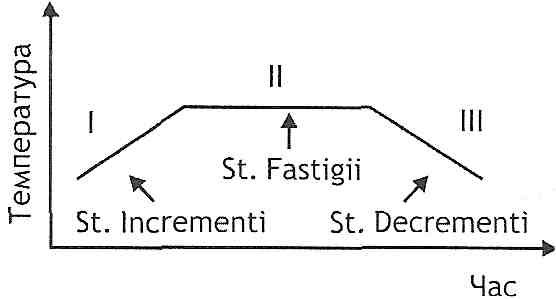
Рис. 47. Стадії гарячки
I — підвищення температури (st. incrementi);
II - стояння підвищеної температури (st. fastigii);
III - зниження температури (st. decre-menti).
15.13. Поясніть, як відбувається підвищення температури тіла на І стадії гарячки.
На самому початку різко зменшується тепловіддача. У результаті активації сим-патоадреналової системи звужуються кровоносні судини шкіри й кінцівок, скорочуються гладкі м'язи, що піднімають волосся (у тварин піднімається шерсть, у людини виникає ознака "гусячої шкіри").
Ці зміни мають два наслідки. З одного боку, різке обмеження тепловіддачі саме по собі веде до підвищення температури ядра. З другого боку, зменшується температура шкіри, що викликає збудження холодових терморецепторів. Інформація про зниження температури "оболонки" надходить у центр терморегуляції, а звідти в кору головного мозку — у людини виникає відчуття холоду. Крім того, відбувається збудження підкіркових рухових центрів, у результаті чого підвищується тонус скелетних м'язів, розвивається тремтіння (озноб). Збільшується скоротливий термогенез.
Поряд із цим відбувається активація нескоротливого термогенезу, пов'язаного з підвищенням швидкості окисних процесів. Велике значення при цьому має підвищення інтенсивності клітинного дихання в бурій жировій тканині під дією катехо-ламінів.
Таким чином, підвищення температури тіла спочатку обумовлене зменшенням тепловіддачі, а потім збільшенням теплопродукції.
15.14. Поясніть механізми зниження температури тіла при завершенні гарячки. Які існують варіанти такого падіння?
Як тільки припиняється дія інтерлейкіну-1 на центр терморегуляції, у нейронах "установочної точки" зменшується вміст простагландинів Е, що веде до відновлення чутливості клітин до сигналів, які надходять від "термостата". Температура ядра починає сприйматися як підвищена, у результаті чого активується центр тепловіддачі й пригнічується центр теплопродукції. Найбільше значення при цьому мають дві фізіологічні реакції: розширення кровоносних судин шкіри й кінцівок та збільшення потоутворення і потовиділення. Ці реакції ведуть до збільшення тепловіддачі і, як наслідок, до зменшення температури тіла.
Буває два варіанти падіння температури:
1) критичне падіння — різке зменшення температури протягом кількох годин;
2) літичне падіння - поступове зменшення температури протягом кількох діб.
Критичне падіння може бути небезпечним, особливо у літніх і у хворих на серцево-судинні недуги людей у зв'язку з можливим падінням артеріального тиску і розвитком колапсу.
15.15. Які типи температурних кривих можуть бути характерні для гарячки? Які фактори впливають на динаміку зміни температури тіла при гарячці?
До основних типів температурних кривих відносять:
1) febris intermittent- температура нормалізується один або кілька разів на добу
(гнійна інфекція, абсцеси, туберкульоз);
2) febris remittens — коливання температури становлять більше 1 °С на добу, однак вона не повертається до норми (більшість вірусних і багато бактеріальних інфекцій);
3) febris continua - добові коливання температури становлять менше 1 °С (черевний і висипний тиф);
4) febris recurrens — приступи підвищення температури чергуються з періодами її нормалізації, що тривають кілька діб (поворотний тиф, малярія).
Динаміка змін температури при гарячці визначається, з одного боку, властивостями й особливостями життєвого циклу збудника, з другого - добовими (циркадни-ми) біологічними ритмами організму.
15.16. Чому гарячку називають патологічним процесом?
Гарячка є патологічним процесом, тому що в ній сполучаються два типи протилежних явищ: власне патологічні й захисно-компенсаторні. їх співвідношення залежить від величини підвищення температури.
15.17. У чому полягає захисно-пристосувальне значення гарячки?
При гарячці в організмі створюються несприятливі умови для розвитку збудників інфекційних хвороб і підвищується активність механізмів неспецифічної й специфічної резистентності організму. Зокрема: а) пригнічується розмноження багатьох вірусів, посилюється утворення інтерферонів;
б) збільшується фагоцитарна активність макрофагів і нейтрофілів;
в) підвищується інтенсивність синтезу антитіл;
г) зростає чутливість багатьох інфекційних збудників до дії лікарських речовин.
15.18. Які власне патологічні зміни можуть виникати при гарячці?
При температурі тіла, що перевищує 39 °С, можуть розвиватися:
а) порушення загального стану - нездужання, головний біль, відчуття жару та ін.;
б) розлади обміну речовин;
в) підвищення навантаження на серце (тахікардія, збільшення серцевого виштовху) або зменшення артеріального тиску при критичному падінні температури;
г) розлади центральної нервової системи, які можуть виявлятися маренням, галюцинаціями, а в дітей від 5 міс. до 5 років розвитком фебрильних судом. Можуть провокуватися напади епілепсії;
ґ) якщо температура перевищує 40 °С, послаблюється фагоцитоз, порушується життєдіяльність і функціональна активність лімфоцитів, збільшується чутливість організму до дії деяких екзотоксинів;
д) у вагітних можливе порушення розвитку плода, тератогенні ефекти.
15.19. Чим виявляє себе вплив гарячки на обмін речовин?
Насамперед, збільшується основний обмін (на 10-12 % при збільшенні температури на 1°С). Це викликає збільшення споживання кисню і поживних речовин. Оскільки апетит у хворого відсутній, іде використання ендогенних джерел енергії, людина втрачає у вазі.
Зростає потреба організму у воді, оскільки збільшуються нечутливі втрати води через шкіру й дихальні шляхи. У зв'язку з цим у маленьких дітей і важкохворих може розвиватися зневоднення. З метою його попередження особам з гарячкою рекомендують багато пити.
При помірній гарячці (38-39 °С) частота дихання й альвеолярна вентиляція зростають у більшій мірі, ніж утворення вуглекислого газу. У зв'язку із цим може розвиватися гіпокапнія й, як наслідок, газовий алкалоз. З метою його попередження рекомендують пити підкислену рідину.
При високій гарячці (понад 39 °С) відбувається мобілізація вільних жирових кислот і посилюється утворення в печінці кетонових тіл. Кетонемія може вести до розвитку негазового ацидозу.
При гарячці розвивається негативний азотистий баланс, обумовлений переважанням катаболізму білків над їх синтезом.
15.20. У чому полягає принципова відмінність гарячки і гіпертермії?
При гарячці має місце не порушення, а перебудова терморегуляції. Організм сам підтримує високу температуру, оскільки "установочна точка" терморегуляторного центру налаштована на більш високий рівень. Якщо тварину з гарячкою охолоджувати, то її температура не зменшується, а продовжує зберігатися високою.
При гіпертермії терморегуляція порушена. Температура тіла підвищується всупереч прагненням організму підтримувати температурний гомеостаз. "Установочна
точка" терморегуляторного центру не міняється. Якщо тварину з гіпертермією охолоджувати, то в результаті різкого збільшення тепловіддачі температура тіла починає зменшуватися (рис. 48).
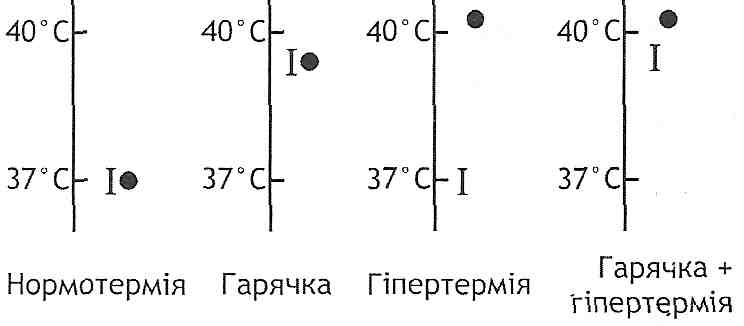
Рис. 48. "Установочна точка " і температура прирізних видах порушень терморегуляції
15.21. Що є основним патогенетичним принципом жарознижувальної терапії?
Основним патогенетичним принципом жарознижувальної терапії є зменшення "установочної точки" центру терморегуляції, що досягається пригніченням утворення простагландинів Е з допомогою інгібіторів циклооксигенази (ацетилсаліцилова кислота, індометацин, парацетамол) та інгібіторів фосфоліпази А2.
15.22. Наведіть приклади використання гарячки з лікувальною метою.
Нині з лікувальною метою застосовують високоочищені препарати пірогенів — пірогенал, пірексаль та ін. Піротерапію використовують для лікування пізніх стадій сифілісу, кістково-суглобного туберкульозу й інших інфекційних захворювань.
Застосування пірогенів при лікуванні сифілісу ефективне завдяки тому, що на пізніх стадіях хвороби збудник перебуває в головному мозку, куди утруднений доступ лікарських препаратів і антитіл через наявність гематоенцефалічного бар'єра. Проникність цього бар'єра збільшується при підвищенні температури тіла. Як наслідок, піднімається загальна реактивність і поліпшується перебіг хвороби.
16» Патологія клітинного росту. Пухлини
16.1. Назвіть основні види змін клітинного росту.
Розрізняють два види змін клітинного росту: гіпербіотичні і гіпобіотичпі процеси. До перших відносять:
а) гіпертрофію і гіперплазію;
б) регенерацію;
в) пухлини.
До других:
а) атрофію;
б) дистрофію і дегенерацію.
16.2. Що таке пухлинний процес? Які існують види пухлин?
Пухлинним називається типовий патологічний процес, сутність якого полягає в безмежному, нерегульованому розростанні тканини, не пов'язаному із загальною структурою ураженого органа та його функціями.
Виділяють два види пухлин: доброякісні і злоякісні.
16.3. Чому пухлинний процес вважають загальнобіолопчним
явищем?
Пухлинний процес носить загальнобіологічний характер, оскільки пухлини виникають як у тварин, так і у рослин. У всіх видів багатоклітинних живих організмів виявляють пухлини (у комах, риб, амфібій, рептилій, ссавців).
16.4. Чи існують особливості в характері пухлинного процесу у людини й у різних видів тварин?
У людини 90 % усіх злоякісних пухлин мають епітеліальне походження, тобто є раком. У той же час у великої рогатої худоби, коней, свиней 80 % злоякісних пухлин походять із клітин крові, тобто є гемобластозами, а у собак 50 % злоякісних новоутворень являють собою саркоми — пухлини із клітин сполучної тканини.
16.5. У чому полягають основні відмінності доброякісних і злоякісних пухлин?
1. Доброякісні пухлини складаються з добре диференційованих клітин. Ці пухлини зберігають типову структуру тканини, з якої утворилися. У той же час злоякісні пухлини характеризуються втратою диференціювання клітин, спрощенням і ати-повістю своєї будови.
2. Доброякісні пухлини часто ростуть повільно, їх ріст може зупинитися, а іноді спостерігається й зворотний розвиток (регресія). Для злоякісних пухлин, як правило, характерний швидкий ріст, що спонтанно не зупиняється. Мимовільна регресія таких пухлин невідома.
3. Доброякісні пухлини мають капсулу й ростуть експансивно, тобто не проростають у навколишні здорові тканини, а розсовують їх. Ріст злоякісних пухлин інвазивний (інфільтративний). Вони не мають капсули і проростають у навколишні тканини.
4. Доброякісні пухлини не метастазують, у той час як злоякісні зазвичай дають метастази.
5. Доброякісні пухлини добре лікуються хірургічними методами, летальних наслідків, як правило, не буває. Злоякісні пухлини при відсутності лікування призводять до смерті.
16.6. Які існують методи експериментального вивчення пухлин?
Методами експериментального моделювання пухлин є індукція, трансплантація і експлантація.
Метод індукції передбачає відтворення злоякісних пухлин шляхом введення в організм канцерогенних факторів. Найчастіше з цією метою використовують хімічні канцерогенні сполуки й безклітинні фільтрати пухлинної тканини, що містять онкогенні віруси. Крім того, з метою індукції пухлин іноді використовують фізичні впливи (рентгенівське випромінювання, радіонукліди, ультрафіолетове опромінення).
Метод трансплантації - це пересадження пухлини від однієї тварини іншій. Уперше здійснена М. Новинським у 1876 р. Для успішної трансплантації пухлини важливими є такі умови:
а) пересадження має здійснюватися в межах одного виду тварин;
б) пересаджувати треба живі життєздатні пухлинні клітини;
в) трансплантацію слід робити в стерильних умовах, щоб уникнути запального процесу в тканині.
Метод експлантації— це вирощування пухлини в культурі тканини поза організмом. Цей метод дає можливість вивчати вплив різних факторів на пухлинний ріст, здійснювати пошук засобів терапії злоякісних пухлин.
16.7. Назвіть основні причини виникнення злоякісних пухлин.
Існує три групи етіологічних факторів: хімічні (канцерогенні речовини), фізичні (іонізуюча радіація, ультрафіолетове випромінювання, висока температура, механічний вплив), біологічні (онкогенні віруси).
16.8. Хто і як уперше довів роль хімічних факторів у виникненні злоякісних пухлин?
У 1775 р. англійський лікар П. Ttomm уперше припустив, що хімічні сполуки можуть бути причиною раку. До такого висновку він дійшов на підставі своїх спостережень численних випадків раку шкіри мошонки у молодих людей, які багато років до виникнення хвороби, будучи дітьми, працювали сажотрусами. Було вперше встановлено зв'язок між виникненням раку сажотрусів і дією на шкіру сажі та смоли.
Однак тільки в 1915 p. японські вчені Ямагіва та Ішикава підтвердили цей висновок в експерименті. Вони вперше відтворили злоякісну пухлину (рак шкіри) шляхом тривалого, протягом 6-ти місяців утирання кам'яновугільної смоли в шкіру вуха кролів.
На початку 30-х років з кам'яновугільної смоли був виділений 3,4-бензпірен — речовина, з дією якої й був пов'язаний канцерогенний ефект продуктів неповного згоряння вугілля.
16.9. Як класифікують хімічні канцерогени?
Хімічні канцерогени — це речовини, здатні викликати розвиток злоякісних пухлин. ї. За походженням розрізняють канцерогени природні і штучні. її. За хімічною будовою канцерогени можуть бути представлені:
а) поліциклічними ароматичними вуглеводнями (ПАВ);
б) ароматичними амінами;
в) нітрозосполуками;
г) мікотоксинами;
ґ) гетероциклічними вуглеводнями;
д) аміноазосполуками;
є) простими сполуками (миш'як, азбест та ін.).
III. Відносно організму хімічні канцерогени можуть бути екзогенними й ендогенними.
IV. За механізмом канцерогенного впливу розрізняють канцерогени прямої дії й канцерогени непрямої дії.
16.10. Наведіть приклади канцерогенів природного і штучного походження.
До канцерогенів природного походження відносять продукти життєдіяльності деяких грибів (мікотоксини) і продукти вулканічної діяльності.
Джерелами канцерогенів штучного походження є:
1) викиди промислових підприємств;
2) вихлопні гази автомобілів;
3) тютюновий дим;
4) продукти неправильної кулінарної обробки їжі (використання пересмажених жирів, порушення технології копчення та ін.).
16.11. Охарактеризуйте канцерогенну дію поліциклічних ароматичних вуглеводнів (ПАВ).
ЛАВ - це сполуки, які мають у структурі своїх молекул кілька сполучених бензольних кілець. Найпоширенішими і найбільш вивченими є такі їхні представники, як 3,4-бензпірен, диме-
тилбензантрацен (рис. 50). їх можна виявити в продуктах вулканічної діяльності, викидах промислових підприємств, вихлопних газах автомобілів, тютюновому димі, харчових продуктах при неправильному їх копченні та ін.
Головна особливість канцерогенного впливу ПАВ полягає в їх місцевій дії. Це означає, що вони викликають розвиток пухлин у тих органах і тканинах, з якими контактують. Так, при втиранні в шкіру виникає рак шкіри, при підшкірному введенні - саркома, при вдихуванні - рак легень, при надходженні з їжею - рак стравоходу, шлунка і кишок, при виділенні з молоком - рак молочних залоз.
Рис. 50. Деякі канцерогенні ПАВ
Рис. 51. Канцерогенні ароматичні аміни
Рис. 52. Деякі канцерогенні сполуки
16.12. У чому полягає особливість канцерогенної дії ароматичних амінів?
Ароматичні аміни — це сполуки, що містять у своїй структурі бензольні кільця й аміногрупи. Канцерогенні властивості мають анілін та його похідні, зокрема, 2-на-фтиламін, диметиламіноазобензол (ДАБ) (рис. 51). Ці сполуки широко використовуються в промисловості як основа барвників, вихідні речовини для синтезу лікарських препаратів і виробництва вибухових речовин.
Характерною рисою канцерогенної дії ароматичних амінів є їх органотропність. Незалежно від шляху надходження або введення речовини пухлини виникають у певних органах. Так, при введенні 2-нафтиламіну розвивається рак сечового міхура, а при введенні ДАБ - рак печінки.
16.13. Чим характеризується канцерогенний вплив нітрозосполук?
До нітрозосполук належать нітрозаміни і нітрозаміди (рис. 52). У дослідах на тваринах показано, що 90 % всіх вивчених нітрозосполук мають канцерогенну дію. Багато речовин цього класу викликають утворення пухлин після одноразового вве-
дення в порівняно невеликих дозах. Не виявлено жодного виду тварин (вивчалося 40 видів, від риб до приматів), які були б стійкі до канцерогенної дії нітрозосполук. Дуже чутливим до їх впливу є й організм людини.
Нітрозаміни можуть утворюватися в шлунку людини з неканцерогенних попере-дників (нітритів, амінокислот, амідопірину) при наявності хлороводневої (соляної) кислоти.
Для канцерогенної дії нітрозосполук характерна органотропність. Є речовини, які вибірково викликають розвиток пухлин головного мозку, нирок, печінки, шлунка та ін.
16.14. Наведіть приклади канцерогенно)'дії продуктів життєдіяльності грибів.
Серед продуктів життєдіяльності грибів (цвілі), що мають канцерогенну дію, найбільш вивчено афлашоксин В. Цей мікотоксин є гетероциклічною сполукою і продукується грибом Aspergillus flavum (див. рис. 52). Канцерогенна дія афлатоксину виявляє себе в дуже малих дозах - 0,02 мг/кг маси. Для порівняння, ціанід калію спричиняє токсичну дію в дозах, що перевищують зазначену в кілька десятків разів.
Із впливом мікотоксинів пов'язують розвиток первинного раку печінки у представників племені Банту в Африці. Клімат території, на якій проживає це плем'я, дуже вологий, тому продукти харчування швидко покриваються цвіллю. За традицією, що існує в тих місцях, такі продукти придатні до використання. Аналізи показали, що у споживаній їжі міститься афлатоксин В.
16.15. Який зміст вкладають у поняття "ендогенні канцерогени"?
Ендогенними називають канцерогени, які утворюються в організмі з його нормальних компонентів.
Остаточно не доведено, однак є підстави думати, що в організмі при порушенні обміну речовин можуть утворюватися канцерогенні поліциклічні ароматичні вуглеводні, їхніми джерелами можуть бути холестерол, жовчні кислоти, стероїдні гормони. Зокрема, показано, що обробка in vitro жовчних кислот призводить до появи ме-тилхолантрену — представника ПАВ, що має сильну канцерогенну дію.
Крім того, ендогенні канцерогени можуть з'являтися при порушенні обміну деяких амінокислот, зокрема, триптофану.
16.16. Які досліди доводять можливу участь гормонів у виникненні злоякісного пухлинного росту?
При порушенні регуляції секреції тронних гормонів аденогіпофіза (при порушенні механізмів зворотного зв'язку) їхня кількість у крові може істотно зростати. Впливаючи на органи-мішені, вони можуть стимулювати проліферацію клітин і розвиток пухлини.
Подібну можливість демонструють у такому досліді. У тварини видаляють один яєчник, а другий пересаджують у селезінку. Кров від селезінки надходить у печінку, де естрогени, що утворилися в яєчнику, руйнуються. До гіпофіза надходить кров, у якій мало естрогенів. Це викликає стимуляцію утворення гонадотропних гормонів. Останні, діючи на яєчник, посилюють секрецію статевих гормонів, однак їх, як
і раніше, мало в крові, що надходить у гіпофіз, оскільки відбувається руйнування естрогенів у печінці. Постійна стимулятивна дія гонадотропних гормонів на яєчник викликає проліферацію його клітин і у великій кількості випадків - розвиток злоякісних пухлин.
16.17. Що таке хімічні канцерогени прямої і непрямої дії? Дайте їх порівняльну характеристику.
Канцерогени прямої дії— це високоактивні хімічні сполуки, зокрема лактони, хлоретиламіни, епоксиди. Вони здатні безпосередньо взаємодіяти зі структурами клітин і викликати розвиток пухлини. Ці сполуки не вимагають жодних перетворень в організмі для прояву своєї канцерогенної дії.
Маючи високу реакційну здатність, прямі канцерогени не можуть накопичуватися в навколишньому середовищі (при взаємодії з компонентами зовнішнього середовища вони руйнуються). Тому, з погляду гігієни, вони не являють великої небезпеки для людини як фактори канцерогенезу.
Канцерогени непрямої дії— це інертні за своїми хімічними властивостями сполуки. До них, зокрема, належать поліциклічні ароматичні вуглеводні, ароматичні аміни, нітрозосполуки, афлатоксини. Маючи низьку реакційну здатність, зазначені канцерогени можуть накопичуватися в навколишньому середовищі, а тому становлять велику небезпеку для людини.
Канцерогенними ці сполуки стають в організмі тільки після ряду хімічних ферментативних перетворень, у результаті яких утворюються їхні активні форми - власне канцерогени (рис. 53). Подібним чином з ПАВ утворюються епоксиди, з ароматичних амінів- гідроксиламіни, з нітрозамінів - алкильний радикал. Ці форми канцерогенів впливають на генетичний апарат клітини й викликають її перетворення на пухлинну.
Рис. 53. Перетворення канцерогенів непрямої дії в печінці
16.18. Які властивості хімічних речовин обумовлюють їхню канцерогенну дію ?
У 60-ті роки подружжя Міллерів показало, що канцерогенна дія хімічних сполук пов'язана з наявністю в їхній структурі позитивно зарядженої (електрофільної) групи. У молекулах канцерогенів прямої дії така група з'являється відразу ж при розчиненні речовини у воді. У випадку канцерогенів непрямої дії електрофільна група утворюється в процесі ферментативних перетворень речовини в організмі.
Так, продуктами перетворення ПАВ, що містять позитивно заряджену групу, є епоксиди. Подружжя Пульманів показало, що канцерогенні властивості мають епоксиди, у молекулі яких атом кисню приєднаний до так званої К-зони (від нім. слова
Krebs - рак). Пізніше було встановлено, що найбільш канцерогенними є епоксиди з атомом кисню в так званій бей-ділящі молекули ПАВ (від англ. bay - затока) (рис. 54).
Таким чином, знаючи хімічну будову молекули ПАВ, можна передбачити канцерогенні властивості речовини.
Рис. 54. Хімічні властивості канцерогенних ПАВ
16.19. Які стадії проходить хімічний канцерогенез? У чому їхня сутність ?
У хімічному канцерогенезі виділяють три стадії: ініціацію, промоцію і пухлинну прогресію.
Сутність ініціації полягає в тому, що під впливом хімічного канцерогену відбувається
трансформація клітини, тобто перетворення її з нормальної на пухлинну.
Під час стадії промоціїтрансформована клітина отримує стимул до розмноження, пухлина починає рости. Речовини, що стимулюють розмноження трансформованих клітин, отримали назву промоторів. Самі вони можуть бути й неканцерогенни-ми. Дуже сильними промоторами є форболові ефіри, які активують протеїнкіназу С, що бере участь у регуляції клітинного поділу.
Між ініціацією і промоцією може пройти багато часу (іноді роки). Більшість вивчених хімічних канцерогенів є повними, тобто виявляють властивості й ініціаторів, і промоторів.
Пухлинна прогресія — це якісні зміни властивостей пухлини в процесі її розвитку. Як правило, згодом пухлина набуває усе більш і більш злоякісних властивостей.
16.20. Чим пояснюється явище пухлинної прогресії?
Пухлинним клітинам притаманна висока мінливість, тому їхня популяція неоднорідна. Іде постійна боротьба клітин за виживання в несприятливих умовах існування (дефіцит субстратів, кисню і т. д.). Це є основою природного їх добору - виживають тільки ті, що найбільш пристосовані до існування в таких умовах. А найбільш пристосованими є найпростіше влаштовані пухлинні клітини, які втратили свої спеціалізовані функції й зберегли тільки властивість безмежного поділу. У такий спосіб іде подальше збільшення злоякісності пухлини (малігнізація) — головний напрям пухлинної прогресії.
16.21. Які фізичні фактори можуть мати значення у виникненні злоякісних пухлин?
Серед факторів фізичної природи до етіології пухлин можуть мати стосунок:
1) іонізуюча радіація;
2) ультрафіолетове випромінювання;
3) механічний вплив (тривалий тиск на тканини);
4) висока температура.
Роль іонізуючоїрадіації доводять в експериментах і численними клінічними спостереженнями. Радіаційний канцерогенез було виявлено вже через 7 років після відкриття рентгенівського випромінювання. Першою його жертвою став перший виробник рентгенівських трубок Фрікен, що перевіряв якість своєї продукції на власних руках. У нього виник рак шкіри.
При тривалому перебуванні білих щурів на сонці у них часто розвивається рак шкіри, що пов'язують із впливом ультрафіолетового випромінювання.
Розвиток пухлини в ділянці імплантації тваринам пластинок, виготовлених з хі-Вачно інертного матеріалу ("пластмасовий " канцерогенез), виникнення раку в місці тривалого тиску на шкіру кантрі (спеціальної сумки, у якій жителі Індії носять вугілля) - це приклади можливого значення механічних факторів у канцерогенезі.
Нарешті, часте виникнення пухлин порожнини рота у пастухів гірських районів, які п'ють дуже гарячий чай, наводить на думку про можливу роль високої температури в розвитку пухлин.
16.22. Назвіть основні закономірності канцерогенної дії іонізуючого випромінювання
Хоча для розвитку пухлин мають значення доза й вид випромінювання, немає лінійної залежності між частотою виникнення пухлинного росту і поглиненою дозою. Немає також і мінімальної порогової дози, так що будь-яке випромінювання в будь-якій дозі є потенційно небезпечним. За інших рівних умов тривалий і постійний вплив низьких доз, з погляду канцерогенезу, значно небезпечніший, ніж короткочас-ний вплив великих доз.
16.23. Що таке "пластмасовий" канцерогенез? У чому його особливості?
В експерименті було показано, що після імплантації пластмасових пластинок під шкіру в цій ділянці часто розвивається злоякісна пухлина. Таке явище отримало назву "пластмасового" канцерогенезу. Було встановлено такі його закономірності. 1. Для виникнення пухлини матеріал, з якого зроблено пластинки, не має особливого значення. Такий висновок зроблено на підставі результатів дослідження багатьох речовин, серед яких целофан, поліетилен, тефлон, скло, золото, платина та ін. | 2. Якщо з речовини зробити пластинку й імплантувати її - пухлина виникає, якщо пластинку подрібнити і ввести в тканину отриманий порошок - пухлина не розвивається.
3. Вирішальне значення має розмір пластинки. Пухлина виникає при імплантації пластинок розмірами не менше 0,5 х 0,5 см.
4. Пластинка має бути суцільною, тобто не мати отворів (перфорацій).
Відповідно до однієї з гіпотез, навколо пластинки утворюється сполучнотканинна капсула, серед колагенових волокон якої перебувають одиничні клітини. Вони є відірваними від інших клітин і сигналів, що регулюють інтенсивність проліфератив-них процесів. У зв'язку з цим створюються умови, які сприяють переродженню нормальних клітин у пухлинні.
16.24. Ким і в яких експериментах було доведено роль вірусів у виникненні пухлин?
Експериментальними доказами вірусного походження пухлин вважають їх виникнення після введення тваринам безклітинних фільтратів пухлинної тканини. Такі фільтрати готують із суспензії пухлинних клітин, пропускаючи її через порцелянові фільтри, що затримують бактерії і клітини тканини (рис. 55).
Уперше в 1908 р. В. Елерман і О. Банг у такий спосіб відтворили лейкоз у курей, уводячи безклітинні фільтрати лейкозних клітин.
У 1910 р. П. Раус викликав саркому у курей введенням безклітинного фільтрату саркоматозної тканини. За ці дослідження його в 1966 р. було удостоєно Нобелівської премії.
У 1932 p. Р. Шоуп повідомив про виділення вірусу з доброякісної пухлини кролів - фіброми, а трохи пізніше - з папіломи.
У 1934 р. Б. Люке виявив у ядрах ракових клітин нирок леопардової жаби тільця-включення, що нагадують такі при вірусних інфекціях. Потім він відтворив пухлину введенням тваринам висушеного екстракту пухлинних клітин.
У 1936 р.Дж. Біттнер відкрив "фактор молока" (фактор Біттнера), що викликав рак молочних залоз у мишей. Подальші дослідження показали, що цей фактор є ві- . русом, який передається від матері дитинчатам з молоком.
У 1951 p. JT. Гросс уперше відтворив лейкоз у мишей введенням безклітинних фільтратів пухлинних клітин новонародженим тваринам. Ці дослідження довели можливість вірусної етіології злоякісних пухлин і у представників ссавців.
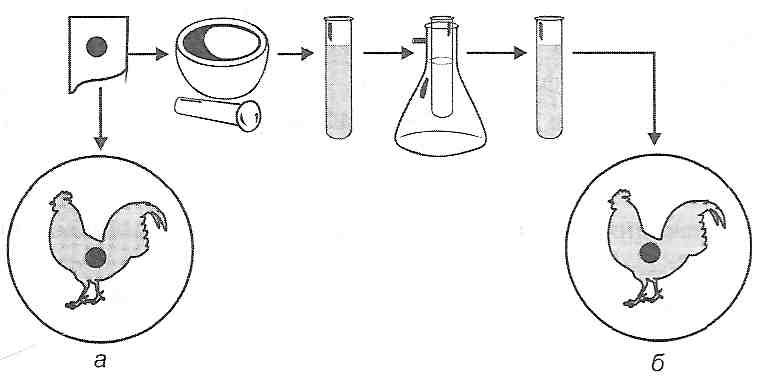
Рис. 55. Перевивання пухлини клітинним і безклітинним матеріалом: а — клітинне перевивання; б - перевивання фільтратом, що не містить клітин
16.25. Які ДНК-вмісні віруси є онкогенними для тварин і людини?
До онкогенних ДНКчвмісних вірусів відносять:
а) nanoea-eipycu. Вони викликають розвиток у тварин трьох видів пухлин: папіломи, поліоми і пухлини, що виникає під дією вакуолізуючого вірусу мавп SV-40;
б) аденовіруси. Онкогенними для тварин є аденовіруси 12,18 і 31-го типів;
в) герпес-віруси, зокрема, вірус Епшгпейна — Барр.
Що стосується людини, то в одних випадках доведено, а в інших є підстави думати, що причиною деяких пухлин є:
а) вірус Епштейна - Барр (викликає лімфому Беркіта, назофарингеальний рак);
б) вірус гепатиту В (може бути причиною раку печінки);
в) вірус папіломи людини (викликає доброякісні пухлини шкіри, жіночих геніталій, гортані).
16.26. Які РНК-вмісні віруси є онкогенними для тварин і людини?
Всі онкогенні РНК-вмісні віруси належать до сімейства pempoeipycie. Загальною їхньою властивістю є наявність у вірусному геномі гена, що кодує структуру ферменту ревертази, відомого ще як зворотна транскриптаза, або РНК-залежна ДНК-полімераза. Цей фермент забезпечує синтез двоспіральної ДНК на матриці односпіральноїРНК. Унаслідок цього утворюється ДНК-копія ретровірусу, що отримала назву ДНК-провірусу.
Залежно від онкогенності ретровіруси поділяють на дві групи.
I. Гостротрансформуючі ретровіруси. Дуже онкогенні, викликають розвиток пухлин після короткого латентного періоду. Ці віруси мають у своєму геномі онкоген, тому в основі трансформації клітин у пухлинні лежить епігеномнш механізм. До зазначеної групи, зокрема, відносять віруси гострих лейкозів у птахів, мишей і саркоми Рауса у курей.
II. Повільнотрансформуючі ретровіруси. Викликають розвиток пухлин після тривалого латентного періоду. Ці віруси не мають у своєму складі онкогена, тому основний механізм їх трансформуючої дії — мутаційний. До вірусів цієї групи відносять віруси лімфолейкозів.
Онкогенним ретровірусом людини є вірус Т-клітинноїлімфоми-лейкемії. Він передається від людини до людини при тривалих інтимних контактах, переливанні крові. Слід зазначити, що цей лімфотропний вірус дуже схожий на віруси імунодефіциту людини (ВІЛ), що викликають СНІД.
16.27. Назвіть етапи вірусного онкогенезу.
I. Рецепція вірусу. Відбувається взаємодія вірусної частки з певними структурами плазматичної мембрани клітини (рецепторами). Відсутністю відповідних рецепторів пояснюється видовий імунітет до вірусної інфекції.
II. Роздягання й проникнення вірусу в клітину (інтернолізація).
III. Об'єднання (інтеграція) вірусного геному з геномом клітини. Це центральний і обов'язковий етап вірусного онкогенезу. У випадку ДНК-вмісних онковірусів відбувається вбудовування вірусної ДНК у ДНК клітини; у випадку РНК-вмісних вірусів - вбудовується ДНК-провірус, який утворюється під впливом ферменту ревертази.
IV. Постійне перебування (персистенція) вірусу в геномі клітини. При цьому вірус розмножується разом з клітиною. Такий перебіг вірусної інфекції називається абортивним. Абортивний перебіг є неодмінною умовою перетворення клітини в пухлинну під впливом вірусу.
V. Трансформація клітини.
VI. Промоція.
VII. Пухлинна прогресія (див. запит. 16.19 і 16.20).
16.28. Назвіть фактори, від яких залежить трансформуюча дія вірусів на клітину.
I. Фактори, що їх визначають властивості вірусу. До них, зокрема, відносять так звану структурну дефектність вірусу. Структурно дефектними називаються віруси, які в процесі розмноження й формування вірусних часток втратили частину свого геному. Вони здатні проникати в клітину й об'єднуватися з її геномом, однак не здатні до репродукції. У цьому випадку єдино можливим варіантом вірусної інфекції є абортивний її перебіг. Це збільшує ймовірність трансформації клітини під впливом структурно дефектних вірусів.
Крім того, для механізмів онкогенної дії вірусів має значення наявність або відсутність у їхньому геномі вірусного онкогена.
II. Фактори, що їх визначають властивості клітин. До них відносять наявність віщіовіщтх. рецепторів на поверхні клітин, період клітинного циклу, під час якого вірус потрапляє в клітину. Інтеграція геному вірусу з геномом клітини можлива тільки в синтетичну фазу (фазу S) або в період, що безпосередньо передує їй (кінець фази G,). Цим, зокрема, пояснюється та обставина, що високодиференційова-ні клітини, які втратили здатність до поділу, є стійкими до дії онкогенних вірусів. Крім того, клітина стає чутливою до онкогенних вірусів, якщо в ній нема умов для синтезу всіх білків, необхідних для формування вірусних часток. За таких умов неможлива репродукція вірусу й вірусна інфекція має абортивний перебіг, при якому збільшується ймовірність трансформації клітини. Таке явище отримало назву функціональної дефектності вірусу.
16.29. Що таке вірусні онкогєни?
Вірусні онкогєни - це гени вірусу, з функціонуванням яких пов'язане перетворення нормальних клітин у пухлинні. Білки-продукти вірусних онкогенів порушують регуляцію процесів клітинного поділу і в такий спосіб викликають трансформацію клітини (епігеномний механізм канцерогенезу).
16.30. Що таке протоонкогени?
Про то онкогєни - це власні гени клітин, які несуть інформацію про структуру білків, що беруть участь у регуляції клітинного поділу. Протоонкогени є клітинними аналогами вірусних онкогенів.
Вважають, що вірусні онкогєни являють собою протоонкогени, які потрапили в геном вірусів у результаті тривалої еволюції останніх. Це пояснюється тим, що віруси, проходячи через клітини, можуть "красти", тобто прихоплювати із собою клітинні гени.
Протоонкогени є у всіх клітинах. Однак в одних клітинах вони все життя репресовані, тобто "мовчать", в інших вони "працюють" тільки в період ембріогенезу, а в інших - функціонують відповідно до регуляторних сигналів, що надходять.
16.31. Як класифікують вірусні онкогени і протоонкогени?
Залежно від продуктів, інформацію про які несуть онкогени вірусів і протоонкогени клітин, їх поділяють на такі групи (рис. 56):
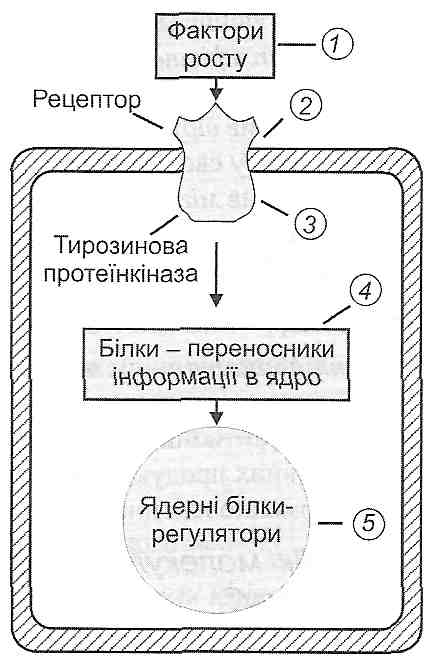
Рис. 56. Продукти протоонкогенів і вірусних онкогенів
1. Онкогени, що кодують фактори росту або їхні аналоги. Наприклад, sis-онкоген вірусу саркоми мавп і аналогічний протоонкоген клітин кодують структуру фактора росту тромбоцитар-ного походження.
2. Онкогени, що кодують клітинні рецептори до факторіє росту.
Наприклад, онкоген вірусу лейкозу курей erb-B несе інформацію про видозмінений рецептор до епідермального фактору росту.
3. Онкогени, що кодують тирозинспецифічні про-теїнкінази. Прикладом може бути scr-онкоген вірусу саркоми Рауса та відповідний протоонкоген клітин.
4. Онкогени, що кодують структуру білків, які переносять інформацію від рецепторів плазматичної мембрани клітини до ядра. Прикладом є ras-онкоген вірусу саркоми щурів, який несе інформацію про цитоплазматичний білок, що зв'язує ГТФ.
5. Онкогени, що кодують структуру ядерних білків-регуляторів.
До них, зокрема, належить mic-онкоген вірусу мієлоцитозу курей. Продукти вірусних онкогенів є аналогами продуктів протоонкогенів, тобто власних білків клітин, що беруть участь у регуляції клітинного поділу. Однак є деякі, хоча й невеликі, відмінності, Вони стосуються певних структурних характеристик вірусних онкогенів і протоонкогенів. Саме ці відмінності і роблять продукти вірусних онкогенів власне онкогенними для клітини.
16.32. Що таке клітинні онкогени? Назвіть основні механізми перетворення протоонкогенів у клітинні онкогени.
Клітинні онкогени (трансформуючі гени) - це протоонкогени, які набули здатності трансформувати клітину, тобто перетворювати її в пухлинну. Перенесення таких генів в інші, здорові клітини викликає трансформацію останніх.
Нині доведено існування таких механізмів перетворення протоонкогенів у клітинні онкогени.
І. Дерепресія протоонкогена. Може відбуватися або в результаті порушення структури, а отже, і функції відповідного антионкогена, або внаслідок мутацій у генах-репресорах, що блокують діяльність (експресію) протоонкогена.
її. Підвищення експресії протоонкогена. Спостерігається тоді, коли білок-про-дукт протоонкогена утворюється і в нормі, однак його дуже мало. Під впливом певних генетичних факторів утворення такого продукту іноді істотно зростає. Зазначене явище може бути обумовлене такими конкретними механізмами:
а) ампліфікація генів (збільшення кількості їхніх копій);
б) хромосомні мутації — транслокація;
в) вплив вірусних промоторів і підсилювачів (так діють ретровіруси, що не мають у своєму складі онкогена);
г) вплив мігруючих генів самої клітини (транспозонів).
Ш. Якісні зміни протоонкогена, що викликають утворення видозміненого продукту. Ці зміни, як правило, обумовлені точковими мутаціями протоонкогена.
16.33. Що таке антионкогени?
AnmuoHKOzenu — це клітинні гени, продукти яких викликають репресію прото-онкогенів.
Втрата антионкогенів (делеція) або мутації в них, що призводять до утворення неактивних продуктів, можуть мати своїм наслідком дерепресію протоонкогенів і трансформацію клітини, тобто утворення злоякісної пухлини.
16.34. Які молекулярні механізми можуть лежати в основі вірусного онкогенезу?
I. Продукти вірусного онкогена порушують регуляцію клітинного поділу й у такий спосіб викликають трансформацію клітини. Цей механізм отримав назву епігеном-ного. Він реалізує себе у випадку дії вірусів, що містять у своєму геномі онкогени. Із цим механізмом, зокрема, пов'язаний вплив на клітини гостротрансформуючих ретровірусів.
II. Під дією промоторів і підсилювачів вірусів, що потрапили в геном клітини, відбувається активація власних протоонкогенів, і вони перетворюються на клітинні онкогени, що трансформують клітину. Це так звання мутаційний механізм вірусного канцерогенезу. Він виявляє себе при дії вірусів, позбавлених онкогенів, зокрема, повільнотрансформуючих ретровірусів (рис. 57).
16.35. З якими молекулярними механізмами може бути пов'язаний канцерогенез, що його викликають хімічні і фізичні фактори?
При дії хімічних і фізичних канцерогенних факторів можливими є такі молекулярні зміни, що призводять до перетворення клітини в пухлинну.
1. Точкові мутації протоонкогенів.
2. Мутації антионкогенів, що регулюють експресію протоонкогенів.
3. Хромосомні аберації.
4. Утворення дефектних вірусів, унаслідок чого вони отримують онкогенні властивості.
5. Вплив на системи природної і специфічної протипухлинної резистентності, зокрема, розвиток стану імунодепресії.
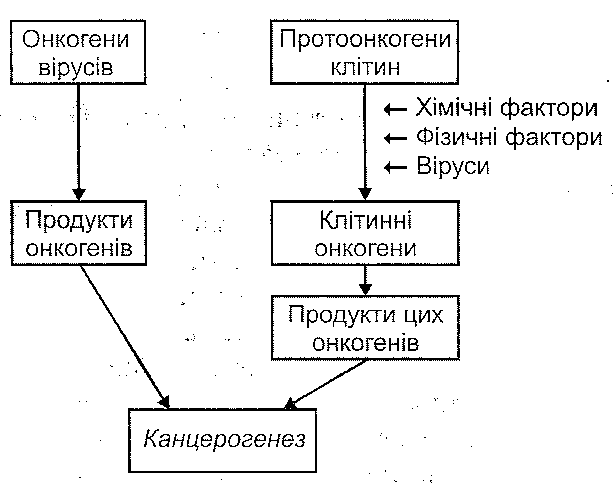
Рис. 57. Механізми канцерогенезу
Таким чином, значення хімічних і фізичних факторів як чинників канцерогенезу може полягати в тому, що вони впливають:
а) на клітини, викликаючи утворення клітинних онкогенів із протоонкогенів (мутаційний механізм канцерогенезу);
б) на віруси, змінюючи їхню структуру і властивості;
в) на організм у цілому, порушуючи механізми протипухлинного захисту.
16.36. Назвіть особливості росту злоякісних пухлинних клітин в умовах in vitro.
1. Відсутність контактного гальмування.
Нормальні клітини в культурі діляться доти, доки не утвориться моношар, що покриває дно судини. При цьому поділ припиняється (контактне гальмування). Пухлинні клітини розмножуються увесь час, утворюючи багатошарову структуру (контактного гальмування нема).
2. Здатність до поділу без прикріплення до якої-небудь поверхні. Пухлинні клітини, на відміну від нормальних, можуть ділитися, плаваючи в рідині і зберігаючи сферичну форму.
3. Для росту пухлинних клітин не обов'язкова наявність у середовищі сироватки крові.
Поділ нормальних клітин вимагає наявності в культуральному розчині не тільки поживних речовин і кисню, але й сироватки крові в концентрації від 10 до 30 %. Вважають, що сироватка містить білки, які є факторами росту для нормальних клітин.
4. Імортилізація (безсмертя) — безмежність у часі клітинного поділу. Нормальні клітини в культурі після певного числа пересаджень із судини в судину поступово втрачають здатність до поділу, культура старіє і клітини зрештою гинуть. Існує так званий ліміт Хейфліка — генетично запрограмована максимальна кількість поділів, яку може здійснити клітина.
У пухлинних клітинах ліміту Хейфліка нема, їхній поділ при створенні відповідних умов не має межі.
16.37. Назвіть основні особливості росту злоякісних пухлин in vivo.
1. Моноклональнісшь походження. Пухлина росте "сама із себе", тобто походить із однієї первинно трансформованої клітини. Інші, розташовані поруч, клітини в процес пухлинного переродження не втягуються:.
2. Автономність, росту — незалежність росту пухлини від регуляторних впливів організму (нервової системи, гормонів, факторів росту, інгібіторів проліферації). Для деяких пухлин така автономність ле абсолютна, а відносна.
3. Анаплазія- набуття пухлинними клітинами властивостей, характерних для ембріонального етапу розвитку організму. Інакше кажучи, пухлинні клітини своїми структурними, функціональними, біохімічними, імунологічними властивостями нагадують ембріональні клітини.
4. Поява в пухлинних клітинах великої кількості хромосомних аберацій. У клітинах злоякісної пухлини закономірно виявляють такі види хромосомних порушень, як транслокація, делеція, інверсія, дуплікація (докладно див. розд. 6).
5. Пухлинна прогресія (див. запит. 16.19 і 16.20).
6. Інвазивність.
7. Метастазування.
16.38. З якою швидкістю ростуть злоякісні пухлини? Якими чинниками вона визначається?
Теоретичні розрахунки показали, що за умови, коли всі пухлинні клітини постійно діляться, можна виділити два періоди розвитку пухлини.
Перший — це період від появи однієї пухлинної клітини до утворення пухлини, що містить 109 клітин. Для цього необхідно ЗО подвоєнь, тобто 90 діб. Маса такої пухлини становить приблизно 1 г - це мінімальна за величиною пухлина, яку можна виявити в організмі за допомогою сучасних методів дослідження.
Другий період — це період, під час якого кількість клітин у пухлині зростає від 109 до 1012. Для цього необхідно тільки 10 подвоєнь, тобто 30 діб. Маса пухлини наприкінці другого періоду має становити близько 1 кг — це максимально можлива за величиною злоякісна пухлина, ще сумісна з життям.
Однак у природних умовах швидкість росту пухлини значно менша, тому що не всі клітини діляться, а багато з них гине. На швидкість росту злоякісної пухлини впливають такі чинники.
1. Тривалість мітотичного циклу пухлинних клітин. Для більшості з них вона становить від 20 до 60 год.
2. Величина проліферативного пулу (ростової фракції) - показника, що відображає відсоток клітин пухлини, які перебувають у стані поділу. У деяких пухлинах він досягає 30 %.
3. Швидкість загибелі пухлинних клітин. Вона досить велика. Є пухлини, у яких на 100 щойно утворених клітин припадає 80-90 таких, що гинуть.
Причинами загибелі клітин у пухлині є:
а) дефіцит поживних речовин;
б) висока чутливість до дії ушкоджувальних факторів;
в) знищення захисними протипухлинними системами організму.
16.39. Якими порушеннями обміну речовин характеризуються злоякісні пухлини?
I. Порушення білкового обміну. У клітинах злоякісної пухлини спотворюється біосинтез білків: зникає багато власних білків, зокрема, деякі поверхневі антигени, у той же час з'являються невластиві клітинам білки вірусного і ембріонального походження, ферменти ектопічного синтезу гормонів. У пухлинній тканині зменшується інтенсивність катаболізму білків, порушується утворення деяких необхідних пухлині амінокислот, зокрема L-аспарагіну. Оскільки пухлина інтенсивно поглинає з крові величезну кількість амінокислот, її називають "пасткою " азоту.
II. Порушення вуглеводного обміну. Для злоякісної пухлини характерна низька інтенсивність клітинного дихання, висока активність гліколізу, ослаблений ефект Пастера. У зв'язку з цим пухлина має потребу в дуже великих кількостях глюкози. Поглинаючи її з крові, вона "обкрадає" організм, що може бути причиною розвитку гіпоглікемії. У зв'язку з цим злоякісну пухлину називають "пасткою " глюкози.
III. Порушення жирового обміну. У пухлинній тканині зменшується синтез жирових кислот із глюкози й ацетату. Всі необхідні ліпіди пухлина отримує з крові, поглинаючи ліпопротсїни дуже низької густини і комплекси альбумінів з жировими кислотами.
16.40. Що таке інвазивність пухлини? Як злоякісні клітини проростають у навколишню тканину?
Інвазивність - це здатність злоякісної пухлини проростати у навколишні тканини. Основу інвазії становлять три процеси.
1. Прикріплення пухлинних клітин до колагенових волокон інтерстиціальної тканини. Це відбувається завдяки наявності на поверхні пухлинних клітин рецепторів до фібронектину, що сприяє утворенню "фібронектинових містків" між клітинами пухлини і колагеном І і III типів.
2. Деградація інтерстиціальної сполучної тканини. Після прикріплення до колагену пухлинні клітини активуються й починають виділяти ферменти: колагенази І і III типів, еластазу, катепсини, плазмін. Усі вони спричиняються до гідролітичного розщеплення волокнистих структур сполучної тканини і компонентів основної інтерстиціальної речовини. Цим створюється шлях для міграції клітин, для просування їх у здорову тканину.
3. Власне міграція пухлинних клітин. Тиск, що створюється в процесі поділу, направляє пухлинні клітини в інтерстиціальну тканину, що зазнала змін. Крім того, частина клітин пухлини має власну рухливість і має властивість хемотаксису. їхній хемотаксис стимулюється продуктами деградації сполучної тканини.
16.41. Що таке метастазування? Як воно здійснюється?
Метастазування — це здатність злоякісної пухлини давати вторинні пухлинні вузли (метастази) у віддалених від первинної пухлини частинах організму.
Процес метастазування складається з трьох етапів. І. Вихід пухлинних клітин із тканини в лімфатичні або кровоносні судини - інтра-
вазація. Цьому передує прикріплення пухлинних клітин до компонентів базаль-
ної мембрани судин. В основі такого прикріплення лежить утворення "ламініно-вих містків" між поверхнею клітин пухлини і колагеном IV типу базальної мембрани. Глікопротеїд ламінін є компонентом інтерстиціальної речовини базальної мембрани. Деякі найбільш злоякісні клітини самі здатні продукувати цю речовину. Ламінін, з одного боку, взаємодіє з так званими ламініновими рецепторами пухлинних клітин, а з другого - з хімічними групами колагену IV типу.
II. Транспорт пухлинних клітин лімфою й кров'ю - дисемінація. Із клітин пухлини, що проникли в судини, утворюються пухлинні емболи. їх утворення пов'язане з агрегацією пухлинних клітин. У цьому процесі обов'язково беруть участь тромбоцити.
III. Вихід пухлинних клітин з судин у тканини й утворення вторинних вогнищ (метастазів)— екстравазація. Для локалізації метастазів мають значення місцезнаходження первинної пухлини, шлях метастазування (лімфогенний чи гематогенний), рецепторна взаємодія пухлинних клітин з ендотелієм судин. Останнім, зокрема, пояснюється факт специфічної локалізації метастазів деяких пухлин (раку простати в кістки, бронхокарциноми в головний мозок і надниркові залози, не-йробластоми в печінку і т. д.).
16.42. Як впливає пухлина на організм у цілому? Чому розвивається ракова кахексія?
Розвиток злоякісної пухлини може виявляти себе трьома групами загальних порушень в організмі.
ї. Ракова кахексія - загальне виснаження. Характеризується різким зменшенням маси тіла, слабкістю, втратою апетиту, анемією. Виникнення ракової кахексії пояснюють такими явищами:
а) пухлина захоплює із крові великі кількості глюкози ( "пастка глюкози"), у результаті чого розвивається гіпоглікемія і відбувається "енергетичне обкрадання" організму;
б) пухлина захоплює із крові великі кількості амінокислот ("пастка азоту"). Відбувається "пластичне обкрадання" організму;
в) з пухлинних клітин, що гинуть, у кров надходять токсичні продукти — ток-согормони. Вони обумовлюють явища загальної інтоксикації;
г) з пухлинних клітин виходить багато недоокиснених продуктів - розвивається негазовий ацидоз;
ґ) у результаті виходу ферментів із загиблих пухлинних клітин у кров розвивається ферментемія;
д) при локалізації пухлини у травному каналі порушуються функції травної системи.
II. Загальні прояви, пов'язані з місцевими змінами тканин. До цієї групи відносять утворення виразок, вторинні інфекції, кровотечі, больовий синдром.
III. Паранеонластичні синдроми. Вони часто супроводжують розвиток пухлин, однак їх патогенез і зв'язок зі злоякісним пухлинним ростом залишаються нез'ясованими.
До цієї групи відносять:
а) ендокринопатії;
б) гіперкальціємію;
в) нервово-м'язовий синдром (міастенію, порушення центральної й периферичної нервової системи);
г) дерматологічні порушення; ґ) ураження кісток і суглобів;
д) судинні й гематологічні порушення (тромбози, анемію, лейкемоїдну реакцію).
Великий внесок у вивчення складних механізмів впливу пухлини на організм зробив Р. Є. Кавецький- учень
0. О. Богомольця.
16.43. Які фактори організму впливають на розвиток злоякісних пухлин?
Розвиток пухлини визначається не тільки властивостями самих пухлинних клітин, але й впливом організму на цей процес. Найбільше значення мають:
а) васкулярілзація пухлини. Показано, що максимальна віддаленість пухлинних клітин від просвіту кровоносних судин не може бути більше 1—2 мм. Якщо відстань перевищує цю величину, клітини пухлини гинуть.
У злоякісну пухлину, як правило, інтенсивно проростають кровоносні судини. Це пов'язане з тим, що пухлинні клітини вивільнюють так званий ангіогенетичний фактор (ангіогенін), що стимулює ріст капілярів і розмноження ендотеліальних клітин;
б) гормони. Хоча пухлинний процес є автономним, існують, однак, пухлини, які виявляють високу чутливість до гормонів. Це, зокрема, рак молочної залози, матки, яєчників, передміхурової залози. Одні гормони посилюють ріст зазначених пухлин, інші, навпаки, гальмують;
в) стан механізмів протипухлинного захисту організму.
16.44. Які механізми протипухлинного захисту існують в організмі?
1. Механізми природної неспецифічної резистентності організму до пухлин. Не мають імунологічної специфічності і не вимагають попередньої імунізації. Вони здійснюються такими клітинами:
а) NK-wiimuHOMU (природними кілерами). Це великі гранулярні лімфоцити, що є різновидом О-лімфоцитів. Вони розпізнають пухлинні клітини і знищують їх;
б) LAK-wiimuHaMU (лімфокін-активованими кілерами). Вони, як і NK-клітини, здійснюють цитоліз пухлинних клітин;
в) макрофагами. Знищення клітин пухлини макрофагами здійснюється за допомогою фагоцитозу й механізмів позаклітинної цитотоксичності. Механізми природного неспецифічного протипухлинного захисту ефективні, якщо кількість пухлинних клітин в організмі не перевищує 103.
II. Реакції набутого (специфічного) протипухлинного імунітету. Обумовлені специфічними пухлинними антигенами і складаються як з клітинних, пов'язаних з функцією Т-лімфоцитів, так і гуморальних, пов'язаних з утворенням антитіл, імунних реакцій.
Зазначені реакції ефективні, якщо кількість клітин у пухлині становить від 103 до 10б. Якщо ж їхня кількість перевищує 106, то розвивається стан імунологічної депресії й описані вище механізми протипухлинного захисту пригнічуються.
16.45. Назвіть основні патогенетичні підходи до лікування пухлин.
1. Хірургічне видалення.
2. Променева терапія (використання радіоактивного опромінення).
3. Хіміотерапія.
4. Імунотерапія.
5. Підвищення неспецифічної резистентності (наприклад, введення БЦЖ).
17. Порушення енергетичного обміну
17.1. Що таке позаклітинні і внутрішньоклітинні порушення енергетичного обміну?
Залежно від причин розвитку розрізняють поза- і внутрішньоклітинні розлади енергетичного обміну.
Позаклітинними називають розлади енергетичного обміну, пов'язані з первинними порушеннями забезпечення клітин поживними речовинами і киснем. Це голодування і більшість видів гіпоксії.
При внутрішньоклітинних розладах енергетичного обміну доставка поживних речовин і кисню не страждає, а порушується їх використання клітинами.
7 7.2. Назвіть основні причини порушення постачання клітин поживними речовинами.
1. Повна відсутність їжі або дефіцит поживних речовин у ній.
2. Порушення процесів травлення і всмоктування.
3. Порушення мобілізації поживних речовин з депо (наприклад, ураження печінки, розлади нервово-гуморальної регуляції жирового обміну).
4. Порушення транспорту поживних речовин кров'ю (загальні й місцеві розлади кровообігу).
5. Порушення дифузії поживних речовин у тканинах.
6. Втрата поживних речовин або їх використання не за призначенням (протеїнурія, глюкозурія, опікова хвороба, злоякісні пухлини).
17.3. Які порушення хімічного складу крові свідчать про порушення постачання клітин поживними речовинами?
1. Гіпоглікемія - зменшення концентрації глюкози в крові.
2. Гіполіпацидемія й гіполіпопротеїнемія — зменшення вмісту в крові вільних жирових кислот і ліпопротеїнів.
3. Гіпопротеїнемія й гіпоаміноацидемія - зменшення вмісту білків і вільних амінокислот у крові.
17.4. Назвіть причини внутрішньоклітинних порушень енергетичного обміну.
1. Порушення транспорту поживних речовин через клітинну мембрану (наприклад, глюкози при цукровому діабеті).
2. Порушення центральних внутрішньоклітинних катаболічних шляхів.
3. Розлади процесів біологічного окиснення в мітохондріях.
4. Роз'єднання процесів окиснення й фосфорування.
5. Порушення транспорту АТФ із мітохондрій до місць використання.
6. Порушення використання енергії АТФ.
17.5. Порушення яких центральних внутрішньоклітинних катаболічних шляхів можуть призводити до розладів енергозабезпечення клітин?
1. Пригнічення гліколізу.
2. Порушення циклу Кребса.
3. Розлади пентозного циклу.
4. Пригнічення окиснення жирових кислот.
5. Пригнічення процесів дезамінування і окиснення амінокислот.
17.6. Назвіть основні причини порушень центральних катаболічних шляхів у клітинах.
I. Зменшення вмісту вітамінів і вітаміноподібних речовин у клітинах (вітамінна недостатність).
II. Набуте зменшення активності ферментів:
а) зменшення активності окремих молекул ферментів (дія метаболічних отрут);
б) зменшення кількості молекул ферментів (розлади білоксинтетичної функції клітин).
III. Спадково обумовлені ензимопатії. Описано численні генетичні дефекти ферментів гліколізу, пентозного циклу, катаболічних перетворень амінокислот. Нині не відомі які-небудь ензимопатії, безпосередньо пов'язані з циклом Кребса й (3-окис-ненням жирових кислот.
IV. Дефіцит АТФ. АТФ використовується клітиною для активації субстратів, які надходять у центральні катаболічні шляхи (наприклад, фосфорування глюкози й фрук-тозо-6-фосфату, активація жирових кислот). Дефіцит АТФ створює "зачароване коло" — він порушує катаболічні перетворення поживних речовин, що веде до порушення ресинтезу АТФ. Це, у свою чергу, збільшує дефіцит макроергічних сполук.
17.7. Які причини можуть викликати розвиток вітамінної недостатності у клітинах?
Розвиток вітамінної недостатності в клітинах викликають:
а) гіпо- і авітамінози;
б) порушення транспорту вітамінів у клітину;
в) порушення перетворення вітамінів у коферменти;
г) порушення утворення холоферментів — комплексів коферментів з апоферментами.
17.8. Назвіть основні причини порушення біологічного окиснення в мітохондріях клітин.
1. Дефіцит коферментів: НАД, ФМН, убіхінону.
2. Дефіцит мікроелементів: Fe (входить до складу залізо-сірчаних центрів НАДН-дегі-дрогеназного комплексу і цитохромів) і Си (входить до складу цитохромоксидази).
3. Блокада транспорту електронів по дихальному ланцюгу (дія отрут— ротенону, антиміцину А, аміталу, ціанідів, оксиду вуглецю (II), сірководню; рис. 58).
Рис. 58. Блокада транспорту електронів по дихальному ланцюгу
4. Порушення акцепторного контролю дихання (АДФ не контролює швидкість транспорту електронів по дихальному ланцюгу).
17.9. Що таке роз 'єднання окиснення й фосфорування? Які його механізми?
Роз'єднання окиснення й фосфорування — це стан, при якому енергія, що вивільняється в процесі транспорту електронів по дихальному ланцюгу, не здатна акумулюватися в макроергічних зв'язках АТФ і тому виділяється у вигляді теплоти.
Для цього стану характерне зменшення ресинтезу АТФ і збільшення споживання кисню клітинами.
В основі роз'єднання окиснення й фосфорування можуть лежати такі механізми:
а) зменшення градієнта концентрацій іонів водню між матриксом мітохондрій і цитоплазмою;
б) зменшення трансмембранного електричного потенціалу на внутрішній мітохон-дріальній мембрані;
в) порушення АТФ-синтетазного ферментного комплексу;
г) використання енергії градієнта концентрацій іонів водню не на синтез АТФ, а на інші цілі (транспорт іонів кальцію із цитоплазми в мітохондрії, транспорт фосфату, АДФ, АТФ та ін.).
17.10. При порушенні яких біохімічних реакцій зменшується ресинтез АТФ у клітинах?
Утворення АТФ у клітинах зменшується за умов пригнічення: а) гліколітичного (субстратного) фосфорування:
![]()
б) окисного фосфорування в мітохондріях:
![]()
в) креатинкіназної реакції:
![]()
г) аденілаткіназної реакції:
![]()
ґ) нуклеозиддифосфокіназної реакції:
![]()
17.11. Які наслідки для клітини має дефіцит АТФ?
Дефіцит АТФ у клітині призводить до:
1. Порушення механічної роботи - скорочення контрактильних структур клітин. Це виявляє себе розладами елементарних клітинних функцій: скорочення, міграції, екзо- і ендоцитозу, клітинного поділу, руху війок, джгутиків (рис. 59).
2. Порушення осмотичної роботи — процесів активного транспорту іонів. При дефіциті АТФ страждають механізми як первинного, так і вторинного активного транспорту: натрій-калієвий і кальцієвий насоси, натрій-кальцієвий і натрій-водневий обмінні механізми. Це викликає порушення внутрішньоклітинного гомеостазу й ушкодження клітин.
3. Порушення хімічної роботи — біосинтезу речовин. Наслідком цього є порушення самовідновлення й самовідтворення клітин, механізмів їх довгострокової адаптації до дії факторів навколишнього середовища. В остаточному підсумку відбувається повільна загибель клітин.
4. Порушення реакцій клітинного метаболізму.
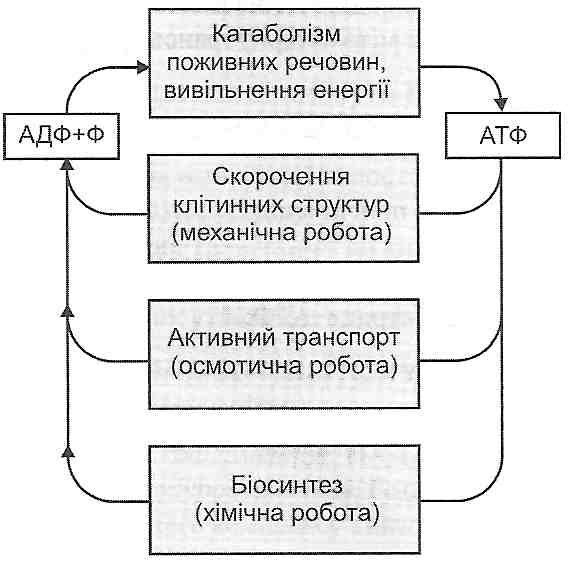
Рис. 59. Шляхи використання АТФ у клітинах
17.12. Які порушення метаболізму в клітинах можуть бути пов'язані з первинним дефіцитом АТФ?
В умовах дефіциту АТФ порушуються не тільки реакції біосинтезу речовин (анаболізм), але й реакції їх розщеплення (катаболізм).
Це пов'язано з тим, що при дефіциті АТФ безпосередньо порушуються:
а) активація субстратів (фосфорування) і залучення їх у катаболічні шляхи (гліколіз, Р-окиснення);
б) активація багатьох ферментів, здійснювана протешкіназами;
в) утворення циклічного АМФ.
17.13. Наведіть приклади "зачарованих кіл" у розвитку енергодефіцитного стану клітин.
Зменшення вмісту АТФ у клітині призводить до пригнічення функції Са-насосів, внаслідок чого збільшується концентрація іонів кальцію в цитоплазмі. Це викликає роз'єднання окиснення й фосфорування - у результаті дефіцит АТФ зростає (див. розд. 11).
Недостатність АТФ у клітині є причиною порушень її білоксинтетичних функцій. Страждає синтез усіх білків, у тому числі й білків-ферментів. Це веде до порушення катаболічних перетворень поживних речовин у клітині й процесів біологічного окиснення - дефіцит АТФ збільшується (рис. 60).
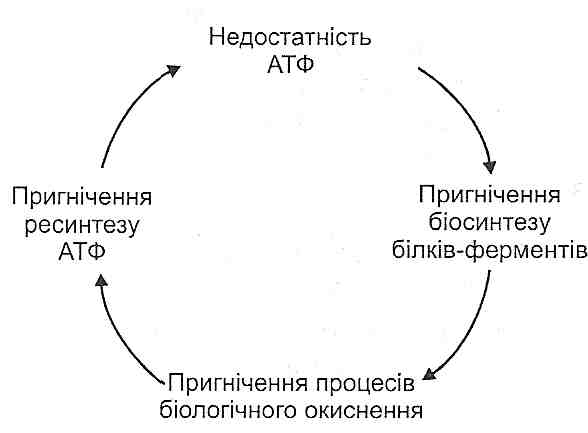
Рис. 60. Одне з "зачарованих кіл" у розвитку енергодефіцитного стану клітини
17.14. Що таке основний обмін?
Основний обмін — це мінімальні енергетичні витрати організму, визначені ранком у стані повного спокою, натще (через 12-14 год. після останнього прийому їжі), в умовах температурного комфорту (t = 18-20 °С, вологість повітря - 60-80 %, швидкість його руху - 0,1-0,2 м/с).
17.15. Які зовнішні і внутрішні фактори впливають на величину основного обміну?
Зовнішні фактори:
а) кліматичні умови;
б) час доби;
в) висота над рівнем моря;
г) характер харчування; ґ) характер виробничої діяльності.
Внутрішні фактори:
а) ріст і маса тіла;
б) площа поверхні тіла;
в) вік;
г) стать.
17.16. Наведіть приклади збільшення і зменшення основного обміну в умовах патології.
Збільшення основного обміну характерне для гіперфункції щитоподібної залози, аденогіпофіза, статевих залоз.
Зменшення основного обміну реєструють при гіпофункції щитоподібної залози, аденогіпофіза, статевих залоз; при хронічній недостатності кори надниркових залоз, при голодуванні.
18с Голодування
18.1. Дайте визначення поняття "голодування".
Голодування - це типовий патологічний процес, що виникає внаслідок повної відсутності їжі або недостатнього надходження в організм поживних речовин, а також в умовах різкого порушення складу їжі та її засвоєння.
-J v- , •'
18.2. Як класифікують голодування?
За походженням виділяють фізіологічне, патологічне і лікувальне голодування. Фізіологічне голодування характерне для деяких видів тварин під час зимової (бабаки, ховрашки) або літньої сплячки (плазуни).
Залежно від змісту виділяють такі види голодування.
1. Повне голодування:
а) із уживанням води;
б) без уживання води (абсолютне).
2. Неповне голодування (недоїдання).
3. Часткове голодування (якісне).
18.3. На які періоди поділяють патогенез повного голодування із уживанням води?
I. Період неекономного витрачання енергії.
II. Період максимального пристосування.
III. Термінальний період.
18.4. Чим характеризується період неекономного витрачання енергії?
Його тривалість— 2-4 доби. Характерне сильне відчуття голоду, обумовлене збудженням харчового центру. При повному голодуванні воно триває до 5-ти діб, а потім зникає. Відбувається швидке падіння маси тіла (схуднення). Основним джерелом енергії у цей період є вуглеводи, про що свідчить дихальний коефіцієнт, який дорівнює 1,0. Виникає гіпоглікемія, яка збільшує секрецію глюкокортикоїдів корою надниркових залоз. Наслідком цього є посилення катаболізму білків у периферичних тканинах, зокрема м'язовій, і активація глюконеогенезу в печінці.
Основний обмін спочатку трохи збільшується, а потім поступово зменшується і стає на 10—20 % менше вихідного. Розвивається негативний азотистий баланс.
18.5. Що характерно для другого періоду голодування - періоду максимального пристосування?
Середня його тривалість — 40-50 діб. Темпи зменшення маси тіла уповільнюються і становлять 0,5-1 % на добу. Відчуття голоду зникає. Основним джерелом енергії є жири, про що свідчить дихальний коефіцієнт, який дорівнює 0,7.
Гіпоглікемія збільшує надходження в кров ліполітичних гормонів (адреналіну, глюкокортикоїдів, глюкагону). Унаслідок цього відбувається мобілізація жиру з депо — розвивається гіперліпацидемія. Вона, у свою чергу, є причиною посиленого утворення кетонових тіл у печінці. Кетонемія, що виникає, може призводити до не-газового ацидозу.
Основний обмін у цей період на 10-20 % нижче вихідного рівня. Азотистий баланс негативний.
18.6. Як втрачають масу різні органи і тканини в другому періоді голодування?
Втрата маси в другому періоді голодування становить: жирова тканина — 97 %,' селезінка — 60 %, печінка — 54 %, сім'яники — 40 %, м'язи - 31 %, кров - 26 %, нирки - 26 %, нервова система - 4 %, серце - 3,6 %.
18.7. Дайте характеристику третього періоду голодування.
Цей період називають термінальним, тому що він передує смерті. Його тривалість 2—3 доби. Відбувається інтенсивний розпад тканин, розвивається інтоксикація. Основним джерелом енергії є білки, про що свідчить дихальний коефіцієнт, який дорівнює 0,8. Збільшується виділення із сечею азоту, калію, фосфатів (ознаки деструкції клітин і тканинних білків).
Смерть настає при зменшенні маси тіла до 50 % від вихідної.
18.8. Які фактори визначають максимально можливу тривалість повного голодування із уживанням води?
І. Ендогенні фактори:
а) вид тварини-
б) вік;
в) стать;
г) кількість і якість жирових та білкових резервів організму; ґ) загальний функціональний стан організму;
д) м'язова робота.
Усі зазначені чинники впливають на тривалість голодування, змінюючи величину основного обміну. Що вищий рівень основного обміну, то менша тривалість голодування, і навпаки. її. Екзогенні фактори. Це чинники зовнішнього середовища, які збільшують енер-говитрати організму. Такими є:
а) низька температура навколишнього середовища;
б) висока вологість повітря;
в) велика швидкість руху повітря.
18.9. Як розрахувати максимально можливу тривалість повного голодування із вживанням води?
Для такого розрахунку слід пам'ятати, що, по-перше, дорослий організм гине при втраті 50 % вихідної маси тіла й, по-друге, втрата маси в другому, самому тривалому періоді голодування становить 0,5—1 % на добу.
Так, якщо вихідна маса тіла дорівнює 70 кг, то смерть настає при втраті 35 кг. За умови, що кожної доби втрачається 0,5 % вихідної маси (0,35 кг), максимально можлива тривалість другого періоду, а отже й голодування в цілому, становить 100 днів.
18.10. Що таке абсолютне голодування? У чому його особливість?
Абсолютним називають повне голодування без уживання води. Його тривалість у 2-3 рази менша, ніж тривалість повного голодування з водою.
При абсолютному голодуванні відбувається посилене розщеплення жирів для утворення ендогенної (оксидаційної) води, у результаті чого швидко розвивається кетонемія і негазовш ацидоз. Важкість протікання абсолютного голодування обумовлена також накопиченням великої кількості кінцевих продуктів обміну й інших токсичних продуктів, для виведення яких з організму потрібна вода.
18.11. Назвіть особливості неповного голодування.
Неповне голодування (енергетична недостатність) розвивається, коли енергетична цінність їжі не задовольняє енергетичні потреби організму.
Неповне голодування від повного відрізняється такими особливостями:
1) тривалістю (неповне голодування може тривати місяці, роки);
2) більш вираженими деструктивними змінами в тканинах;
3) значно більшим зменшення основного обміну (на 30—40 %);
4) розвитком виражених набряків унаслідок зменшення вмісту білків у плазмі крові;
5) великого падіння маси тіла не відбувається через затримку рідини в організмі;
6) відновити життєдіяльність систем організму після неповного голодування набагато складніше.
18.12. Що таке білково-енергетична недостатність? Наведіть приклади.
Білково-енергетична недостатність — це стан, що виникає як результат поєднання неповного і якісного білкового голодування. Прикладами є:
а) аліментарна дистрофія. Описана в Ленінграді під час облоги в роки другої світової війни. У її патогенезі, крім білкової й енергетичної недостатності, мають значення й додаткові чинники: холод, фізичне стомлення, нервово-психічна напруга;
б) аліментарний маразм. Розвивається в дітей віком до одного року. На перше місце виступає енергетична недостатність;
в) квашіоркор. Розвивається в дітей віком 3-6 років. Головним у патогенезі є білкова недостатність. Енергетичний дефіцит компенсується надлишковим споживанням вуглеводів.
18.13. Якими клінічними синдромами виявляє себе білково-енергетична недостатність?
І. Недостатнє надходження в організм білків призводить до порушення білоксин-тетичної функції печінки. Це є причиною гіпопротеїнемії, що, у свою чергу, обумовлює розвиток онкотичних набряків.
II. Енергетична недостатність є причиною зменшення основного обміну. Це виявляється зниженням температури тіла (гіпотермією).
III. Атрофічні синдроми. їх розвиток пов'язаний з порушеннями пластичного й енергетичного забезпечення клітин.
18.14. Якими порушеннями фізіологічних функцій виявляють себе атрофічні зміни в органах і тканинах при білково-енергетичній недостатності?
Атрофічні зміни розвиваються у всіх тканинах, органах і системах організму.
Проявом атрофічних змін у ЦНС є уповільнення розумового розвитку, у травній системі - розлади всмоктування і діарея, у серцево-судинній системі — гіпотензія, в імунній системі — зменшення синтезу антитіл і підвищення чутливості до інфекцій, у червоному кістковому мозку — розвиток анемії, у скелетних м'язах — гіподинамія та м'язова слабкість, у кістках — затримка росту скелета.
18.15. Що таке часткове (якісне) голодування? Назвіть його види.
Частковим (якісним) голодуванням називають недостатнє надходження з їжею одної або кількох поживних речовин при нормальній енергетичній цінності їжі.
Видами часткового голодування є білкове, жирове, вуглеводне, вітамінне, мінеральне, водне голодування.
19о Гіпоксія
19.1. Дайте визначення поняття "гіпоксія".
Гіпоксія (кисневе голодування) — це типовий патологічний процес, що виникає внаслідок недостатнього постачання тканин киснем або в результаті порушення його використання клітинами.'
19.2. Як класифікують кисневе голодування?
I. Етіологічна класифікація:
а) гіпоксична (екзогенна);
б) дихальна (респіраторна);
в) серцево-судинна (циркуляторна);
г) кров'яна (гемічна); ґ) тканинна (гістотоксична) гіпоксія.
II. За темпами розвитку і тривалістю виділяють:
а) блискавичну;
б) гостру;
в) підгостру;
г) хронічну гіпоксію Ні. Залежно від поширеності процесу гіпоксія може бути загальною і місцевою.
19.3. У яких біохімічних процесах, що відбуваються в організмі, використовується кисень?
I. Тканинне (клітинне) дихання. Здійснюється в мітохондріях клітин за участю дегідрогеназ та інших компонентів дихального ланцюга, що забезпечують транспорт електронів від субстрату на кисень. Основна функція клітинного дихання - вивільнення енергії поживних речовин і акумулювання її в макроергічних зв'язках АТФ (окисне фосфорування).
На ці процеси клітинами використовується не менше 90 % кисню, що поглинається.
II. Мікросомне окиснення. Здійснюється в ендоплазматичному ретикулумі клітин. Особливо інтенсивно проходить у печінці та деяких ендокринних залозах (надниркових, статевих).
Каталізаторами мікросомного окиснення є ферменти оксигенази, які приєднують кисень безпосередньо до субстрату. Залежно від кількості атомів кисню, який приєднується, розрізняють монооксигенази і діоксигенази. Перші забезпечують реакції гідроксилювання (утворення стероїдних гормонів, перетворення проліну в оксипролін), другі - реакції детоксикації в печінці.
III. Пероксидгенеруючі реакції. Здійснюються в пероксисомах, а також у гранулах нейтрофілів і макрофагів. їхніми каталізаторами є ферменти оксидази, що забез-
печують утворення пдропероксидш цілого ряду сполук і пероксиду водню. Зазначені процеси мають значення в катаболізмі деяких сполук (амінокислот, пуринів), а в лейкоцитах є одним з механізмів бактерицидності. IV. Пероксидне окиснсння ліпідів (див. розд. 11). У нормі інтенсивність цього процесу незначна. Він істотно активується при ушкодженні клітин.
19.4. Які механізми можуть лежати в основі зменшення напруги кисню в тканинах?
1. Зменшення доставки кисню кров'ю.
2. Порушення дифузії кисню від кровоносних капілярів до клітин.
3. Посилене використання кисню клітинами.
19.5. Назвіть причини зменшення доставки кисню кров'ю.
![]()
де С. — вміст кисню в артеріальній крові; Q — об'ємна швидкість течії крові. Причинами порушення доставки кисню кров'ю можуть бути:
а) зменшення вмісту кисню в артеріальній крові;
б) зменшення об'ємної швидкості течії крові в тканині (порушення кровопостачання);
в) зменшення віддачі кисню гемоглобіном (зменшення дисоціації оксигемоглобіну).
19.6. Чим може бути обумовлене зменшення вмісту кисню в артеріальній крові?
![]()
де - Ск - вміст кисню в артеріальній крові; [НЬ] - концентрація гемоглобіну в крові; S — насичення гемоглобіну киснем; 1,34 - число Хюфнера.
Причиною зменшення вмісту кисню в артеріальній крові можуть бути:
а) зменшення концентрації гемоглобіну, здатного зв'язувати кисень (зменшення кисневої ємності крові). Це може бути обумовлено або анемією (зменшується загальний вміст гемоглобіну), або інактивацією гемоглобіну;
б) зменшення насичення гемоглобіну киснем. Закономірно виникає при зменшенні напруги кисню в артеріальній крові нижче 60 мм рт. ст.
19.7. Які зміни можуть зменшувати об'ємну швидкість течії крові в тканинах і призводити до гіпоксії?
Оскільки
![]()
де Q — об'ємна швидкість течії крові; Ра — артеріальний тиск на початку, а Рвсн — венозний тиск наприкінці перфузованої ділянки; (Р т — Р ) перфузійний тиск; R — гемодинамічний опір, то зменшення кровопостачання тканин може бути обумовлене такими групами причин.
І. Зменшення перфузійного тиску в судинах органа або тканини:
а) зменшення артеріального тиску;
б) збільшення венозного тиску.
IJ. Збільшення гемодинамічного опору судин даної ділянки:
а) звуження судин;
б) підвищення в'язкості крові.
19.8. Які фактори викликають зміщення кривої дисоціації оксигемоглобіну?
Крива дисоціації оксигемоглобіну відображує залежність між напругою кисню в артеріальній крові і насиченням гемоглобіну киснем (рис. 61).
Рис. 61. Крива дисоціації оксигемоглобіну
Зміщення цієї кривої вліво відбувається при:
а) зниженні температури;
б) алкалозі;
в) гіпокапнії;
г) зменшенні в еритроцитах вмісту 2,3-дифос- -фогліцерату;
ґ) отруєннях оксидом вуглецю (II);
д) появі спадково обумовлених патологічних форм гемоглобіну, які не віддають кисень тканинам.
При зміщенні кривої вліво гемоглобін легше приєднує кисень у капілярах легень, але гірше віддає його тканинам.
Причиною зміщення кривої дисоціації оксигемоглобіну вправо можуть бути:
а) підвищення температури;
б) ацидоз;
в) гіперкапнія;
г) збільшення вмісту в еритроцитах 2,3-дифосфогліцерату.
Вплив ацидозу і гіперкапнії на дисоціацію оксигемоглобіну відомий як ефект Бора.
При зміщенні кривої вправо гемоглобін гірше приєднує кисень у капілярах легень, але краще віддає його тканинам. З цим, зокрема, пов'язане захисно-компенсаторне значення ефекту Бора при кисневому голодуванні.
19.9. Які фактори зумовлюють порушення дифузії кисню в тканинах? Відповідно до закону Фіка
![]()
де m — кількість газу, що дифундує (дифузійний потік); k — коефіцієнт дифузії; S - загальна площа поверхні, через яку здійснюється дифузія; 1 - відстань дифузії; (Pj —Р2) — різниця між напругою О, у капілярах і клітинах.
Звідси випливає, що причинами порушення дифузії кисню в тканинах можуть бути:
1) зменшення коефіцієнта дифузії кисню (наприклад, при відкладеннях у тканині ліпідів, гіаліну, амілоїду, солей кальцію);
2) зменшення загальної площі поверхні кровоносних капілярів при зменшенні їхньої кількості;
3) збільшення дифузійної відстані (наприклад, при набряку);
4) зменшення напруги кисню в капілярах;
5) збільшення напруги кисню в клітинах.
19.10. Що таке гіпоксія навантаження?
Гіпоксія навантаження — це кисневе голодування, що виникає при збільшенні функціонального навантаження. Воно пов'язане з посиленим використанням кисню клітинами. При цьому доставка кисню тканинам може навіть зростати.
Якщо все-таки доставка кисню не покриває його посиленого використання клітинами, напруга кисню в тканинах падає й розвивається гіпоксія.
19.11. Що таке гіпоксична гіпоксія? Коли вона виникає?
Тіпоксичною (екзогенною) називають гіпоксію, причиною якої є зменшення парціального тиску кисню у вдихуваному повітрі.
Гіпоксична гіпоксія може наставати при: а) зниженні атмосферного тиску (при підніманні на висоту в гори);
6) зменшенні вмісту кисню у вдихуваному повітрі (робота в шахтах, несправність систем кисневого забезпечення в літальних апаратах, на підводних човнах, у скафандрах).
19.12. Назвіть патогенетичні фактори розвитку гірської хвороби.
Гірська хвороба виникає при підніманні неадаптованого організму в гори. Вона являє приклад підгострої і хронічної гіпоксії.
Провідне значення в патогенезі гірської хвороби має зменшення парціального тиску кисню у вдихуваному повітрі (гіпоксична гіпоксія).
Крім того, при підніманні в гори патогенну дію чинять й інші фактори, зокрема, зменшення власне атмосферного тиску (синдром декомпресії), сонячна радіація, зниження температури зовнішнього середовища, сухість вдихуваного повітря, збільшення фізичного навантаження.
19.13. Які зони виділяють при підніманні в гори з урахуванням ознак гіпоксії, що розвивається?
I. Нейтральна зона (висота від 0 до 2000 м над рівнем моря). Функції організму не страждають.
II. Зона повної компенсації (висота від 2000 до 4000 м над рівнем моря). Відзначається збільшення частоти пульсу, дихання, підвищення артеріального тиску. У той же час зменшується фізична й розумова працездатність, розвивається ейфорія, порушується тонка координація рухів, послаблюється увага.
III. Зона неповної компенсації (висота від 4000 до 6000 м над рівнем моря). Розвиваються важкі, але оборотні зміни. Тахікардія змінюється брадикардією, падає артеріальний тиск, дихання стає частим і поверхневим, іноді розвивається дихання Чейна-Стокса, характерні сонливість, млявість, нудота.
IV. Критична зона (понад 7000 м над рівнем моря). Розвиваються необоротні зміни. Артеріальний тиск падає до 0, пульс стає ниткоподібним, з'являється термінальне дихання, людина непритомніє, розвиваються судоми й настає смерть.
19.14. Що таке висотна хвороба?
Висотна хвороба - це гостра або блискавична форма гіпоксичної гіпоксії, що виникає під час висотних польотів у літальних апаратах з кабінами відкритого типу або при порушенні герметичності кабін закритого типу.
19.15. Які зміни показників газового стану крові характерні для гіпоксичної гіпоксії?
Зменшення р02 і рС02 артеріальної крові, розвиток газового алкалозу.
19.16. Що таке дихальна гіпоксія?
Дихальна гіпоксія - це кисневе голодування, причиною якого є недостатність зовнішнього дихання. Причини її розвитку див. розд. 29.
19.17. Які зміни показників газового стану крові характерні для дихальної гіпоксії?
Зменшення р02 і збільшення рС02 артеріальної крові, розвиток газового ацидозу.:
19.18. Що таке циркуляторна гіпоксія?
Циркуляторна гіпоксія — це кисневе голодування, причиною якого є розлади загальної гемодинаміки або порушення місцевого кровообігу.
В основі порушень системного кровообігу можуть лежати недостатність серця й недостатність судин (шок, колапс).
До місцевої гіпоксії призводять ішемія, тромбоз, емболія, венозна гіперемія.
Залежно від механізмів розвитку деякі автори виділяють дві форми циркулятор-ної гіпоксії: ішемічну і застійну.
19.19. Які зміни показників газового стану крові характерні для циркуляторної гіпоксії?
Збільшення артеріовенозної різниці за киснем за рахунок повнішого вилучення його з артеріальної крові.
19.20. Що таке кров'яна (гемічна) гіпоксія? Назвіть її види.
Кров 'яна (гемічна) гіпоксія — це кисневе голодування, що виникає внаслідок зменшення кисневої ємності крові.
Виділяють дві форми кров'яної гіпоксії:
а) анемічну — виникає як наслідок анемії (див. розд. 26);
б) гіпоксію, пов'язану з інактивацією гемоглобіну.
19.21. Назвіть форми інактивованого гемоглобіну.
1. Карбоксигемоглобін - продукт взаємодії гемоглобіну з оксидом вуглецю (II) (чадним газом, CO).
2. Метгемоглобін — гемоглобін, у якому залізо перебуває в окисненому, тривалентному стані.
3. Сульфгемоглобін — сполука гемоглобіну із сірководнем.
19.22. Які механізми обумовлюють розвиток порушень в організмі при отруєнні оксидом вуглецю (II)?
У патогенезі порушень, спричинюваних оксидом вуглецю (II), мають значення такі фактори: |< -*•
а) інактивація гемоглобіну, що зменшує кисневу ємність крові, — розвивається кров 'яна гіпоксія;
б) зміщення кривої дисоціації оксигемоглобіну вліво - навіть той гемоглобін, що не зазнав інактивації, погано віддає кисень тканинам;
в) зв'язування оксиду вуглецю (II) із залізом інших білків, що містять у собі гем, зокрема цитохромів — розвивається тканинна гіпоксія.
19.23. Які фактори можуть бути причиною утворення метгемоглобіну, а отже й розвитку кров'яної гіпоксії?
Причини утворення метгемоглобіну:
1) екзогенніречовши-окислювачі (метгемоглобіноутворювачі). До них відносять:
а) нітросполуки (оксид азоту (II), нітрити, нітрати);
б) аміносполуки (анілін, фенілгідразин);
в) окислювачі (хлорати, перманганати, хінони);
г) окисно-відновні барвники (метиленовий синій у високих концентраціях); ґ) лікарські препарати (нітрогліцерин, амілнітрит, сульфаніламіди, барбітурати);
2) недостатність антиоксидантних систем еритроцитів, що відновлюють метгемоглобін. Це спостерігається при порушеннях пентозного циклу і глютатіон-редуктази. Описано генетично обумовлений дефект ферменту - НАДФ-залежної метгемоглобінредуктази.
19.24. Які зміни показників газового стану крові характерні для кров 'яної гіпоксії?
Зменшення кисневої ємності крові.
19.25. Що таке тканинна гіпоксія?
Тканинна гіпоксія — це кисневе голодування, що виникає в результаті порушення утилізації кисню клітинами.
В її основі лежать два типи порушень:
а) пригнічення біологічного окиснення;
б) роз'єднання окиснення й фосфорування (див. розд. 17).
19.26. Які зміни показників газового стану крові характерні для тканинної гіпоксії?
Зменшення артеріовенозної різниці за киснем і збільшення рО, венозної крові.
19.27. Дайте порівняльну характеристику основних показників газового стану крові при різних видах гіпоксії.
р02 — напруга кисню в артеріальній крові; рС02 — напруга вуглекислого газу в артеріальній крові; AV-різниця — артеріовенозна різниця за киснем; КЄК — киснева ємність крові; КОС — кислотно-основний стан.
19.28. На які групи можна поділити всі захисно-компенсаторні реакції, що виникають при гіпоксії?
I. Реакції, спрямовані на збільшення доставки кисню кров 'ю.
II. Місцеві (тканинні) реакції, спрямовані на поліпшення забезпечення клітин киснем.
III. Реакції в системах утилізації кисню.
19.29. Назвіть захисно-компенсаторні реакції організму, спрямовані на збільшення доставки кисню тканинам.
1. Реакції зовнішнього дихання. Спрямовані на збільшення р02 артеріальної крові, тому можуть бути ефективними тільки при гіпоксичній і дихальній гіпоксії. Вони виявляють себе:
а) збільшенням глибини дихання;
б) збільшенням частоти дихання;
в) мобілізацією резервних альвеол.
Комплекс зазначених змін отримав назву гіпервенпгиляції.
2. Реакції системи кровообігу. Спрямовані на збільшення кровопостачання тканин і є ефективними при всіх видах гіпоксії, крім тканинної. До цих реакцій відносять:
а) збільшення хвилинного об'єму крові за рахунок збільшення сили й частоти серцевих скорочень;
б) підвищення артеріального тиску;
в) перерозподіл течії крові - зменшення кровообігу в шкірі, скелетних м'язах, органах черевної порожнини і збільшення — у серці й головному мозку.
3. Реакції системи крові. Спрямовані на збільшення кисневої ємності крові і виявляють себе збільшенням кількості еритроцитів та концентрації гемоглобіну в периферичній крові. Це досягається за рахунок:
а) виходу додаткової кількості еритроцитів з депо;
б) активації еритропоезу (при гіпоксії посилюється утворення ниркових ери-тропоетинів).
Крім того, захисне значення має зміщення кривої дисоціації оксигемоглобіну вправо — ефект Бора (див. запит. 19.8).
19.30. Які небажані наслідки може мати гіпервентиляція при гіпоксичній гіпоксії?
Гіпервентиляція, що виникає, веде до зменшення рС02 артеріальної крові - гіпо-капнії. Це має ряд негативних наслідків:
а) відбувається пригнічення дихального центру;
б) розвивається газовий алкалоз;
в) настає спазм мозкових і вінцевих судин;
г) крива дисоціації оксигемоглобіну зміщується вліво - кров погано віддає кисень тканинам.
19.31. Назвіть місцеві (тканинні) реакції, спрямовані на поліпшення забезпечення клітин киснем в умовах гіпоксії.
1. Посилення місцевого кровообігу — артеріальна гіперемія. Розвивається як наслідок безпосереднього впливу зменшення р02 на гладкі м'язи судин і дії на судини вазо-активних метаболітів (аденозину, молочної кислоти, іонів калію й водню та ін.)
2. Збільшення кількості капілярів, що функціонують. У результаті зменшується відстань дифузії кисню і збільшується загальна площа дифузійної поверхні.
3. Підвищення вмісту в клітинах міоглобіну, який у м'язах є внутрішньоклітинним депо кисню.
19.32. Назвіть захисно-компенсаторні реакції в системах утилізації кисню при гіпоксії.
1. Зниження функціональної активності клітин, унаслідок чого зменшується їхня потреба в кисні.
2. Збільшення кількості дихальних ферментів і мітохондрій у клітинах.
3. Збільшення спорідненості цитохромоксидази з киснем.
4. Підвищення ступеня спряженості окиснення й фосфорування до максимально можливої величини, що дорівнює 3.
5. Активація гліколізу.
19.33. При яких значеннях напруги кисню в тканинах починає зменшуватися утворення АТФ у клітинах?
При зменшенні р02 у тканинах нижче ЗО мм рт. ст. знижується інтенсивність споживання кисню клітинами, а отже, падає інтенсивність утворення АТФ.
"Критична" напруга 02 в мітохондріях - 0,1—1 мм рт. ст. При зменшенні рО, нижче цієї величини цитохромоксидаза не здатна передавати електрони на кисень — тканинне дихання повністю припиняється.
19.34. Які механізми складають основу гіпоксичного ушкодження клітин?
І. Гіпоксія, викликаючи дефіцит АТФ у клітинах, призводить до порушення роботи іонних насосів. Наслідком цього є збільшення концентрації іонів кальцію й на-
трію в цитоплазмі, що започатковує кальцієві й електролітно-осмотичні механізми ушкодження клітини (див. розд. 11).
II. При гіпоксії відбувається активація анаеробного гліколізу. Це призводить до накопичення молочної кислоти й розвитку внутрішньоклітинного ацидозу. Як наслідок, вмикаються ацидотичні механізми ушкодження клітини (див. розд. 11).
III. При значному дефіциті кисню компоненти дихального ланцюга перебувають у відновленому стані. Вони "скидають" свої електрони безпосередньо на молекули кисню, що залишився, обминаючи дихальний ланцюг. Це веде до одноелектронно-го відновлення кисню й утворення супероксидних радикалів - активуються реакції вільнорадикального окиснення. Результатом є ініціація пероксидного окиснення ліпідів (ПОЛ) і реалізація "ліпідноїтріади" ушкодження клітин (див. розд. 11).
19.35. Від яких факторів залежить чутливість клітин до гіпоксії?
Існує закономірність: що вища інтенсивність споживання кисню клітинами, то вища їхня чутливість до кисневого голодування.
Оскільки споживання кисню визначається енергетичними потребами, а останні — функціональною активністю клітин, стає зрозумілим, чому головний мозок, серце, печінка, нирки дуже чутливі, а кістки, хрящі, сухожилля резистентні до гіпоксії.
Крім того, чутливість до гіпоксії залежить від швидкості окисних процесів в організмі й від температури тіла. При зменшенні температури чутливість тканин до кисневого голодування падає. Цю обставину використовують у медицині при проведенні тривалих операцій на серці (штучна гіпотермія).
7 9.36. Які періоди характерні для гострої гіпоксії клітин?
I. Латентний період. Триває кілька секунд. Клітини функціонують нормально.
II. Період порушення функцій. Триває до повного припинення функцій органа, тканини.
III. Період оживлення. Охоплює час від моменту повного припинення функції до початку розвитку необоротних структурних змін.
IV. Період необоротного ушкодження клітин. Триває до загибелі клітин.
і9.37. Яка динаміка змін у ЦНС при гострій гіпоксії?
Головний мозок є "критичним органом" при гострій гіпоксії. Це означає, що гі-поксичне ушкодження структур ЦНС, розвиваючись дуже швидко, призводить до смерті організму, незважаючи на те, що інші органи й тканини ще якийсь час зберігають свою життєздатність.
Латентний період гострої гіпоксії для ЦНС становить 4 с. Через 8-12 с. після повного припинення постачання головного мозку киснем функції ЦНС припиняються і людина непритомніє. Через 20-30 с. зникає спонтанна електрична активність кори головного мозку (на електроенцефалограмі - ізоелектрична лінія). Межа оживлення головного мозку становить 8-10 хв. У той же час межа оживлення організму в цілому - 4-5 хв. після зупинки серця. Це пояснюється тим, що після відновлення роботи серця необхідно ще 4-5 хв., щоб створити артеріальний тиск, достатній для кровопостачання мозку.
20o Порушення вуглеводного обміну
20.1. Назвіть основні причини порушень вуглеводного обміну.
1. Порушення перетравлювання вуглеводів та їх усмоктування в кишках (див. розд. ЗО).
2. Порушення вуглеводної функції печінки (див. розд. 31).
3. Порушення катаболізму глюкози в периферичних клітинах (див. розд. 17).
4. Порушення нейрогормональної регуляції вуглеводного обміну.
20.2. Які гормони беруть участь у регуляції вуглеводного обміну?
Гормони, що беруть участь у регуляції вуглеводного обміну, поділяють на дві групи: інсулін і контрінсулярні гормони.
Контрінсулярними називають гормони, які за своїми біологічними ефектами є антагоністами інсуліну. До них належать адреналін, глюкагон, глюкокортикоїди, соматотропний гормон.
20.3. На які органи і тканини діє інсулін?
Залежно від чутливості до інсуліну всі структури організму поділяють на три групи.
I. Абсолютно залежні від інсуліну. Такими є печінка, м'язи (скелетні, міокард), жирова тканина.
II. Абсолютно нечутливі. Це головний мозок, мозкова речовина надниркових залоз, еритроцити, сім'яники.
III. Відносно чутливі (всі інші органи й тканини).
20.4. Назвіть основні біологічні ефекти інсуліну.
1. Гіпоглікемична дія. Інсулін зменшує вміст глюкози в крові за рахунок:
а) пригнічення процесів, що забезпечують вихід глюкози з печінки в кров (глі-когенолізу і глюконеогенезу);
б) посиленого використання глюкози інсулінозалежними тканинами (м'язовою, жировою).
2. Анаболічна дія. Інсулін стимулює ліпогенез у жировій тканині, глікогенез у печінці і біосинтез білків у м'язах.
3. Мітогенна дія. У великих дозах інсулін стимулює проліферацію клітин in vitro та in vivo.
Залежно від швидкості виникнення ефекти інсуліну поділяють на:
а) дуже швидкі (виникають протягом секунд) - зміна мембранного транспорту глюкози, іонів;
б) швидкі (тривають хвилини) — алостерична активація анаболічних ферментів і пригнічення ферментів катаболізму;
0:#:«
в) повільні (тривають від кількох хвилин до кількох годин) - індукція синтезу анаболічних ферментів і репресія синтезу ферментів катаболізму;
г) дуже повільні (від кількох годин до кількох діб) — мітогенна дія.
20,5. Які зміни вуглеводного обміну викликає адреналін?
Під впливом адреналіну збільшується вміст глюкози в крові. В основі цього ефекту лежать такі механізми:
а) активація глікогенолізу в печінці. Вона пов'язана з активацією аденілатциклазної системи гепатоцитів і утворенням, в кінцевому підсумку, активної форми фосфорилази;
б) активація глікогенолізу в м'язах з наступною активацією глюконеогенезу в печінці. При цьому молочна кислота, що вивільняється з* м'язової тканини в кров, іде на утворення глюкози в гепатоцитах (рис. 62);
в) пригнічення поглинання глюкози інсулінозалежними тканинами з одночасною активацією ліпрлізу в жировій тканині;
г) пригнічення секреції інсуліну β-клітинами і стимуляція секреції глюкагону а-клі-тинами острівців підшлункової залози.
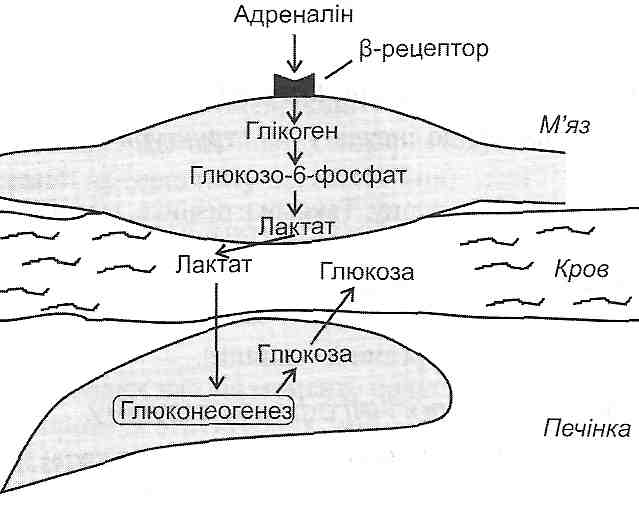
Рис. 62. Один з механізмів гіперглікемічної дії адреналіну
20.6. Які механізми лежать в основі гіперглікемічної дії глюкагону?
1. Активація глікогенолізу в печінці.
2. Активація глюконеогенезу в гепатоцитах.
Обидва механізми є цАМФ-опосередкованими.
20.7. Яким чином глюкокортикоїди підвищують рівень глюкози в крові?
Глюкокортикоїди активують процеси глюконеогенезу в печінці, збільшуючи:
а) синтез відповідних ферментів (вплив на транскрипцію);
б) вміст у крові субстратів глюконеогенезу — амінокислот — за рахунок посилення протеолізу в м'язах.
Крім того, глюкокортикоїди зменшують поглинання глюкози інсулінозалежними тканинами.
20.8. Чому при дії великих доз соматотропного гормону підвищується вміст глюкози в крові?
Тривалий вплив великих доз соматотропного гормону супроводжується розвитком інсулінорезистентності м'язів і жирової тканини — вони стають нечутливими до дії інсуліну. Результат цього — гіперглікемія.
20.9. Яка тривалість гіперглікемічного ефекту контрінсулярних гормонів?
Гіперглікемічна дія адреналіну триває до 10 хв, глюкагону—3 0-60 хв, глюкокортико-ї'дів — від кількох годин до кількох діб, соматотропного гормону—тижні, місяці, роки.
20.10. Як змінюється вміст глюкози в крові при порушенні балансу між інсуліном і контрінсулярними гормонами?
При збільшенні вмісту інсуліну розвивається гіпоглікемія, а при зменшенні його концентрації - гіперглікемія.
При збільшенні вмісту контрінсулярних гормонів розвивається гіперглікемія, а . при зменшенні їх концентрації - гіпоглікемія.
20.11. Як здійснюється нервова регуляція вуглеводного обміну?
Існує ряд доказів того, що нервова система бере участь у регуляції вмісту глюкози в крові.
Так, Клод Бернар уперше показав, що укол у дно IV шлуночка спричиняється до гіперглікемії ("іскровийукол"). Збільшення концентрації глюкози крові може виникати внаслідок подразнення сірого горба гіпоталамуса, сочевицеподібного ядра і смугастого тіла базальних ядер великого мозку. Кеннон спостерігав, що психічне перенапруження, емоції можуть підвищувати рівень глюкози в крові. Гіперглікемія виникає також при больових відчуттях, під час нападів епілепсії і т. д.
Нині доведено, що вплив нервової системи на рівень глюкози крові опосередковується гормонами. Можливі такі варіанти:
1) ЦНС → симпатична нервова система → мозкова речовина надниркових залоз → адреналін → гіперглікемія (укол К. Бернара);
2) ЦНС →• парасимпатична нервова система → острівці підшлункової залози → інсулін і глюкагон;
3) ЦНС → симпатична нервова система → мозкова речовина надниркових залоз → адреналін → Β-клітини острівців підшлункової залози → пригнічення секреції інсуліну;
4) ЦНС щ гіпоталамус → аденогіпофіз кі АКТГ → глюкокортикоїди → гіперглікемія.
20.12. Дайте визначення поняття "гіпоглікемія".
Гіпоглікемія - це зменшення концентрації глюкози в плазмі крові до рівня, який обумовлює появу клінічних симптомів, що зникають після нормалізації вмісту цієї речовини.
Ознаки гіпоглікемії виникають, як правило, при зменшенні вмісту глюкози нижче 4 ммоль/л.
20.13. Які механізми можуть лежати в основі розвитку гіпоглікемії?
1. Зменшення надходження глюкози в кров. Це буває при голодуванні, порушеннях травлення (недостатність амілолітичних ферментів, розлади всмоктування), при спадкових і набутих порушеннях глікогенолізу і глюконеогенезу в печінці.
2. Посилене використання глюкози на енергетичні потреби організму (наприклад, важка фізична робота).
3. Втрата глюкози (глюкозурія) або використання її не за призначенням (злоякісні пухлини).
20.14. Якими клінічними ознаками виявляє себе гіпоглікемія? Що таке гіпоглікемічна кома?
Клінічні ознаки гіпоглікемії пов'язані з двома групами порушень в організмі.
I. Порушення постачання глюкозою тканин головного мозку. Залежно від ступеня гіпоглікемії розвиваються такі симптоми, як головний біль, неможливість зосередитися, стомлюваність, неадекватна поведінка, галюцинації, суцоми, гіпоглікемічна кома.
II. Активація симпатоадреналової системи. Цим обумовлені серцебиття, посилене потовиділення, тремтіння, почуття голоду.
Гіпоглікемічна кома є найтяжчим проявом гіпоглікемії, і якщо вчасно не надати допомогу (введення глюкози), призводить до смерті. Вона характеризується втратою свідомості, зникненням рефлексів, порушеннями життєво важливих функцій (див. розд. 12).
20.15. Що таке гіперглікемія?
Гіперглікемія — це збільшення вмісту глюкози в плазмі крові (понад 6,66 ммоль/л при визначенні методом Хагедорна - Йєнсена).
20.16. Які механізми можуть лежати в основі гіперглікемії?
1. Збільшення надходження глюкози в кров. Це буває після приймання їжі (аліментарна гіперглікемія), при посиленні глікогенолізу й глюконеогенезу в печінці (зменшення вмісту інсуліну або збільшення концентрації контрінсулярних гормонів).
2. Порушення використання глюкози периферичними тканинами.
Так, при зменшенні вмісту інсуліну порушується надходження й утилізація глюкози в інсулінозалежних тканинах (м'язах, жировій тканині, печінці).
20.17. Дайте визначення поняття "цукровий діабет".
Цукровий діабет - це хвороба, яка у нелікованому стані виявляє себе хронічним збільшенням вмісту глюкози в крові - гіперглікемією (визначення ВООЗ, 1987).
20.18. Які існують експериментальні моделі цукрового діабету?
1. Панкреатичний цукровий діабет- видалення в собак 9/10 підшлункової залози (Мерінг і Мінковський, 1889).
2. Алоксановий цукровий діабет: однократне введення тваринам алоксану — речовини, що вибірково ушкоджує Β-клітини острівців підшлункової залози.
3. Стрептозотоциновий цукровий діабет: введення тваринам антибіотика - стреп-тозотоцину, що вибірково ушкоджує )3-клітини острівців.
4. Дитизоновий цукровий діабет: введення тваринам дитизону — речовини, що зв'язує цинк і в такий спосіб порушує депонування й секрецію інсуліну.
5. Імунний цукровий діабет - введення тваринам антитіл проти інсуліну.
6. Метагіпофізарний цукровий діабет — тривале введення тваринам гормонів адено-гіпофіза - соматотропного гормону, АКТГ.
7. Метастероїдиий цукровий діабет — тривале введення тваринам глюкокортикоїдів.
8. Генетичні моделі цукрового діабету — виведення чистих ліній мишей та інших тварин зі спадково обумовленою формою хвороби.
,у' НА 10***111 V,.'!1'-'
20.19. Наведіть патогенетичну класифікацію цукрового діабету.
1. Спонтанний цукровий діабет (первинний) - являє собою самостійну нозологічну одиницю. На його частку припадає близько 90 % усіх випадків цукрового діабету. Виділяють два різновиди спонтанного діабету: тип І, або інсулінозалеоісний (юнацький), і тип II, або інсулінонезолежний (діабет дорослих).
2. Вторинний цукровий діабет — є тільки ознакою інших захворювань. Він, зокрема, розвивається при ураженнях підшлункової залози, ендокринних хворобах, що супроводжуються збільшенням секреції контрінсулярних гормонів, при складних спадково обумовлених синдромах (наприклад, атаксія-телеангіектазія).
20.20. Дайте порівняльну характеристику цукрового діабету і і II типів.
Цукровий діабет І типу — інсулінозалежшй. Він характеризується абсолютною інсуліновою недостатністю, що виникає в результаті загибелі β-клітин панкреатичних острівців. Розвивається в осіб молодого віку, зазвичай до 30 років. Тому його ще називають ювенільним, або юнацьким. Супроводжується кетозом — накопиченням кетонових тіл. Необхідне лікування інсуліном.
Цукровий діабет II типу — інсулінонезолежний. Він характеризується відносною недостатністю інсуліну або інсулінорезистентністю. Розвивається у дорослих, найчастіше після 40 років. Не супроводжується кетозом. У його лікуванні інсулін не застосовують.
20.21. Які причини розвитку цукрового діабету І типу?
Цукровий діабет І типу — захворювання з генетичною схильністю, у виникненні якого важливу роль відіграють фактори зовнішнього середовища (рис. 63).
Спадкова схильність до діабету І типу обумовлена антигенасоційованим характером цього захворювання. Так, доведений зв'язок між виникненням цукрового діабету І типу і наявністю певних HLA-генів (В8, Bwl5, Dw3, Dw4) у головному комплексі гістосумісності (МНС). Це наводить на думку про значення аутоімунних механізмів у розвитку цієї хвороби.
Серед факторів зовнішнього середовища, здатних ушкоджувати β-клітини панкреатичних острівців, велике значення мають р-цитотропні віруси (віруси Коксакі,
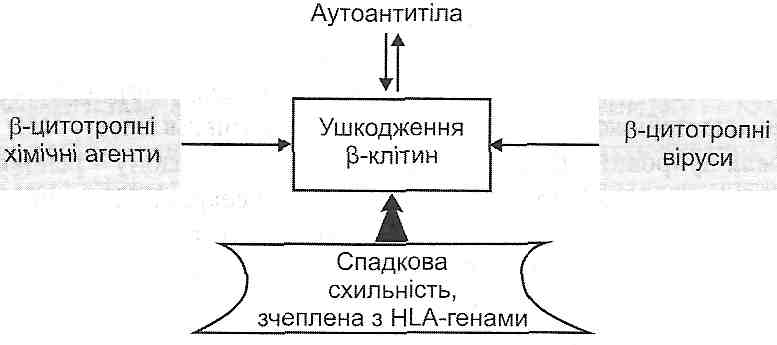
Рис. 63. Причини цукрового діабету І типу
епідемічного паротиту, кору) і fi-цитотропні хімічні агенти (наприклад, алоксан, стрептозотоцин).
20.22. Які механізми лежать в основі розвитку абсолютної інсулінової недостатності при цукровому діабеті і типу?
Генетична схильність, пов'язана з генами МНС, виявляє себе, з одного боку, утворенням аутоантитіл, здатних вибірково ушкоджувати Β-клітини острівців підшлункової залози, з другого - зменшенням резистентності клітин до дії екзогенних ушкоджувальних факторів (Р-цитотропних вірусів і хімічних агентів).
Усе це разом призводить до ушкодження інсулярного апарату й загибелі β-клітин (рис. 64). Утворення інсуліну припиняється.
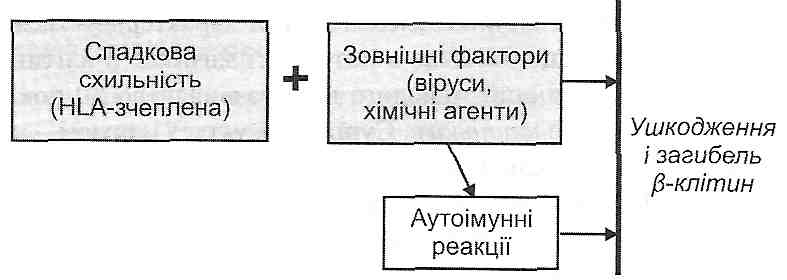
Рис. 64. Механізми абсолютної інсулінової недостатності при цукровому діабеті І типу
20.23. Які причини розвитку цукрового діабету // типу?
1. Спадкова схильність. Вона, однак, на відміну від діабету І типу, не пов'язана з генами головного комплексу гістосумісності (МНС).
2. Ожиріння. Його відзначають у 80 % хворих. Тому виділяють дві форми цукрового діабету II типу:
а) з ожирінням;
б) без ожиріння.
На відміну від діабету І типу фактори зовнішнього середовища не мають особливого значення в етіології діабету II типу.
20.24. Опишіть патогенез цукрового діабету II типу з ожирінням.
Умовно виділяють два етапи патогенезу. І. Гіперінсулінемічний етап (рис. 65). Споживання великої кількості їжі особами з ожирінням викликає збільшення секреції інсуліну (гіперінсулінемія). Ця реакція спрямована на активацію процесів депонування поживних речовин у жировій тканині.
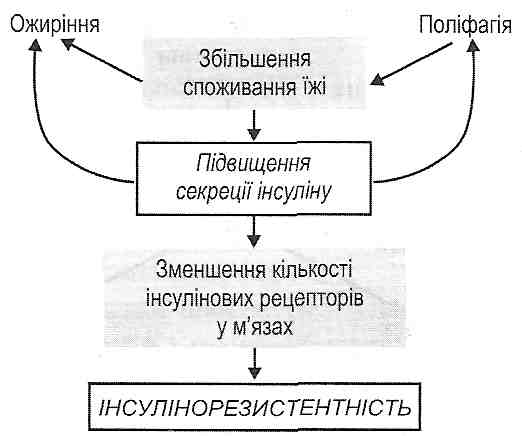
Рис. 65. Гіперінсулінемічний етап патогенезу цукрового діабету II типу з ожирінням
М'язи ж не мають потреби в дії інсуліну. Тому вони оберігають себе від надлишку цього гормону зменшенням кількості рецепторів на поверхні м'язових клітин. Розвивається явище інсулінорезистентності м'язової тканини — її чутливість до дії інсуліну падає. II. Гіпоїнсулінемічний етап (рис. 66). Підвищене навантаження на інсулярний апарат може призводити до функціонального виснаження β-клітин. Цьому сприяють генетично обумовлені їхні дефекти і надлишок в організмі контрінсулярних гормонів. Як наслідок, секреція інсуліну падає й розвивається його відносна недостатність. При цьому дія інсуліну на жирову тканину зберігається (на жирових клітинах багато рецепторів до інсуліну), а на м'язову тканину - зменшується внаслідок інсулінорезистентності.
Клінічно це виявляє себе розвитком гіперглікемії (немає дії інсуліну на м'язову тканину) і відсутністю кетозу (зберігається дія інсуліну на жирову тканину).
20.25. Назвіть можливі причини позапанкреатичної недостатності інсуліну.
Позапанкреатичну недостатність інсуліну можуть викликати такі причини:
а) порушення перетворення проінсуліну в інсулін;
б) утворення аномального інсуліну;
в) висока активність печінкових інсуліназ;
г) зв'язування інсуліну сироватковими білками;
ґ) утворення антитіл проти інсуліну;
д) аномалії інсулінових рецепторів на поверхні периферичних клітин.
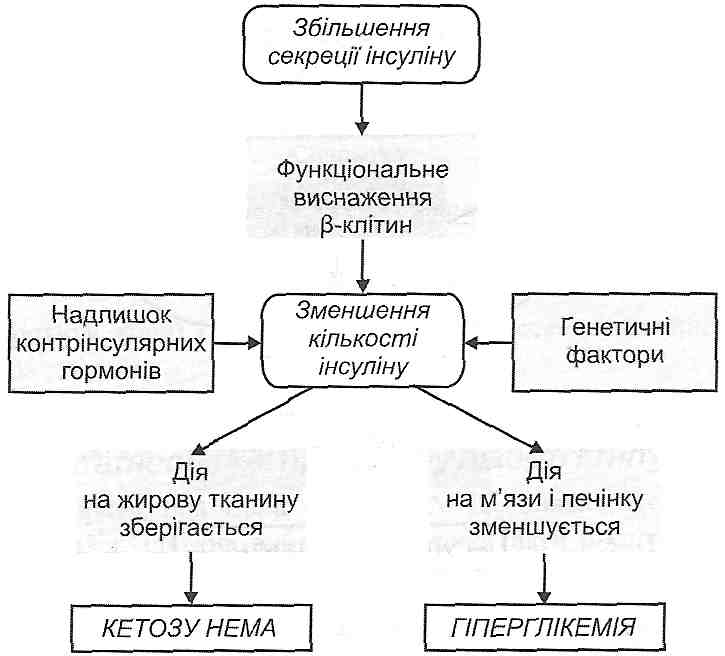
Рис. 66. Гіпоінсулінемічний етап патогенезу цукрового діабету II типу з ожирінням
20.26. Які види обміну речовин порушуються при цукровому діабеті?
Цукровий діабет — це захворювання, при якому порушуються всі види обміну речовин: вуглеводний, жировий, білковий, водно-електролітний обмін, кислотно-основний стан.
20.27. Поясніть механізми розвитку гіперглікемії при цукровому діабеті.
Абсолютна або відносна недостатність інсуліну при цукровому діабеті викликає розвиток гіперглікемії, в основі якої лежать такі механізми.
I. Збільшення надходження глюкози в кров із печінки. Це пояснюється тим, що знімається гальмівний вплив інсуліну на ферменти глікогенолізу й глюконеогенезу, унаслідок чого збільшується інтенсивність цих процесів у печінці.
II. Зменшення використання глюкози інсулінозалежшми тканинами. Це пов'язане з тим, що при дефіциті інсуліну:
а) зменшується проникність клітинних мембран для глюкози у м'язовій (при обох типах цукрового діабету) і жировій (тільки при діабеті І типу) тканині;
б) зменшується утворення глікогену в печінці і м'язах;
в) падає активність пентозного циклу в печінці й жировій тканині;
г) зменшується активність гліколізу в усіх інсулінозалежних тканинах; ґ) відбувається пригнічення ферментів циклу Кребса в печінці і м'язах;
д) порушується перетворення глюкози в жири у печінці й жировій клітковині.
20.28. Які клінічні ознаки цукрового діабету обумовлені гіперглікемією ?
Можна виділити три групи таких ознак.
I. Гіперглікемія, глюкозурія, поліурія, полідипсія (спрага). Глюкозурія виникає тоді, коли концентрація глюкози в крові перевищує так званий "нирковий поріг", тобто 10 ммоль/л.
Унаслідок появи глюкози у вторинній сечі в ній збільшується осмотичний тиск.
Це викликає осмотичний діурез і поліурію.
Як результат поліурії розвивається зневоднення й спрага.
II. Висока гіперглікемія (понад ЗО ммоль/л) викликає збільшення осмотичного тиску крові, унаслідок чого розвивається дегідратація тканин, особливо мозку. Це є причиною так званої гіперосмолярної коми.
III. При гіперглікемії істотно зростає швидкість неферментпативного глікозіїлювання білків (хімічної взаємодії білків з глюкозою крові). Це спричиняється до структурних та функціональних порушень багатьох білків — як наслідок, виникають різні зміни в організмі, серед яких деформація й гемоліз еритроцитів, порушення зсідання крові, підвищення проникності судинної стінки, помутніння кришталика та ін.
20.29. Які порушення свідчать про розлади жирового обміну при цукровому діа б є ті?
1. Гіперліпацидемія — збільшення вмісту в крові вільних жирових кислот. Пов'язана з активацією ліполізу і пригніченням ліпогенезу в жировій тканині внаслідок порушення балансу між інсуліном і контрінсулярними гормонами (рис. 67).
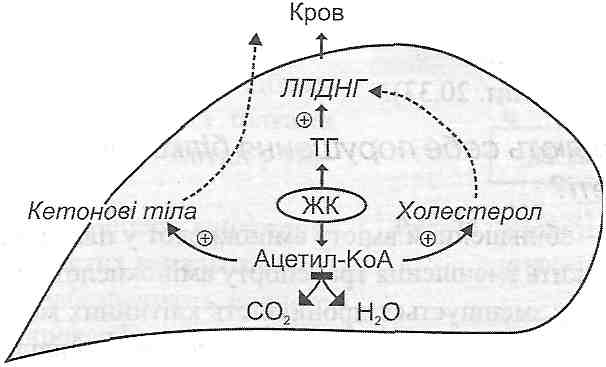
Рис. 67. Перетворення жирових кислот (ЖК) у печінці при цукровому діабеті: ТГ — тригліцериди; ЛПДНГ - ліпопротеїди дуже низької густини
2. Кетоз (гіперкетонемія й кетонурія). Збільшення вмісту кетонових тіл у крові й поява їх у сечі пов'язані з гіперліпацидемією (див. розд. 21).
3. Гіперліпопротеїнемія. Характеризується збільшенням вмісту в крові ліпопротеї-дів дуже низької густини (ЛПДНГ) (див. розд. 21).
4. Жирова інфільтрація печінки. Як і гіперліпопротеїнемія, вона є наслідком надмірного надходження в печінку вільних жирових кислот. Останні виводяться з печінки, перетворюючись у тригліцериди, з наступним утягуванням у формування ЛПДНГ, — розвивається гіперліпопротеїнемія. Якщо можливості гепатоцитів утворювати міцели ЛПДНГ вичерпуються, надлишок тригліцеридів відкладається в печінкових клітинах.
5. Схуднення. При нелікованому цукровому діабеті порушується здатність жирової тканини перетворювати вільні жирові кислоти плазми крові в тригліцериди. Це пов'язано з гальмуванням ліпогенезу при відсутності інсуліну й пригніченням реакцій гліколізу, які необхідні для цього процесу (рис. 68).
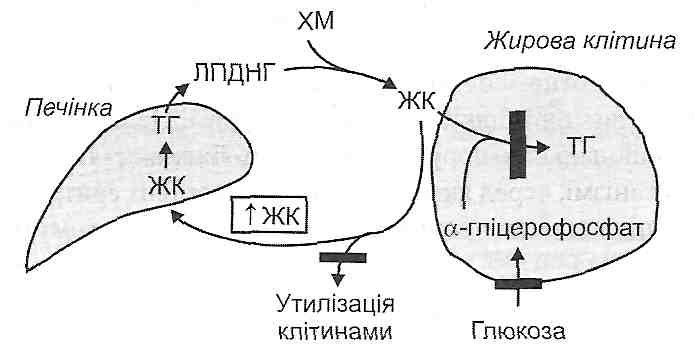
Рис. 68. Порушення депонування жирових кислот (ЖК) у жировій тканині при цукровому діабеті: ТГ— тригліцериди; ХМ — хіломікрони; ЛПДНГ'—ліпопротеїди дуже низької
густини
Посилення ліполізу під дією контрінсулярних гормонів також сприяє зменшенню маси жирової клітковини. 6. Атеросклероз (див. запит. 20.33).
20.30. Чим виявляють себе порушення білкового обміну при цукровому діабеті?
1. Аміноацидемією — збільшенням вмісту амінокислот у плазмі крові (рис. 69).
В основі цього лежить зменшення транспорту амінокислот у м'язові клітини (при відсутності інсуліну зменшується проникність клітинних мембран для амінокислот) і посилення протеолізу в м'язах, унаслідок чого вивільнені амінокислоти надходять у кров.
Надлишок вільних амінокислот поглинається печінкою, де посилюються процеси їх перетворення в глюкозу (глюконеогенез). Це, в кінцевому підсумку, призводить до подальшого збільшення рівня гіперглікемії.
2. Порушеннями біосинтезу білків. Це прямо пов'язано з випадінням анаболічної дії
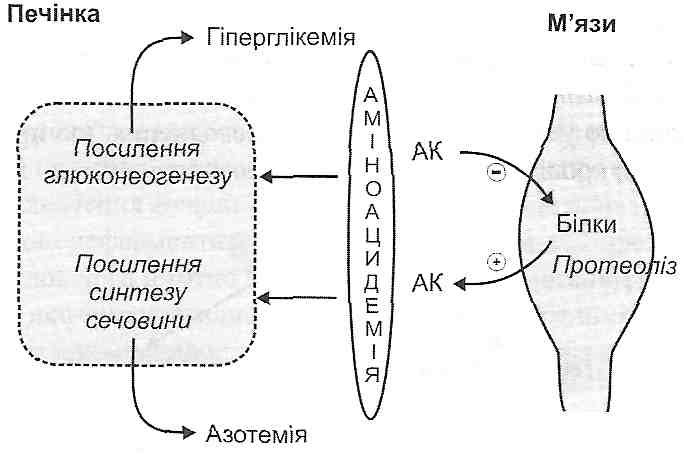
Рис. 69. Порушення обміну білків при цукровому діабеті: АК—амінокислоти
інсуліну.
Клінічно пригнічення білоксинтетичних процесів виявляє себе порушеннями фізичного й розумового розвитку дітей, уповільненням загоєння ран, порушеннями утворення антитіл, унаслідок чого збільшується чутливість до інфекцій, часто розвивається фурункульоз.
20.31. Які порушення водно-електролітного обміну характерні
для цукрового діабету? Який їх
патогенез?
1. Зневоднення (дегідратація). Є наслідком поліурії. Посилює дегідратацію блювота, яка часто супроводжує ацидоз, що розвивається у хворих на цукровий діабет (рис. 70).
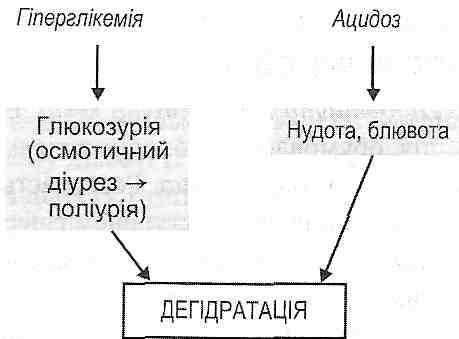
Рис. 70. Механізми дегідратації при цукровому діабеті
2. Гіперкаліємія. Є наслідком активації внутрішньоклітинного протеолізу. Відбувається вивільнення зв'язаного з білками калію, і його іони виходять з клітин у тканинну рідину і кров.
3. Гіпонатріємія. Якщо процеси ацидогене-зу в дистальних звивистих канальцях ниркових нефронів не забезпечують повного відтитровування гідрокарбонатного буфера, то якась частина іонів натрію втрачається із сечею разом з аніонами органічних кислот (ацетооцтової, р-оксимасля-ної).
20.32. Які порушення кислотно-основного стану розвиваються при цукровому діабеті?
Для цукрового діабету характерний негазовий ацидоз. Залежно від механізмів
його розвитку виділяють:
а) кетонемічний метаболічний ацидоз - пов'язаний з накопиченням кетонових тіл;
б) лактацидемічний метаболічний ацидоз — пов'язаний з накопиченням молочної кислоти. Причиною утворення останньої є зневоднення, що призводить до гіпо-волемії, згущення крові (гемоконцентрації) і, як наслідок, — до гіпоксії (рис. 71).
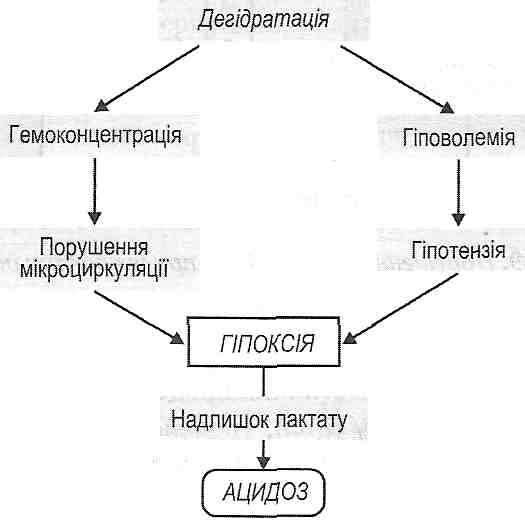
Рис. 71. Механізми лактацидеміїпри цукровому діабеті
20.33. Які варіанти коматозних станів можуть розвиватися при цукровому діабеті?
1. Діабетична кетонемічна кома. В основі її розвитку лежать ацидоз та інтоксикація, обумовлені кетоновими тілами.
2. Гіперосмолярна кома. Розвивається внаслідок дегідратації головного мозку, обумовленої високим ступенем гіперглікемії (див. запит. 20.26).
3. Лактацидемічна кома. Обумовлена накопиченням молочної кислоти й пов'язаним із цим ацидозом.
4. Гіпоглікемічна кома. Може розвиватися в результаті передозування інсуліну при лікуванні цукрового діабету.
20.34. Які ускладнення характерні для цукрового діабету?
... . . . 'Лі
Макроангюпатії, мікроангюпатії, нейропатії.
20.35. Які механізми можуть лежати в основі розвитку макроангіопатій при цукровому діабеті?
Макроангіопшпії характеризуються прискореним розвитком атеросклерозу в артеріях хворих на цукровий діабет. Найчастіше вражаються вінцеві артерії серця, артерії головного мозку й нижніх кінцівок. Це може призводити до розвитку таких
ускладнень, як інфаркт міокарда, інсульт, гангрена пальців ніг і всієї стопи. Існують дві концепції, що пояснюють патогенез макроангіопатій.
I. Концепція порушеного гомеостазу (власне діабетична). Головне значення в розвитку атеросклерозу при цукровому діабеті надається загальним порушенням обміну речовин в організмі, а саме: гіперглікемії, гіперліпопротеїнемії й ацидозу. Патогенетичне значення гіперглікемії полягає в тому, що вона:
1) є причиною неферментативного глікозилювання ліпопротеїдів плазми крові, унаслідок чого істотно збільшується їхня атерогенність;
2) викликає неферментативне глікозилювання мембранних білків ендотеліаль-них клітин і, як наслідок, призводить до підвищення проникності судинної стінки;
3) активує сорбітоловий шлях перетворення глюкози в гладких м'язових клітинах судин. Останнє відбувається, якщо концентрація глюкози в крові перевищує 20 ммоль/л.
Результатом активації сорбітолоеого шляху є утворення в клітинах фруктози. , Оскільки плазматична мембрана непроникна для цієї речовини, вона накопичується в цитоплазмі, підвищуючи осмотичний тиск внутрішньоклітинної рідини, викликаючи набряк і ушкодження клітин.
Гіперліпопротеїнемія при цукровому діабеті характеризується збільшенням вмісту в крові ліпопротеїдів дуже низької густини (ЛПДНГ) і появою "модифікованих" ліпопротеїдів (ЛП): глікозильованих і ацетоацетильованих ЛІХ Про значення цих порушень у розвитку атеросклерозу див. розд. 28.
З виникненням ацидозу пов'язане підвищення проникності судинної стінки й ушкодження її гладком'язових і ендотеліальних клітин - фактори, що сприяють атеросклерозу.
II. Інсулінова концепція. її прихильники вважають, що провідною ланкою в патогенезі діабетичних макроангіопатій є гіперінсулінемія. Збільшення вмісту інсуліну в крові може бути ендогенним, як при цукровому діабеті II типу, і екзогенним, як результат передозувань інсуліну при лікуванні діабету І типу.
Інсулін у великих кількостях, маючи мітогенну дію, викликає проліферацію гладком'язових клітин артеріальної стінки, що призводить до формування фіброзних атеросклеротичних бляшок.
20.36. Як пояснюють розвиток мікроангіопатій при цукровому діабеті? Чим вони можуть виявляти себе?
Мікроангіопатїї— це ураження судин мікроциркуляторного русла (артеріол, капілярів), що виникають як ускладнення цукрового діабету. Сутність цих уражень полягає в значному збільшенні товщини базальної мембрани мікросудин, що утруднює обмін речовин між кров'ю й тканинами.
Серед механізмів розвитку мікроангіопатій велике значення мають збільшення синтезу глікопротеїнів базальної мембрани і неферментативне глікозилювання її компонентів.
Мікроангіопатії найчастіше виявляють себе ураженням судин нирок (діабетич-
на нефропатія) і сітківки очей (діабетичнаретинопатія). Як наслідок, можуть розвиватися хронічна ниркова недостатність, відшарування сітківки.
20.37. Який патогенез нейропатій при цукровому діабеті?
Heuponamu — це специфічні ураження нервових провідників у хворих на цукровий діабет. Вони виявляють себе розладами чутливості, вегетативних і рухових функцій, нервової трофіки.
В основі патогенезу діабетичних нейропатій лежать процеси демієлінізації нервів і порушення аксоплазматичного транспорту.
Суть демієлінізації полягає в руйнуванні мієлінової оболонки нервових волокон і порушенні утворення мієліну. Ці розлади пов'язують із:
а) активацією сорбітолового шляху перетворення глюкози у шваннівських клітинах, що спричиняє їхнє ушкодження й загибель;
б) пригніченням міоінозитолового шляху, внаслідок чого порушується утворення мі-оінозитолу - речовини, необхідної для побудови мієліну.
20.38. Назвіть основні патогенетичні принципи лікування цукрового діабету.
1. Уведення інсуліну при інсулінозалежному цукровому діабеті І типу. Перспективними в цьому плані є трансплантація (3-клітин острівців підшлункової залози й застосування автоматизованих систем дозування і введення інсуліну.
2. Уведення фармакологічних препаратів, що усувають гіперглікемію, - гіпоглікемічних засобів (бігуаніди, похідні сульфонілсечовини).
3. Дієтотерапія, що забороняє вживати харчові продукти з високим вмістом цукру, регулює енергетичну цінність їжі та режим її споживання.
4. Фізичні навантаження (тренування). Вони зменшують рівень гіперглікемії й збільшують чутливість м'язової тканини до інсуліну.
21. Порушення жирового обміну
21.1. Назвіть основні причини порушень жирового обміну в організмі.
Причинами розладів жирового обміну можуть бути порушення:
1) перетравлювання і всмоктування ліпідів у тонкій кишці;
2) транспорту ліпідів кров'ю;
3) депонування ліпідів у жировій тканині;
4) жирової функції печінки (див. розд. 31);
5) проміжного обміну ліпідів у периферичних тканинах;
6) нервової й гормональної регуляції жирового обміну.
27.2. Що може бути причиною порушень перетравлювання і всмоктування ліпідів у кишках?
1. Порушення емульгування жирів:
а) недостатнє надходження жовчі в кишки (механічна жовтяниця);
б) передчасне руйнування жовчних кислот бактеріальною флорою при порушенні моторної функції кишок.
2. Порушення гідролітичного розщеплення жирів:
а) недостатнє надходження панкреатичної ліпази у дванадцятипалу кишку;
б) порушення активації цього ферменту за умов недостатньої секреції жовчі.
3. Порушення утворення ліпідних міцел у порожнині тонкої кишки:
а) недостатнє надходження жовчі в тонку кишку;
б) зв'язування жовчних кислот деякими лікарськими препаратами (холестира-мін, неоміцин та ін.);
в) утворення кальцієвих солей жирових кислот при надмірному надходженні кальцію з їжею й водою.
4. Порушення всмоктування міцел:
а) швидка евакуація вмісту тонкої кишки (проноси);
б) ушкодження епітелію слизової оболонки тонкої кишки (ентерити, радіаційні ураження).
5. Порушення ресинтезу тригліцеридів і формування хіломікронів в епітеліальних клітинах кишок:
а) зменшення кількості або пригнічення активності відповідних ферментів;
б) дефіцит АТФ.
21.3. Які зміни складу крові можуть бути проявом порушень транспорту ліпідів в організмі?
Гіперліпопротеїнемія і гіполіпопротеїнемія — відповідно збільшення й зменшення вмісту ліпопротеїдів у плазмі крові; дисліпопротеїнемія — порушення співвідно-
шення між окремими класами ліпопротеїдів плазми крові; гіперліпацидемія — збільшення вмісту вільних жирових кислот у крові.
21.4. Які існують класи ліпопротеїдів плазми крові?
У плазмі крові містяться хіломікрони (ХМ), ліпопротеїди дуже низької густини (ЛПДНГ), ліпопротеїди проміжної густини (ЛППГ), ліпопротеїди низької густини (ЛПНГ), ліпопротеїди високої густини (ЛПВГ).
21.5. Дайте порівняльну характеристику різних класів ліпопротеїдів плазми крові.
Показник |
хм |
ЛПДНГ |
ЛПНГ |
ЛПВГ |
Діаметр міцел, нм |
Близько 500 |
Близько 50 |
15-20 |
6-9 |
Основний ліпідний компонент |
Тригліцериди |
Тригліцериди |
Холестерол |
Фосфоліпіди |
Місце утворення |
Епітелій тонкої кишки |
Печінка |
Кровоносні капіляри печінки |
Печінка, тонка кишка |
Функції |
Транспорт екзогенних тригліцеридів у/Ьмнтіітло .ж |
Транспорт ендогенних тригліцеридів Of'V |
Транспорт холестеролу до периферичних тканин |
Транспорт холестеролу від периферичних тканин до печінки |
21.6. Як класифікують гіперліпопротеїнемії?
За походженням гіперліпопротеїнемії бувають первинними (спадковими) і вторинними (набутими).
Класифікація ВООЗ передбачає поділ гіперліпопротеїнемій на типи залежно від того, вміст ліпопротеїдів якого класу збільшений у крові (див. запит. 21.9).
Залежно від механізмів розвитку гіперліпопротеїнемія може бути продукційною і ретенційною.
21.7. Які генетичні дефекти можуть бути причиною розвитку первинних (спадкових) гіперліпопротеїнемій?
1. Генетично обумовлені зміни структури апопротеїнів — білкової частини ліпопро-теїдних міцел, унаслідок чого ліпопротеїди плазми крові не можуть взаємодіяти з відповідними рецепторами або зазнавати ферментативних перетворень.
2. Спадкові дефекти ферментів, що беруть участь в обміні ліпопротеїдів, зокрема, дефіцит ліпопротеїнліпази, печінкової ліпази, лецитинхолестеролацилтрансфера-зи (ЛХАТ).
3. Аномалії клітинних рецепторів до ліпопротеїдів.
21.8. Назвіть можливі причини розвитку вторинних (набутих) . гіперліпопротеїнємій.
1. Ендокринні хвороби (цукровий діабет, гіпотиреоз).
2. Метаболічні розлади (ожиріння, подагра).
3. Хвороби нирок (нефротичний синдром).
4. Хвороби печінки (обтураційна жовтяниця).
5. Інтоксикації (алкоголізм).
21.9. Наведіть класифікацію гіперліпопротеїнємій, запропоновану експертами ВООЗ.

Умовні позначення див. запит. 21.4.
21.10. Наведіть приклади розвитку різних типів гіперліпопротеїнємій за класифікацією ВООЗ.
Тип гшерліпопротеїнемії |
Спадкові порушення |
Набуті порушення |
Типі |
Дефіцит ліпопротеїнліпази |
Системний червоний вовчак |
Тип Па |
Сімейна гіперхолестеролемія (дефіцит рецепторів до ЛПНГ) |
Гіпотиреоз |
Тип ПЬ |
Комбінована сімейна гіперхолестеролемія |
Нефротичний синдром |
Тип III |
Сімейна гіперліпопротеїнемія ПІ типу |
Ожиріння |
Тип IV |
Комбінована сімейна гіперліпідемія |
Цукровий діабет |
ТипУ |
Сімейна гшертриглщеридемія |
Алкогольна інтоксикація |
21.11. Що таке продукційна і ретенційна гіперліпопротеїнемія?
Продукційна гіперліпопротеїнемія розвивається внаслідок збільшення утворення ліпопротеїдів, а ретенційна — у результаті порушення їх утилізації.
21.12. У чому полягає патогенетичне значення гіперліпопротеїнемій ?
Гіперліпопротеїнемії сприяють розвитку атеросклерозу (див. розд. 28).
21.13. Наведіть приклади гіполіпопротеїнемій. Дайте коротку характеристику.
Зменшення вмісту в крові ліпопротеїдів (гіполіпопротеїнемія) спостерігається значно рідше, ніж гіперліпопротеїнемія. Причиною гіполіпопротеїнемій найчастіше є спадково обумовлені порушення. Серед них:
1) абеталіпопротеїнемїя. В організмі нема апопротеїну В, унаслідок чого не утворюються хіломікрони і ЛПНГ. Клінічно виявляє себе стеатореєю (поява жиру в калі) і авітамінозами жиророзчинних вітамінів. Успадковується аутосомно-рецесивно;
2) гіпобеталіпопротеїнемія. Зменшено вміст ЛПНГ. Вважають, що це доброякісний стан, який сприяє довголіттю, оскільки перешкоджає розвитку атеросклерозу й ішемічної хвороби серця. Успадковується аутосомно-домінантно;
3) танжерська хвороба (від назви острова Танжер на східному узбережжі США). Характеризується повною відсутністю ЛПВГ або наявністю їх аномальних форм. Порушується транспорт холестеролу від тканин у печінку, він накопичується в периферичних клітинах. Клінічно це виявляє себе гепатомегалією, спленомегалією, збільшенням лімфатичних вузлів.
21.14. Що таке "модифіковані" ліпопротеїди? Наведіть приклади. Яке їх патогенетичне значення?
"Модифікованими" називають якісно змінені ліпопротеїди (ЛП). До них, зокрема, відносять глікозильовані ЛП (зв'язані з глікозильними групами, утворюються при гіперглікемії), ацетоацетильовані ЛП (зв'язані з ацетооцтовою кислотою, утворюються при цукровому діабеті); ЛП, зв'язані з продуктами пероксидного окиснення ліпідів; ліпопротеїн X (з'являється при обтураційній жовтяниці); комплекси ліпопро-теїд-антитіло.
"Модифіковані" ЛП є атерогенними. їх накопичення сприяє розвитку атеросклерозу.
21.15. Що таке первинне і вторинне ожиріння?
Первинним називають ожиріння, що являє собою самостійний патологічний процес.
Вторинне ожиріння є ознакою тих чи тих захворювань. Найпоширенішими варіантами вторинного ожиріння є церебральне і гормональне ожиріння.
21.16. Назвіть основні причини первинного ожиріння.
1. Надмірне споживання їжі, що перевищує енергетичні витрати організму.
2. Гіподинамія — обмеження фізичної активності людини.
3. Генетична схильність. Може виявляти себе особливостями харчової поведінки людини або особливостями регуляції жирового обміну.
21.17. У яких випадках виникає церебральне ожиріння?
Церебральне ожиріння виникає при ураженнях гіпоталамуса, де зосереджені центри, що регулюють харчову поведінку. Такі ураження виникають при травмах, пухлинах, енцефаліті. Провідним механізмом ожиріння в цьому випадку є поліфагія (підвищення апетиту).
21.18. Які гормональні порушення можуть бути причиною вторинного ожиріння?
Гормональне ожиріння розвивається як одна з ознак ендокринних хвороб. Воно супроводжує розвиток:
а) гіпотиреозу;
б) аденоми острівців підшлункової залози (гіперінсул інізм);
в) синдрому Іценка—Кушинга;
г) гіпофункції статевих залоз.
21.19. Чим відрізняється гіперпластичний тип ожиріння від гіпертрофічного?
Гіперпластичне ожиріння пов'язане з гіперплазією жирових клітин, тобто зі збільшенням їхньої кількості. Для нього характерними є початок у ранньому дитячому віці і велика надлишкова вага.
В основі гіпертрофічного ожиріння лежить збільшення маси окремих жирових клітин, при цьому їхня кількість не міняється. Ожиріння цього типу має більш пізній початок і не настільки виражене, як у попередньому випадку.
21.20. Які існують експериментальні моделі ожиріння?
I. Експериментальні моделі первинного ожиріння. Отримано чисті лінії мишей і щурів з генетично обумовленим ожирінням, що є ознакою, яка передається від потомства до потомства.
II. Експериментальні моделі вторинного церебрального ожиріння:
а) руйнування вентромедіальних ядер гіпоталамуса, що утворюють "центр насичення";
б) електростимуляція латеральних ядер гіпоталамуса, що становлять "центр апетиту".
III. Експериментальні моделі вторинного гормонального ожиріння:
а) вимикання функції деяких ендокринних залоз (видалення щитоподібної залози, кастрація);
б) введення в організм великої кількості деяких гормонів (інсуліну, глюкокор-тикоїдів).
IV. Експериментальні моделі місцевого ожиріння — перетинання симпатичних нервів. При цьому в тканині з порушеною іннервацією збільшується маса жирової клітковини, оскільки припиняється ліполітична дія катехоламінів.
21.21. Які механізми лежать в основі збільшення маси жирової тканини при ожирінні?
I. Посилення ліпогенезу. Цей механізм є провідним при:
а) посиленому надходженні жирових кислот у жирові клітини з хіломікронів і ліпопротеїдів дуже низької густини (переїдання, церебральне ожиріння, гіперінсулінізм);
б) посиленому утворенні жирових кислот в адипоцитах із глюкози (надмірне споживання вуглеводів, гіперінсулінізм, гіперфункція кори надниркових залоз);
в) збільшенні активності ферментів ліпогенезу (гіперінсулінізм).
II. Пригнічення ліполізу. В основі цього механізму зменшення активності гормончут-ливої ліпази адипоцитів. Це буває при:
а) гіподинамії;
б) гіпотиреозі;
в) порушенні симпатичної іннервації.
21.22. У чому полягає патогенетичне значення ожиріння?
При ожирінні значно зростає ризик виникнення багатьох соматичних хвороб. Серед них — атеросклероз, ішемічна хвороба серця, інфаркт міокарда, цукровий діабет II типу.
21.23. Що таке гіперкетонемія? Які причини й механізми лежать в основі її розвитку?
Гіперкетонемія - це накопичення кетонових тіл у крові. До них відносять ацетооцтову кислоту, р-оксимасляну кислоту, ацетон.
Основною причиною гіперкетонемії є збільшення вмісту в крові вільних жирових кислот - гіперліпацидемія. Остання виникає при посиленні ліполізу в жировій тканині, наприклад, при цукровому діабеті І типу, гарячці, у другому періоді голодування.
Надлишок вільних жирових кислот надходить у печінку, де під дією ферментів р-окиснення" утворюється велика кількість ацетил-КоА. Якась його частина зазнає подальших перетворень у циклі Кребса (інтенсивність цього процесу залежить від енергетичних потреб гепатоцитів), а невикористана кількість, що залишилася, іде на
утворення кетонових тіл, які надходять у кров і можуть бути утилізовані як цінний ! енергетичний субстрат периферичними клітинами.
При деяких захворюваннях, наприклад, при цукровому діабеті І типу, використання кетонових тіл клітинами порушується. Це збільшує гіперкетонемію.
Гіперкетонемія завжди супроводжується кетонурією (ацетонурією) — появою кетонових тіл (ацетону) у сечі.
21.24. Які гормони посилюють ліполіз у жировій тканині і можуть викликати гіперліпацидемію з наступною гіперкетонемією?
Ліполітичну дію мають контрінсулярні гормони, зокрема, катехоламіни, глюкагон, глюкокортикоїди, тироксин. Вони через активацію аденілатциклазної системи підвищують активність гормончутливої ліпази клітин жирової тканини.
21.25. Які порушення в організмі обумовлює гіперкетонемія?
1. Метаболічний ацидоз. Пов'язаний з накопиченням кислих продуктів (ацетооцтової, Р-оксимасляної кислот).
2. Інтоксикація. Обумовлена головним чином ацетоном. Ця речовина легко проникає в багаті ліпідами тканини, зокрема, у структури ЦНС і, "розчиняючи" ліпіди мембран, порушує їх бар'єрну і транспортну функції.
3. Кома. Є найтяжчим для організму порушенням. Розвивається як наслідок декомпенсованого метаболічного ацидозу та інтоксикації.
22о Порушеним білкового обміну, обміну амінокислот і азотистих основ
22.1. Що таке позитивний і негативний азотистий баланс? Наведіть приклади.
У дорослої здорової людини кількість азотистих речовин, що виводяться з організму, дорівнює кількості, що він її отримує з їжею. Такий стан називається азопшс- \ тою рівновагою.
В організмі, що росте, при вагітності, при введенні або надмірному утворенні ! анаболічних гормонів, при посиленому годуванні після виснажливих хвороб азоту виводиться менше, ніж його надходить. У цьому випадку йдеться про позитивний азотистий баланс.
І навпаки, якщо азоту виводиться більше, ніж надходить, то розвивається негативний азотистий баланс. Це може бути при голодуванні, втраті білків через нирки (протеїнурія), шкіру (опіки), кишки (проноси); при тиреотоксикозі, інфекційній гарячці.
22.2. Назвіть основні причини аліментарної білкової недостатності.
Аліментарнії білкова недостатність розвивається внаслідок порушень надходження в організм білків, їх перетравлювання і всмоктування.
Основними її причинами є голодування, незбалансоване за амінокислотним і складом харчування, запальні й дистрофічні зміни різних відділів кишок, що супроводжуються порушеннями їх секреторної та моторної функцій.
22.3. Назвіть основні причини порушення біосинтезу білків у клітинах.
1. Порушення структури генів, що кодують інформацію про будову білків (мутації).
2. Отрути й специфічні інгібітори мультиферментних комплексів, що забезпечують ] процеси транскрипції, трансляції й посттрансляційної модифікації білків.
3. Дефіцит незамінних амінокислот.
4. Дефіцит АТФ.
5. Порушення утворення транспортних і рибосомної РНК, білків рибосом.
22.4. Порушення яких етапів можуть спричиняти розлади біосинтезу білків у клітинах?
I. Порушення транскрипції- утворення інформаційної РНК на матриці ДНК.
II. Порушення трансляції— синтезу поліпептидних ланцюгів з амінокислот.
III. Порушення посттрансляційної модифікації білків — формування їхньої третин- ; ної й четвертинної структур.
22.5. Які зміни білкового складу крові можуть виникати в умовах патології?
I. Гіпопротеїнемія - зменшення вмісту білків у плазмі крові. Виникає головним чином за рахунок зниження кількості альбумінів і може бути набутою і спадковою. До гіпопротеїнемії спричиняються голодування, аліментарна білкова недостатність, захворювання печінки, вихід білків із кровоносного русла (крововтрата, плазмовтрата, ексудація, транссудація) і втрата білків із сечею (протеїнурія). Гіпопротеїнемія веде до зменшення онкотичного тиску плазми крові, у результаті чого рідина виходить з кровоносних судин в інтерстиціальну тканину - розвиваються набряки.
II. Гіперпротеїнемія — збільшення вмісту білків у плазмі крові. Буває відносною (згущення крові) і абсолютною. Абсолютна гіперпротеїнемія найчастіше обумовлена збільшенням синтезу білків плазми крові і головним чином у-глобулінів (антитіл).
Клінічні прояви гіперпротеїнемії пов'язані зі збільшенням в'язкості крові, зміною її реологічних властивостей і, як наслідок, порушеннями мікроциркуляції.
III. Диспротеїнемія - зміна співвідношення між окремими білковими фракціями крові. Може бути спадковою і набутою. Часто пов'язана зі зміною спектру а- і у-глобулінів. Характерна для гострих запальних процесів ("білки гострої фази запалення"), дифузних захворювань сполучної тканини, аутоімунних захворювань. Іноді в крові з'являються якісно змінені білки, зокрема, парапротеїни і кріогло-буліни. Парапротеїни — це імуноглобуліни, що є продуктами одиничних клонів лімфоцитів. їх поява обумовлена проліферацією окремих антитілосинтезуючих клітин, що буває при патологічних процесах пухлинної природи (мієломна хвороба, макроглобулінемія Вальденстрема).
Кріоглобуліни — це різновид парапротеїнів. Вони являють собою патологічні білки із властивостями імуноглобулінів, що преципітують при охолодженні.
22.6. Що таке продукційна іретенційна гіперазотемія?
Пперазотемія — це збільшення залишкового (небілкового) азоту в крові. У нормі залишковий азот на 50 % складається з азоту сечовини, близько 25 % його припадає на частку амінокислот, інша частина — на інші азотисті продукти.
Продукційна (печінкова) гіперазотемія виникає внаслідок порушення утворення сечовини, детоксикації азотистих продуктів у печінці.
Ретенційна (ниркова) гіперазотемія є наслідком порушення видільної функції нирок.
22.7. Які фактори можуть викликати порушення утворення сечовини в печінці?
Порушення синтезу сечовини може спостерігатися на завершальних етапах розвитку недостатності печінки як одна з ознак порушення її детоксикаційної функції.
Можливі спадково обумовлені порушення сечовиноутворення. їх причиною може бути недостатній синтез цілого ряду ферментів: аргінінсукцинатліази (аргінін-
сукцинатурія), карбамоїлфосфатсинтетази і орнітинкарбамоїлтрансферази (амоніє-мія) і аргінінсукцинатсинтетази (цитрулінурія).
Наслідком порушення синтезу сечовини є накопичення аміаку в крові й цілий ряд пов'язаних з цим тяжких клінічних проявів (докладно див. розд. 31).
22.8. У чому сутність і чим виявляють себе спадково обумовлені порушення обміну фенілаланіну?
Спадково обумовленим порушенням обміну фенілаланіну є фенілкетонурія -за-хворювання з аутосомно-рецесивним типом спадкування. Його причиною є генетичний дефект ферменту фенілаланінгідроксилази, що в нормі перетворює фенілаланін на тирозин (рис. 73). За відсутності зазначеного ферменту окиснення фенілаланіну відбувається шляхом утворення фенілпіровиноградної і фенілмолочної кислот. Однак цей шлях має малу пропускну здатність, і тому фенілаланін накопичується у великій кількості в крові, тканинах і спинномозковій рідині, що в перші ж місяці життя веде до важкого ураження центральної нервової системи й невиліковного слабоумства.
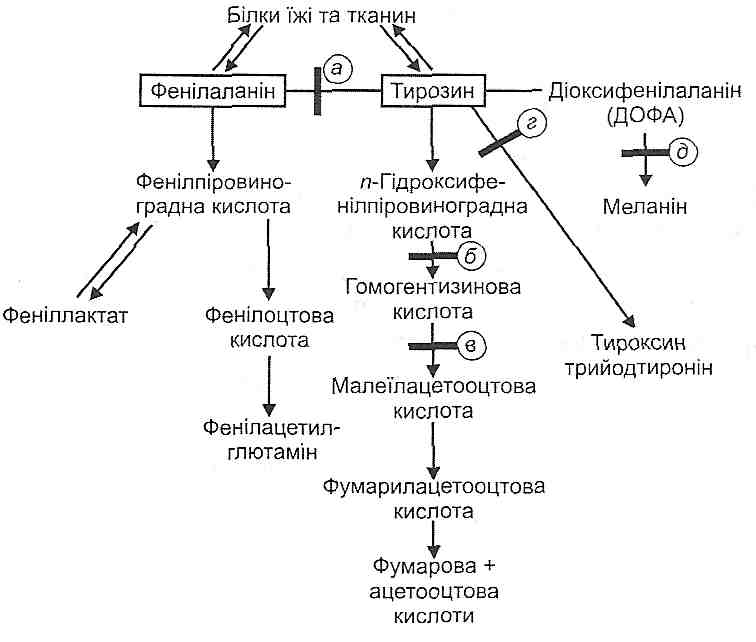
Рис. 73. Блокада шляхів метаболізму фенілаланіну і тирозину: блок а - фенілкетонурія; б — тирозиноз; в — алкаптонурія; г - гіпотиреоз; д — альбінізм
22.9. У чому сутність і чим виявляють себе спадково обумовлені порушення обміну тирозину?
Залежно від рівня генетичних дефектів спадково обумовлені порушення обміну тирозину можуть виявлятися розвитком тирозинозу, алкаптонурії, альбінізму. Усі ці захворювання успадковуються аутосомно-рецесивно.
Тирозиноз виникає внаслідок генетичного дефекту ферменту - оксидази парагі-дроксифенілпіровиноградної кислоти. У результаті вона, будучи першим проміжним продуктом обміну тирозину, не перетворюється в гомогентизинову кислоту, накопичується в крові й разом з тирозином виводиться із сечею.
Алкаптонурія є наслідком порушення синтезу оксидази гомогентизинової кислоти, що перетворює останню в малеїлацетооцтову кислоту. У результаті в крові й сечі з'являється гомогентизинова кислота. Сеча при стоянні на повітрі, а також при додаванні до неї лугу стає чорною, що пояснюється окисненням гомогентизинової кислоти киснем повітря й утворенням алкаптону. Гомогентизинова кислота з крові проникає в тканини — хрящову, сухожилля, зв'язки, внутрішній шар стінки аорти, унаслідок чого з'являються темні плями в ділянці вух, носа, на склерах. Іноді розвиваються тяжкі зміни в суглобах.
Альбінізм обумовлений дефіцитом ферменту тирозинази. Унаслідок цього не утворюється пігмент шкіри й волосся — меланін. Організм, позбавлений пігменту, стає дуже чутливим до дії ультрафіолетового випромінювання.
22.10. Що таке подагра?
Подагра - це хвороба, в основі якої лежить накопичення в організмі сечової кислоти - кінцевого продукту обміну пуринових основ, що входять до структури нуклеїнових кислот.
Для хворих на подагру характерне збільшення рівня сечової кислоти в крові (гіперурикемія). Гіперурикемія може супроводжуватися відкладенням солей сечової кислоти в суглобах і хрящах, де через слабке кровопостачання завжди є тенденція до зменшення рН (кисле середовище сприяє випаданню солей в осад). Відкладення солей викликає гостре подагричне запалення, що супроводжується болем, гарячкою і завершується утворенням подагричних вузлів і деформацією суглобів.
22.11. Що є факторами ризику подагри?
Факторами ризику подагри можуть бути:
1) надмірне надходження пуринів в організм (уживання в їжу великої кількості м'яса, особливо з вином і пивом);
2) надмірне надходження в організм молібдену, що входить до складу ксантинокси-дази, яка перетворює ксантин у гіпоксантин. Останній потім перетворюється в сечову кислоту;
3) стать (частіше хворіють чоловіки);
4) літній вік, для якого характерна вікова гіперурикемія;
5) спадкова схильність. Можливими є домінантно успадковуване підвищення рівня сечової кислоти в крові і зміни факторів, що підтримують сечову кислоту в розчиненому стані.
23о Порушення водно-еольового обміну
23.1. Що таке позитивний і негативний водний баланс?
Якщо надходження води в організм перевищує її виведення, то розвивається позитивний водний баланс.
Негативний водний баланс розвивається тоді, коли виведення води з організму перевищує її надходження. Класифікацію порушень обміну води представлено на рис. 74.
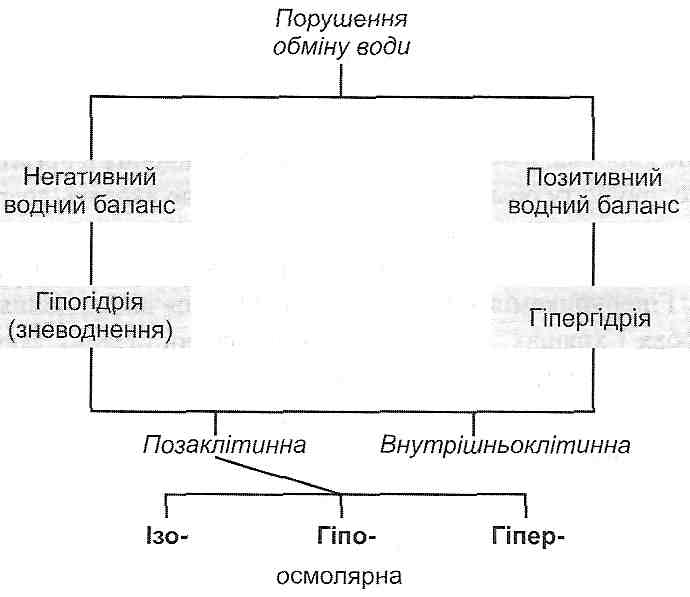
Рис. 74. Класифікація порушень обміну води
23.2. Які гормони беруть участь у захисно-компенсаторних реакціях організму при порушеннях водно-сольового обміну?
Альдостерон, вазопресин (антидіуретичний гормон), передсердний натрійуре-тичний гормон (атріопептин), адреналін.
23.3. Які фактори активують і гальмують утворення й секрецію
альдостерону?
Основним фактором, що стимулює утворення й секрецію альдостерону, є продукти активації ренін-ангіотензинної системи — ангіотензин II і ангіотензин III (рис. 75).
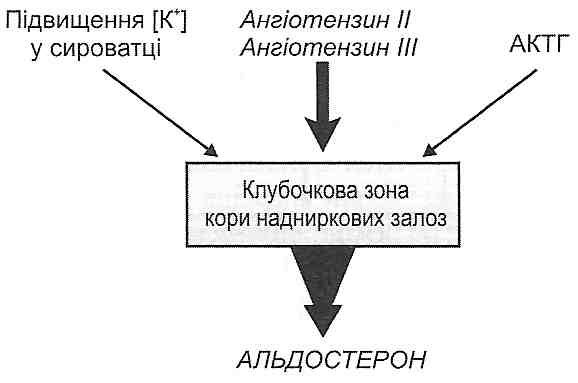
Рис. 75. Фактори — стимулятори секреції альдостерону
їхній стимуляційний ефект максимально виявляє себе за умови нормальної секреції ^КТТ'аденогіпофізом. У цьому випадку АКТГ має так звану пермісивну дію.
Можлива також безпосередня стимуляція секреції альдостерону високими концентраціями іонів калію в плазмі крові.
Гальмують утворення і вивільнення альдостерону в кров передсердний натрійу-ретичний гормон і дофамін.
23.4. Назвіть основні функціональні ефекти альдостерону.
Основні функціональні ефекти альдостерону пов'язані з його впливом на нирки. Діючи на дистальні звивисті канальці нефронів, альдостерон викликає:
1) збільшення реабсорбції іонів натрію;
2) збільшення секреції іонів калію;
3) збільшення секреції іонів водню (посилює ацидогенез).
23.5. Що таке ренін-ангіотензинна система? Як вона активується? Назвіть основні функціональні ефекти ангіотензину II і ангіотензину III.
Ренін-ангіотензинна система пов'язана з функціонуванням юкстагломеруляр-ного апарату нирок (ЮГА). Цілий ряд факторів (порушення кровообігу в нирках, активація симпатоадреналової системи, зменшення концентрації іонів натрію в плазмі крові) викликає вивільнення клітинами ЮГА в кров протеолітичного ферменту — реніну (рис. 76). Ренін діє на а -глобулін плазми крові (ангіотензиноген) і відщеплює від нього пептид, що складається з десяти амінокислотних залишків. Ця речовина, яка ще не має будь-якої біологічної активності, отримала назву ангіотензин І. При проходженні через капіляри легень від ангіотензину І під впливом ферменту кон-вертази, що міститься на поверхні ендотеліальних клітин, відщеплюється дві амінокислоти, у результаті чого утворюється ангіотензин II. Далі під впливом ангіотен-зиназ утворюються ангіотензин III (складається з 7 амінокислотних залишків) та інші пептиди, що містять 6,5 і менше амінокислот і не мають біологічної активності (рис. 77).
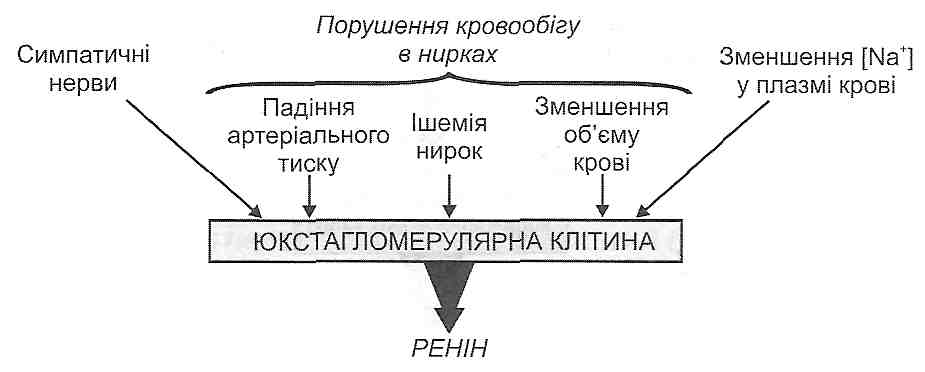
Рис. 76. Фактори - стимулятори секреції реніну
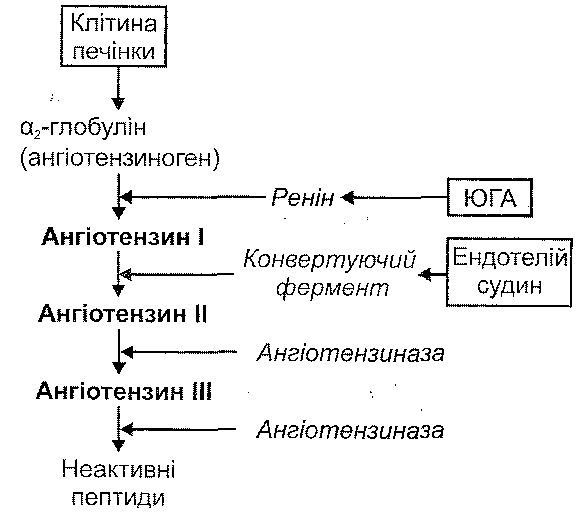
Рис. 77. Ренін-ангіотензинна система
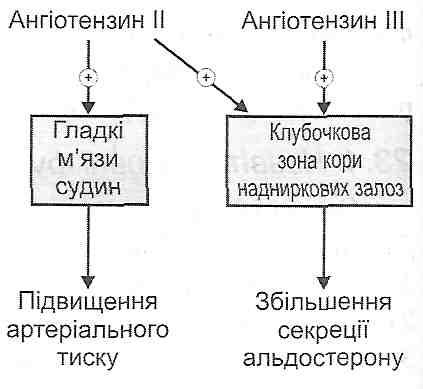
Рис. 78. Функціональні ефекти ангіотензинів II і III
Ангіотензин II має два ефекти:
1) викликає скорочення гладких м'язів артеріол, у результаті чого відбувається їх звуження й підвищується артеріальний тиск;
2) діючи на клубочкову зону кори надниркових залоз, він активує секрецію альдостерону.
Ангіотензин III має тільки одну з двох зазначених дій - збільшує секрецію альдостерону (рис. 78).
23.6. Які фактори стимулюють утворення й секрецію передсердного натрійуретичного гормону (атріопептину)? Яку дію виявляє цей гормон?
Основним фактором, що стимулює утворення й секрецію атріопептину, є збільшення надходження крові в передсердя серця й, зокрема, збільшення об'єму цирку-
люючої крові. При цьому відбувається розтягнення стінок передсердь, у результаті чого міоендо-кринні клітини вивільняють гормон у кров.
Сьогодні відомо два важливих функціональних ефекти атріопептину. Вони пов'язані з його впливом на клітини канальцевого епітелію нирок і гладкі м'язи судин (рис. 79). Діючи на ці структури, передсердний натрійуретичний гормон, з одного боку, зменшує реабсорбцію іонів натрію, у результаті чого збільшується натрійурез і діурез та зменшується об'єм циркулюючої крові, а з другого - викликає розширення артеріол, унаслідок чого зменшується загальний периферичний опір. У результаті відбувається падіння артеріального тиску.
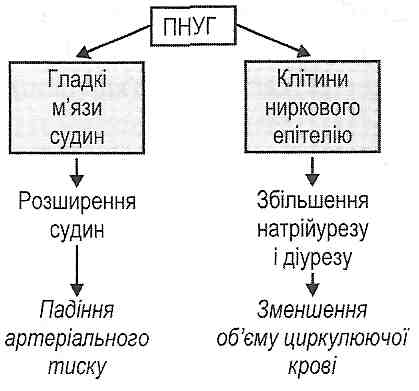
Рис. 79. Функціональні
ефекти передсердного
натрійуретичного гормону
(ПНУГ)
23.7. Що стимулює секрецію вазопресину (антидіуретичного гормону)? Яку дію має цей гормон?
Відомо два механізми активації секреції вазопресину (рис. 80). Перший з них — осмотичний, пов'язаний зі збільшенням осмотичного тиску крові і збудженням центральних та периферичних осморецепторів. Другий механізм — гемодинамічний, починає здійснюватися при зменшенні об'єму циркулюючої крові на 7-15 %. При цьому відбувається збудження волюмо- і барорецепторів, інформація від яких надходить у паравентрикулярне і супраоптичне ядра гіпоталамуса, де відбувається утворення вазопресину. Цей гормон по відростках нейронів опускається в нейрогіпофіз, а звідти надходить у кров.
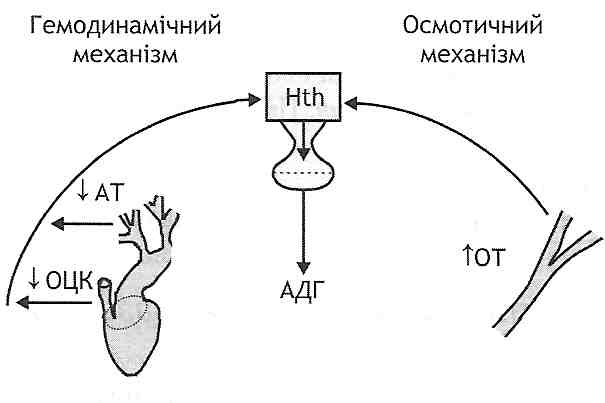
Рис. 80. Механізми активації вивільнення вазопресину (антидіуретичного гормону—АДГ): AT- артеріальний тиск; ОТ- осмотичний тиск; ОЦК—об'єм циркулюючої крові;
Hth - гіпоталамус
Найважливішими ефектами вазопресину є (рис. 81): 1) звуження артеріол і підвищення артеріального тиску;
2) збільшення реабсорбції води в дистальних звивистих канальцях і збірних трубках нирок, що веде до зменшення діурезу, збільшення об'єму циркулюючої крові.
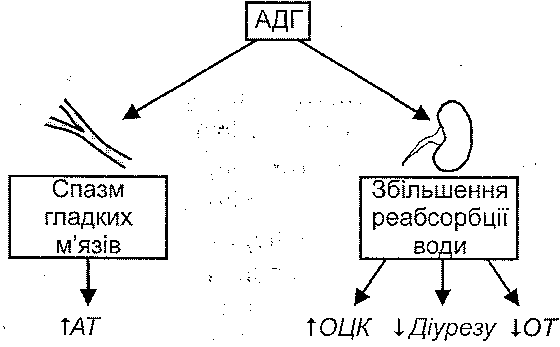
Рис. 81. Функціональні ефекти вазопресину (АДГ)
23.8. Які функціональні ефекти симпатоадреналової системи обумовлюють її участь у захисно-компенсаторних реакціях організму при зневодненні?
1. Активація ренін-ангіотензинної системи. Цей ефект пов'язаний з безпосередньою дією катехоламінів на )3-адренорецептори юкстагломерулярного апарату нирок і опосередкованим впливом на ЮГА через спазм приносних артеріол.
2. Внутрішньонирковий перерозподіл течії крові. При активації симпатоадреналової системи відбувається спазм судин кортикальних нефронів. Унаслідок цього основна частина крові йде через юкстамедулярні нефрони, де площа реабсорбції води й іонів натрію, а також інтенсивність цього процесу значно більша, ніж у кортикальних нефронах. Такий перерозподіл течії крові в нирках веде до значного збільшення реабсорбції натрію й води і сприяє їх збереженню в організмі.
3. Спазм артеріол периферичних тканин. При цьому відповідно до механізму Стар-лінга зменшується фільтрація води з капілярів у тканини, що сприяє підтриманню загального об'єму крові.
4. Зменшення потовиділення. Ця реакція спрямована на зменшення втрати води і солей організмом.
23.9. Що таке позаклітинне зневоднення? Назвіть основні його
причини.
Позаклітинне зневоднення (гіпогідрія, дегідратація, ексикоз) — це зменшення об'єму позаклітинної рідини. В основі його лежить негативний водний баланс.
Причинами позаклітинного зневоднення можуть бути: І. Недостатнє надходження води в організм:
а) екстремальні ситуації, у яких може опинитися людина під час землетрусу, у пустелі (повне водне голодування);
б) неможливість самостійно вгамувати спрагу (важкохворі люди, грудні діти);
в) порушення формування відчуття спраги при ураженнях питного центру в ЦНС. II. Втрата води організмом:
а) виділення великої кількості рідини нирками (поліурія), наприклад, при цукровому й нецукровому діабеті;
б) втрата рідини через травний канал (нестримна блювота, проноси, гіперсалі-вація);
в) посилене потовиділення, наприклад, при інтенсивній фізичній роботі, при дії високої температури;
г) збільшення виділення вологи з видихуваним повітрям при всіх видах задишки; ґ) втрата води з ексудатом при запаленні (набряки, запалення серозних оболонок та ін.);
д) крововтрата.
23.10. Що таке ізоосмолярне, гіпоосмолярне і гіперосмолярне зневоднення? Наведіть приклади.
Ізоосмолярним називають зневоднення, при якому осмотичний тиск плазми крові й міжклітинної рідини не міняється. Воно розвивається у випадках еквівалентної втрати води й електролітів. Це спостерігається іноді при поліурії, розладах діяльності кишок, а також зразу після гострої крововтрати.
Гіпоосмолярне зневоднення характеризується зменшенням осмотичного тиску позаклітинної рідини і виникає у випадках переважної втрати солей. Воно розвивається при втраті секретів шлунка й кишок (пронос, блювота), а також при підвищеному потовиділенні, якщо втрата води відшкодовується питвом без солі.
Гіперосмолярним називають зневоднення, при якому збільшується осмотичний тиск позаклітинної рідини. Це спостерігається в тих випадках, коли втрата води перевищує втрату електролітів (насамперед натрію), наприклад, при гіпервентиляції, профузному потовиділенні, втраті слини (піт і слина гіпотонічні стосовно крові), а також при проносі, блювоті й поліурії, коли відшкодування втрати води її надходженням в організм є недостатнім.
23.11. Які захисно-компенсаторні реакції розвиваються при позаклітинному зневодненні?
1. Відбувається перехід рідини з інтерстиціального сектора в судини. Це пов'язане з тим, що в умовах зневоднення зменшується гідростатичний тиск крові в капілярах, з одного боку, і збільшується онкотичний тиск крові внаслідок її згущення (гемоконцентрації), з другого.
2. Зменшення об'єму циркулюючої крові, пов'язане зі зневодненням, веде до збудження волюморецепторів і, в кінцевому підсумку, до збільшення секреції антиді-уретичного гормону. Останній збільшує реабсорбцію води в нирках, обмежуючи її втрату організмом.
3. Зменшення об'єму циркулюючої крові викликає активацію ренін-ангіотензинної системи і збільшення секреції альдостерону корою надниркових залоз. Це веде до збільшення реабсорбції іонів натрію в нирках і до нормалізації осмотичного тиску позаклітинної рідини.
4. У результаті зменшення артеріального тиску збуджуються барорецептори, що призводить до активації симпатоадреналової системи (див. запит. 23.8).
5. Зневоднення через центральні й периферичні механізми викликає відчуття спраги. У результаті формуються поведінкові реакції, спрямовані на пошук води й поповнення втраченої рідини.
23.12. Що таке синдром ангідремії? Які патогенетичні механізми відіграють провідну роль у його розвитку?
Ангідремія — це зменшення вмісту води в рідкій частині крові. Є крайнім проявом позаклітинного зневоднення й характеризується розвитком ангідремічного шоку (рис. 82). Основне значення в його розвитку мають:
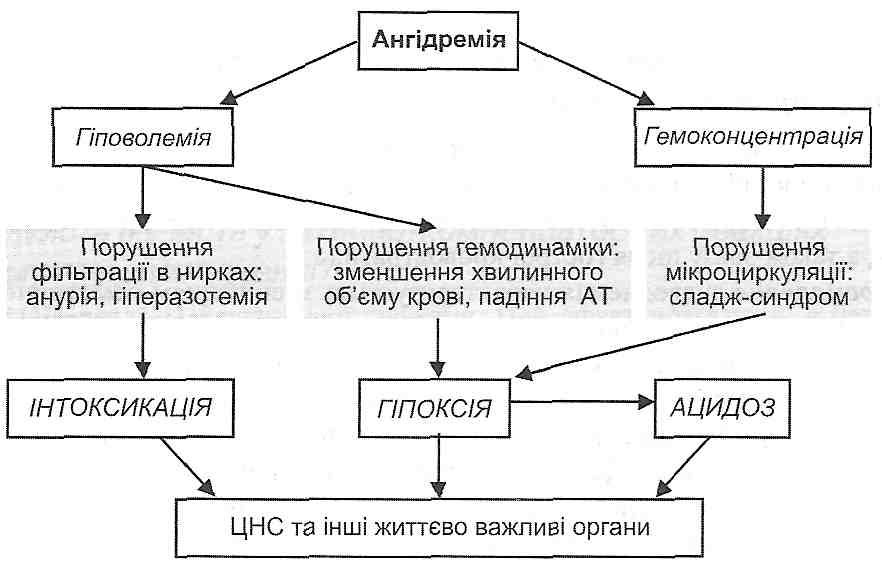
Рис. 82. Патогенез ангідремічного шоку
1) гіповолемія (зменшення об'єму циркулюючої крові). Вона є причиною порушення загальної гемодинаміки. Зменшується хвилинний об'єм крові й артеріальний тиск, що веде до розвитку циркуляторної гіпоксії й метаболічного ацидозу. У результаті гемодинамічних порушень розвивається гостра ниркова недостатність: зменшується фільтраційний тиск і, як наслідок, розвиваються оліго- і анурія, гіпе-разотемія і уремія (інтоксикація);
2) гемоконцентрація (згущення крові, збільшення її в'язкості). Викликає насамперед порушення мікроциркуляції, уповільнюється течія крові в капілярах, розвивається сладж-синдром, істинний капілярний стаз. Наслідком таких розладів є розвиток гіпоксії й ацидозу.
Гіпоксія, ацидоз та інтоксикація є основними факторами, що порушують функції ЦНС та інших життєво важливих органів і призводять до смерті. Ознаки важкої ангідремії й смерть настають у дорослих після втрати 1/3, у дітей — 1/5 об'єму позаклітинної рідини.
23.13. Що є причиною внутрішньоклітинного зневоднення? Які зміни в клітинах виникають при цьому?
Причиною внутрішньоклітинного зневоднення є збільшення осмотичного тиску міжклітинної рідини, пов'язане з розвитком гіпернатріємії (див. запит. 23.29). У цих умовах вода за законами осмосу виходить із клітин у міжклітинний простір.
У результаті зневоднення збільшується внутрішньоклітинна концентрація електролітів, що веде до порушення гідратних оболонок білкових молекул. Зменшується розчинність білків, вони осаджуються, що, в кінцевому підсумку, виявляється порушенням їх функцій.
Крім того, зменшення води в клітинах спричиняється до зменшення їхнього об'єму і, як наслідок, до зменшення активної поверхні клітинних мембран. Результатом цього є порушення функцій, пов'язаних із плазматичною мембраною, - міжклітинних взаємодій, сприйняття регуляторних сигналів, міграції та ін.
23.14. Якими порушеннями на рівні організму виявляє себе внутрішньоклітинне зневоднення?
Серед загальних порушень на перший план виходять розлади функції нейронів ЦНС. Це виявляється розвитком нестерпної спраги, потьмаренням свідомості, галюцинаціями, порушеннями ритму дихання. Підвищується температура тіла, розвивається "сольова гарячка ".
Зневоднення ендотеліальних клітин веде до збільшення проміжків між ними і, як наслідок, до збільшення проникності стінок судин. Це може бути причиною виходу з капілярів у тканини білків плазми крові та її формених елементів — розвиваються геморагії.
23.15. Що таке позаклітинна гіперп'дрія? Назвіть основні її причини.
Позаклітинна гіпергідрія — це збільшення об'єму рідини в позаклітинному секторі організму. Вона є результатом позитивного водного балансу. Причинами позаклітинної гіпергідрії можуть бути:
I. Надмірне надходження води в організм:
а) пиття солоної води, що не вгамовує спрагу;
б) внутрішньовенне введення великої кількості рідини хворим.
II. Затримка води в організмі внаслідок порушення її виведення нирками:
а) ниркова недостатність (оліго- і анурія);
б) порушення регуляції нирок (первинний і вторинний гіперальдостеронізм, гіперпродукція антидіуретичного гормону).
23.16. Що таке ізоосмолярна, гіпоосмолярна і гіперосмолярна гіпергідрія? Наведіть приклади.
При ізоосмолярній гіпергідрії осмотичний тиск позаклітинної рідини не змінюється. Цей вид порушень може спостерігатися протягом деякого часу після введення надлишкових кількостей ізотонічних розчинів.
Гіпоосмолярна гіпергідрія (водне отруєння) характеризується зменшенням осмотичного тиску позаклітинної рідини. Цей вид гіпергідрії в експерименті на тва-
ринах моделюють повторними введеннями води в шлунок, особливо на тлі введення вазопресину, альдостерону або видалення надниркових залоз. У клініці водне отруєння можливе при рефлекторній анурії, а також у другій стадії гострої ниркової недостатності.
Гіперосмолярна гіпергідрія, для якої характерне збільшення осмотичного тиску позаклітинної рідини, може розвиватися при вживанні для пиття солоної морської води.
23.17. Які захисно-компенсаторні реакції розвиваються при позаклітинній гіпергідрії?
1, Позаклітинна гіпергідрія супроводжується збільшенням об'єму циркулюючої крові. Це веде до механічного розтягнення клітин передсердь, які у відповідь на це вивільняють у кров передсердний натрійуретичний гормон (атріопептин). Останній збільшує натрійурез і діурез, унаслідок чого зменшується об'єм циркулюючої крові.
2. Збільшення об'єму циркулюючої крові є причиною зменшення імпульсації від волюморецепторів, у результаті чого зменшується секреція антидіуретичного гормону і зростає діурез.
23.18. Що таке набряки? Як їх класифікують?
Набряки — це накопичення рідини в тканинах організму і серозних порожнинах.
Розрізняють загальні й місцеві набряки. Загальні набряки є проявом позаклітинної гіпергідрії, місцеві — пов'язані з порушенням балансу рідини в обмеженій ділянці тканини або органа.
За етіологією виділяють набряки серцеві, ниркові, печінкові, кахектичні, запальні, алергічні, токсичні та ін.
Залежно від механізмів розвитку набряки можуть бути:
1) гідростатичними;
2) онкотичними;
3) мікседематозними.
Гідростатичні набряки виникають у результаті збільшення гідростатичного тиску в капілярах. Залежно від причин такого збільшення можна виділити:
а) гіперволемічні;
б) застійні;
в) мікроциркуляторні набряки.
Онкотичні набряки виникають унаслідок змін онкотичного тиску в капілярах або інтерстиціальній рідині. У цю групу входять:
а) гіпопротеїнемічні;
б) мембраногенні;
в) лімфогенні набряки.
23.19. У розвитку яких набряків провідна роль належить збільшенню гідростатичного тиску крові в капілярах?
Гідростатичні набряки можуть бути обумовлені такими механізмами: •
1) збільшенням об'єму крові (гіперволемічні набряки);
2) збільшенням венозного тиску (застійні набряки);
3) первинним порушенням мікроциркуляції - розширенням артеріол і спазмом ве-нул (мікроциркуляторні набряки). v^V-V
Гіперволемічнгши є набряки при позаклітинній гіпергідрії і набряки, пов'язані із затримкою в організмі іонів натрію, наприклад, при серцевій недостатності, вторинному гіперальдостеронізмі.
Причиною застійних набряків є порушення відтоку крові по венозних судинах (хронічна серцева недостатність, порушення венозних клапанів, тромбоз вен та ін.).
Розвиток набряків по мікроциркуляторному типу викликає гістамін, який одночасно розширює артеріоли і звужує вени.
23.20. У розвитку яких набряків провідна роль належить зменшенню онкотичного тиску крові?
Онкотичні набряки закономірно розвиваються при зменшенні вмісту білків (альбумінів) у плазмі крові. Це призводить до зменшення онкотичного тиску крові і переходу рідини з капілярів в інтерстиціальний простір.
За таким механізмом розвиваються, зокрема, набряки при голодуванні (кахек-тичні); нефротичні набряки, пов'язані із втратою білка (протеїнурією); печінкові набряки, що виникають унаслідок порушення синтезу альбумінів у печінці.
23.21. Які набряки відносять до мембраногенних?
Мембраногенні набряки виникають унаслідок підвищення проникності стінок судин (див. розд. 14). Збільшення проникності судин є причиною виходу білків плазми крові в міжклітинний простір, унаслідок цього зростає тканинний онкотичний тиск і вода переходить із кровоносних судин в інтерстицій.
Зазначений механізм є провідним у розвитку алергічних, запальних, токсичних набряків.
23.22. Що таке лімфогенні набряки?
Лімфогенні набряки виникають унаслідок порушень лімфоутворення і лімфо-відтоку. При цьому порушується виведення з лімфою білків, що в нормі фільтруються у тканину, і, як наслідок, збільшується тканинний онкотичний тиск.
Серед причин розвитку лімфогенних набряків варто виділити здавлення лімфатичних судин рубцем; збільшення центрального венозного тиску (недостатність серця), що перешкоджає припливу лімфи в систему кровообігу.
23.23. Що таке мікседематозні ("слизові") набряки?
Мікседематозні набряки - це особливий варіант набряків, в основі якого лежить збільшення гідрофільності тканинних колоїдів. При цьому в тканинах зростає кількість зв'язаної води.
Мікседематозні ("слизові") набряки характерні для гіпофункції щитоподібної залози.
23.24. Як відбувається накопичення води в тканинах при розвитку набряків?
У патогенезі набряків розрізняють дві стадії.
Перша стадія — накопичення зв'язаної води. Набрякова рідина зв'язується з тканинними колоїдами й накопичується в основному в гелеподібних структурах (колагенові волокна, основна речовина сполучної тканини). При цьому клінічні ознаки набряку незначні — трохи збільшується тургор тканини.
Друга стадія - накопичення вільної води. Коли маса зв'язаної води збільшується приблизно на ЗО %, а гідростатичний тиск у тканині досягає атмосферного, починає накопичуватися вільна незв'язана вода. Тоді з'являються виражені ознаки набряку: вільна вода переміщається відповідно до сили гравітації, з'являється симптом "ямки" при натискуванні на тканину.
23.25. Як впливає ацидоз на розвиток набряків?
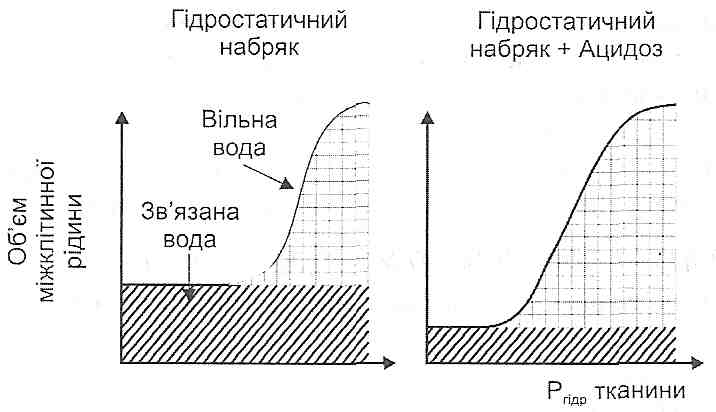
Рис. 83. Вплив ацидозу на розвиток набряків
Ацидоз посилює розвиток набряків. Це пов'язане з тим, що при зміщенні рН у кислий бік зменшується гідрофільність структур сполучної тканини. Тому при надходженні води в тканину менша її кількість буде зв'язуватися з тканинними колоїдами і, отже, буде збільшуватися кількість вільної незв'язаної води. У зв'язку з цим за інших рівних умов клінічні ознаки набряку в кислому середовищі будуть більш вираженими (рис. 83).
23.26. У чому сутність дослідів Старлінга, присвячених вивченню патогенезу набряків?
Е. Старлінг (1896) у дослідах на жабах вивчав роль онкотичного тиску в розвитку набряків. Він показав, що при пер-фузії жаби фізіологічним розчином, що не містить білка, відбувається швидкий перехід рідини з суцин у тканину, внаслідок чого вода затримується в організмі й маса тварини зростає. Якщо потім перемкнути систему на перфузію сироваткою крові, то буде спостерігатися зворотний процес - рідина виходить із тканин у кровоносні судини й маса жаби поступово віднов-
люється до вихідного рівня. Дегідратація тканин відбувається значно повільніше, ніж розвиток набряків. Це пояснюється тим, що вода, яка надійшла в тканину, легко зв'язується із тканинними колоїдами й набагато важче переходить знову у вільний стан.
23.27. Що є причиною розвитку внутрішньоклітинної гіпергідрії? Чим вона виявляє себе на рівні організму?
Основною причиною внутрішньоклітинної гіпергідрії є зменшення осмотичного тиску позаклітинної рідини, пов'язане з розвитком гіпоштріємії (див. запит. 23.30). У цих умовах вода за законами осмосу переходить з міжклітинного простору в клітини - з'являються ознаки генералізованого набряку клітин.
Клінічно виникають явища водного отруєння, серед яких на перший план виступає набряк мозку, що виявляється сильним головним болем, нудотою, блювотою, судомами.
23.28. Які механізми можуть лежати в основі розвитку набряку клітини при її ушкодженні? Яке патогенетичне значення даного явища?
Серед механізмів розвитку набряку клітини основне значення мають:
1) розпад внутрішньоклітинних структур, білків, унаслідок чого вивільняються зв'язані з ними катіони (в основному іони калію) і збільшується внутрішньоклітинний осмотичний і онкотичний тиск;
2) порушення проникності клітинної мембрани, у результаті чого іони натрію й хлору надходять у клітину й збільшують осмотичний тиск цитоплазми;
3) порушення функціонування натрій-калієвих насосів, що призводить до накопичення іонів натрію в клітині.
Набряк клітини посилює процеси її ушкодження. Це пов'язане з тим, що:
а) збільшується проникність клітинних мембран унаслідок їх осмотичного розтягування;
б) можливе явище електричного "пробою" плазматичної мембрани збудливих клітин;
в) відбувається механічний розрив мембран при їх розтягуванні.
23.29. Назвіть причини гіпернатріємп, захисно-компенсаторні реакції, що виникають при цьому, і патогенетичне значення даного порушення.
Залежно від причин розвитку розрізняють первинну і вторинну гіпернатріємію.
Первинна гіпернатріємія (абсолютне збільшення іонів натрію в організмі) може виникати або в результаті збільшення надходження натрію в організм (приймання великої кількості хлориду натрію, введення його гіпертонічного розчину), або внаслідок зменшення виведення натрію з організму (первинний і вторинний гіпераль-достеронізм, ниркова недостатність).
Вторинна (відносна) гіпернатріємія — це збільшення вмісту іонів натрію в крові й міжклітинній рідині внаслідок втрати води організмом. При цьому загальний вміст натрію в організмі може не змінюватися, а іноді навіть зменшується. Такий стан виникає при гіпервентиляції, проносах, підвищеному потовиділенні, нецукро-вому діабеті.
У результаті гіпернатріємії підвищується осмотичний тиск позаклітинної рідини, збуджуються центральні й периферичні осморецептори, збільшується надходження в кров антидіуретичного гормону. Останній посилює реабсорбцію води в нирках, у результаті чого збільшується об'єм позаклітинної рідини і зменшується її осмотичний тиск.
Основним наслідком гіпернатріємії є розвиток внутрішньоклітинного зневоднення (див. запит. 23.13, 23.14).
23.30. Назвіть причини гіпонатріємії, захисно-компенсаторні реакції, що виникають при цьому, і патогенетичне значення даного порушення.
Первинна (абсолютна) гіпонатріємія розвивається в результаті зменшення надходження натрію в організм (безсольова дієта, анорексія) або внаслідок збільшення виведення натрію з організму нирками (гіпофункція кори надниркових залоз, ниркова недостатність).
Причиною вторинної (відносної) гіпонатріємії є. надмірне надходження в організм води або її затримка - гіпонатріємія внаслідок розведення.
Зменшення концентрації іонів натрію в позаклітинній рідині викликає, з одного боку, посилення секреції альдостерону через ренін-ангіотензинний механізм, з другого - зменшення надходження в кров антидіуретичного гормону, оскільки зменшується імпульсація від осморецепторів. Наслідком цього є посилення реабсорбції іонів натрію й пригнічення реабсорбції води в нирках — осмотичний тиск позаклітинної рідини відновлюється.
Патогенетичне значення гіпонатріємії полягає в розвитку генералізованого набряку клітин (див. запит. 23.27).
23.31. Назвіть причини гіперкаліємії, захисно-компенсаторні реакції, що виникають при цьому, і патогенетичне значення даного порушення.
Гіперкаліємією називають збільшення вмісту іонів калію в плазмі крові понад 5,5 ммоль/л.
Причинами її розвитку можуть бути:
1) надмірне надходження калію в організм;
2) перехід іонів калію із внутрішньоклітинного в позаклітинний простір при масивному ушкодженні клітин, при збільшенні інтенсивності катаболічних процесів і ацидозі;
3) порушення виведення калію з організму (оліго- і анурія, недостатність функції кори надниркових залоз).
Збільшення концентрації іонів калію в крові безпосередньо активує клітини клу-бочкової зони кори надниркових залоз і викликає посилення секреції альдостерону. Останній збільшує секрецію іонів калію в ниркових нефронах і в такий спосіб відновлює їх концентрацію в крові.
Наслідками гіперкаліємії є:
1) порушення діяльності збудливих тканин (нервової і м'язової), у результаті чого розвиваються розлади функції ЦНС, серцево-судинної системи, скелетної мускулатури, гладких м'язів травного каналу;
2) розвиток негазового ацидозу (див. розд. 25).
23.32. Назвіть причини гіпокаліємії, захисно-компенсаторні реакції, що виникають при цьому, і патогенетичне значення даного порушення.
Гіпокшііємія — це зменшення концентрації іонів калію в плазмі крові нижче 3,5 ммоль/л.
Причинами її розвитку можуть бути:
1) недостатнє надходження калію в організм з їжею (тривале використання дієти, що не містить продуктів рослинного походження);
2) посилений перехід іонів калію з позаклітинного простору в клітини, що буває при посиленні анаболічних процесів і алкалозі;
3) втрата калію організмом (поліурія, гіперальдостеронізм, тривале використання сечогінних засобів).
При гіпокаліємії розвивається гіперполяризація мембран секреторних клітин і у зв'язку з цим зменшується секреція альдостерону корою надниркових залоз. Це викликає зменшення секреції іонів калію клітинами ниркового епітелію.
Патогенетичне значення гіпокаліємії полягає в тому, що:
а) збільшується поріг збудливості клітин і, як наслідок, з'являються загальна слабкість, метеоризм, гіпотонія скелетних м'язів, зменшується шкірна чутливість;
б) розвивається гіпокаліємічний алкалоз (див. розд. 25).
23.33. Назвіть причини й основні прояви порушень обміну магнію в організмі.
Магній є одним з найважливіших внутрішньоклітинних катіонів. Його концентрація в клітинах у 3—10 разів перевищує його вміст у позаклітинному середовищі. Він є активатором багатьох ферментативних процесів (реакцій фосфорного обміну, гліколізу; ряду етапів синтезу білків і жирових кислот; розпаду нуклеїнових кислот та ін.). У більшості реакцій, у яких бере участь АТФ, обов'язковим є утворення комплексів АТФ з іонами магнію.
Причинами збільшення вмісту магнію в організмі можуть бути: ураження нирок з порушенням їх видільної функції (уремія), отруєння сполуками магнію. При цьому розвивається гіпермагніємія (збільшення концентрації магнію в плазмі крові понад 1 ммоль/л), що виявляє себе пригніченням центральної нервової системи: розвивається депресія і сон (магнезіальний наркоз), відбувається гальмування функцій дихального центру.
До зменшення вмісту магнію в організмі можуть спричинятися: порушення всмоктування цього елемента в кишках (нестримна блювота, проноси, панкреатит); парентеральне введення великих кількостей рідини, що не містить магнію; порушення функції ендокринних залоз (тиреотоксикоз, гіперпаратиреоз, первинний гіпераль-
достеронізм); ураження печінки (хронічний алкоголізм, цироз). Зазначені причини можуть викликати розвиток гіпомагніємії (зменшення концентрації магнію в плазмі крові нижче 0,75 ммоль/л), яка виявляє себе:
а) тетанією (нападами судом), перебіг якої значно важчий, ніж при гіпокальціємії;
б) появою трофічних виразок на шкірі;
в) погіршенням засвоєння їжі, що викликає порушення процесів росту;
г) зниженням температури тіла;
ґ) поширеним кальцинозом тканин, який у першу чергу розвивається в кровоносних судинах, нирках, хрящовій тканині. При цьому вміст кальцію в організмі не міняється.
24. Порушений фосфорно-кальцієвого обміну
24.1. Які гормони здійснюють регуляцію фосфорно-кальцієвого обміну в організмі?
Паратирин (паратгормон), кальцитонін, гормонально активна форма вітаміну D.
24.2. Як відбувається утворення гормонально активної форми вітаміну D?
В організмі існує два джерела вітаміну D:
1) утворення вітаміну D3 (холекальциферолу) у шкірі з 7-дегідрохолестеролу під дією ультрафіолетового випромінювання;
2) надходження в організм вітаміну D2 (ергокальциферолу) у складі харчових продуктів.
У печінці під впливом ферменту 25-гідроксилази утворюється транспортна форма вітаміну — 25-оксивітамін D. Ця форма спочатку в складі жовчі виділяється у дванадцятипалу кишку, а потім разом з жовчними кислотами знову всмоктується в кров у тонкій кишці.
У нирках під дією ферменту la-гідроксилази утворюється гормонально-активна форма вітаміну D - 1,25-(ОН)2-вітамін D. Крім того, можливе утворення і неактивної форми - 24,25-(ОН)2-вітаміну D.
Яка з цих форм утворюється переважно, залежить від концентрації іонів кальцію в плазмі крові. При гіпокальціємії з'являється в основному 1,25-(ОН)2-вітамін D і гальмується утворення 24,25-(ОН)2-вітаміну D; при гіперкальціємії - навпаки.
24.3. Назвіть основні біологічні ефекти гормонально активної форми вітаміну D.
Основний біологічний ефект 1,25-(ОН)2-вітаміну D- збільшення всмоктування кальцію її фосфатів у тонкій кишці. Діючи на епітеліальні клітини слизової кишки, 1,25-(ОН)2-вітамін D, як і інші стероїдні гормони, проникає через плазматичну мембрану в цитоплазму ентероцитів, а потім у комплексі з внутрішньоклітинним білком-рецептором у їхнє ядро. У ядрі відбувається активація транскрипції генів, що кодують структуру функціонально важливих білків: кальбідину (білка, що зв'язує Са) і білків кальцієвих насосів, зокрема, Са-АТФ-ази.
Кальбідин вмонтовується в плазматичну мембрану апікальної частини ентероцитів і забезпечує полегшену дифузію іонів кальцію із просвіту кишок у цитоплазму клітин кишкового епітелію. Кальцієві насоси здійснюють активний транспорт іонів кальцію із цитоплазми ентероцитів через плазматичну мембрану базальної частини клітин в інтерстицій і кров.
Другою "мішенню" для 1,25-(ОН)2-вітаміну D є кісткова тканина, де цей гормон, впливаючи на остеобласти, гальмує синтез колагену, а діючи на остеокласти, стимулює резорбцію кістки.
У кінцевому підсумку дія 1,25-(ОН)2-вітаміну D виявляється збільшенням концентрації іонів кальцію в плазмі крові.
Неактивна форма вітаміну D - 24,25-(ОН)2-вітамін D гальмує секрецію парати-рину прищитоподібними залозами й посилює інактивацію стероїдів у печінці, у тому числі й вітаміну D.
24.4. Які фактори стимулюють і гальмують секрецію паратирину прищитоподібними залозами?
Секрецію napamupuny стимулює зменшення концентрації іонів кальцію в плазмі крові, а гальмує - збільшення вмісту цих іонів у плазмі й 24,25-(ОН)2-вітамін D.
24.5. Назвіть основні біологічні ефекти паратирину.
1. Дія на кісткову тканину - активація функції остеокластів.
Під впливом паратирину відбувається вивільнення остеокластами лізосомних ферментів, які розщеплюють органічну матрицю кісткової тканини. Крім того, остеокласти починають продукувати великі кількості цитрату, що сприяє вимиванню кальцію з кристалів оксіапатиту.
Результатом зазначених ефектів є збільшення концентрації іонів кальцію в крові, з одного боку, і демінералізація, резорбція кісткової тканини, з другого.
2. Пригнічення реабсорбції фосфату в нирках.
3. Активація перетворення в нирках вітаміну D у гормонально активну форму — 1,25-(ОН)2-вітамінВ.
24.6. Які фактори стимулюють і гальмують секрецію кальцитоніну?
Кальцитонін синтезується в парафолікулярних (С-клітинах) щитоподібної залози. Крім того, виявлено клітини, здатні секретувати цей гормон, у вилочковій і при-щитоподібних залозах.
Секрецію кальцитоніну стимулює збільшення концентрації іонів кальцію в плазмі крові й деякі гастроінтестинальні гормони (особливо гастрин). Гальмування відбувається при зменшенні вмісту іонів кальцію в крові.
24.7. Назвіть основні біологічні ефекти кальцитоніну.
1. Кальцитонін перешкоджає резорбції кісткової тканини, пригнічуючи діяльність остеокластів. Це виявляється зменшенням концентрації іонів кальцію в плазмі крові.
2. Гальмує секрецію гастрину. Фізіологічне значення цього ефекту ще не встановлено.
24.8. Що може бути причиною розвитку гіпокальціємічних станів?
Причиною зменшення концентрації іонів кальцію в плазмі крові — гіпокальціємії можуть бути такі фактори (рис. 85).
Рис. 85. Причини розвитку гіпокальціємічних станів
1. Зменшення надходження кальцію з тонкої кишки в кров:
а) зменшення вмісту кальцію в продуктах харчування;
б) порушення співвідношення кальцій/фосфор у харчових продуктах;
в) утворення в кишках нерозчинних кальцієвих сполук (наприклад, з фітовою, інозитфосфорною кислотами, що містяться в продуктах зі злаків);
г) порушення всмоктування кальцію при ураженнях тонкої кишки (ентерити); ґ) гіповітаміноз D.
2. Втрата іонізованого кальцію організмом:
а) із сечею при порушеннях процесів реабсорбції;
б) при вагітності — втрати, пов'язані з формуванням скелета плода.
3. Порушення мобілізації кальцію з кісткової тканини:
а) гіпопаратиреоз;
б) пухлини С-клітин щитоподібної залози, що продукують кальцитонін.
4. Мінералізація м 'яких тканин:
а) гіперфосфатемія;
б) алкалоз.
5. Перехід кальцію плазми крові з іонізованої форми в неіонізовану — у комплекси з білками й органічними кислотами:
а) отруєння щавлевою кислотою, переливання цитратної крові; *б) збільшення концентрації сироваткових білків; в) алкалоз.
24.9. Які захисно-компенсаторні реакції і власне патологічні зміни виникають при гіпокальціємії?
Захисно-компенсаторні реакції в цих умовах спрямовані на збільшення концентрації іонів кальцію в плазмі крові. До них належать:
1) збільшення секреції паратирину;
2) збільшення утворення в нирках 1,25-(ОН)2-вітаміну D;
3) зменшення секреції кальцитоніну. Завдяки цим реакціям збільшується всмоктування кальцію й фосфору в кишках, зростає їхній перехід з костей у кров.
Власне патологічними змінами є:
1) порушення костей скелета - розвиток рахіту у дітей і остеомаляції у дорослих;
2) синдром підвищеної нервово-м'язової збудливості — тетанія.
24.10. Що таке тетанія? Коли вона виникає?
Tema/іія — це синдром підвищеної нервово-м'язової збудливості, що виявляється
розвитком тонічних судом. Виникає при гіпокальціємії.
При зменшенні концентрації іонів кальцію в крові критичний потенціал деполяризації збудливих клітин наближається до рівня потенціалу спокою, а отже, зменшується поріг збудливості. Тому ті подразники, які в нормі є підпорогови-ми, при гіпокальціємії викликають розвиток тонічних скорочень скелетних м'язів.
Особливо небезпечне скорочення дихальних м'язів і м'язів гортані, оскільки можлива периферична зупинка дихання.
24.11. Що таке рахіт? Які виділяють патогенетичні варіанти цього захворювання ?
Рахіт — це хвороба дитячого віку, основний прояв якої — порушення формування кісткового скелета внаслідок порушення утворення оксіа-патиту.
Оскільки основними складовими частинами оксіапатиту є кальцій і фосфат, то порушення утворення цього мінералу може бути пов'язане
Рис. 86. Причини кальципенічного рахіту. УФВ —ультрафіолетове випромінювання; ТК — травний канал
як з первинною недостатністю кальцію, так і з недостатністю фосфату. Тому виділяють два патогенетичних варіанти рахіту: кальципенічний і фосфопенічний.
24.12. Які чинники можуть бути причиною розвитку кальципенічного рахіту?
Основною причиною виникнення даного різновиду рахіту є порушення регуляції фосфорно-кальцієвого обміну вітаміном D.
До цього можуть спричинятися такі фактори (рис. 86).
1. Порушення утворення вітаміну D у шкірі:
а) недостатня інтенсивність ультрафіолетового випромінювання (осінньо-зимовий період, північні широти; забруднення повітря в індустріальних містах);
б) недостатнє перебування дітей на повітрі або перебування на повітрі в одязі, що затримує ультрафіолетове випромінювання;
в) пігментація шкіри.
2. Недостатнє надходження вітаміну D з їжею.
3. Порушення перетворення вітаміну D у печінці в його транспортну форму:
а) набуті порушення діяльності печінки;
б) спадково обумовлений дефіцит ферменту 25-гідроксилази.
4. Порушення зворотного надходження транспортної форми вітаміну D з тонкої кишки в кров.
5. Порушення утворення гормонально активної форми вітаміну D — 1,25-(ОН)2-ві-таміну D у нирках:
а) хронічна ниркова недостатність;
б) спадково обумовлений дефіцит ферменту Іа-гідроксилази.
6. Нечутливість клітин-мішеней до дії гормонально активної форми вітаміну D -генетично обумовлений дефіцит рецепторів до 1,25-(ОН) - вітаміну D.
24.13, Поясніть механізми розвитку основних проявів кальципенічного рахіту.
Порушення регуляції фосфорно-кальцієвого обміну вітаміном D первинно спричиняються до розладів усмоктування кальцію і фосфату у тонкій кишці. У результаті розвивається гіпокальціємія (рис. 87).
Зменшення концентрації іонів кальцію у плазмі крові стимулює секрецію пара-тирину. Останній, активуючи функцію остеобластів, зумовлює перехід іонів кальцію і фосфату із кісткової тканини в кров. Ця реакція, будучи по своїй суті захисно-компенсаторною, поновлює вміст кальцію в крові, однак організм розплачується за це демінералізацією кісток і порушенням окостеніння хрящової тканини. Кістки деформуються, і виникають характерні клінічні ознаки рахіту.
Гіпокальціємія, крім того, є причиною цілого ряду порушень центральної нервової системи: дитина стає лякливою, капризує; порушується сон, з'являється пітливість. Розвивається спазмофілія - напади рахітичної тетанії.
Рис. 87. Патогенез кальципенічного рахіту
і
24.14. Які фактори можуть бути причиною розвитку фосфопенічното рахіту?
Основна причина фосфопенічного рахіту - порушення реабсорбції фосфату в нирках. Найчастіше це спадково обумовлені захворювання:
1) гіпофосфатемічний вітамін D-резистентний рахіт - фосфат-діабет (хвороба зчеплена з Х-хромосомою, однак успадковується домінантно);
2) аутосомно-домінантний гіпофосфатемічний рахіт;
3) синдром Фанконі - складні порушення функції проксимальних звивистих каналь-ців ниркових нефронів.
Перелічені спадкові хвороби характеризуються порушенням реабсорбції фосфату в канальцях нирок.
24.15. Поясніть патогенез основних проявів фосфопенічного рахіту.
Генетично обумовлений дефект ферментів і транспортних білків призводить до порушення реабсорбції фосфату в ниркових канальцях, унаслідок чого розвивається гіпофосфатемія. Добуток концентрації іонів кальцію на концентрацію іонів фосфату в плазмі крові зменшується. Це має два наслідки: 1) вихід кальцію й фосфату з кісток у кров - демінералізація і 2) порушення утворення оксіапатиту в хрящовій тканині - порушення окостеніння хрящів. Усе це, в кінцевому підсумку, призводить до деформації кісток скелета — розвиваються клінічні ознаки рахіту.
24.16. Що може бути причиною розвитку п'перкальціємічних станів?
Збільшення концентрації іонів кальцію в плазмі крові — гіперкальціємія може бути обумовлена такими групами факторів (рис. 88).
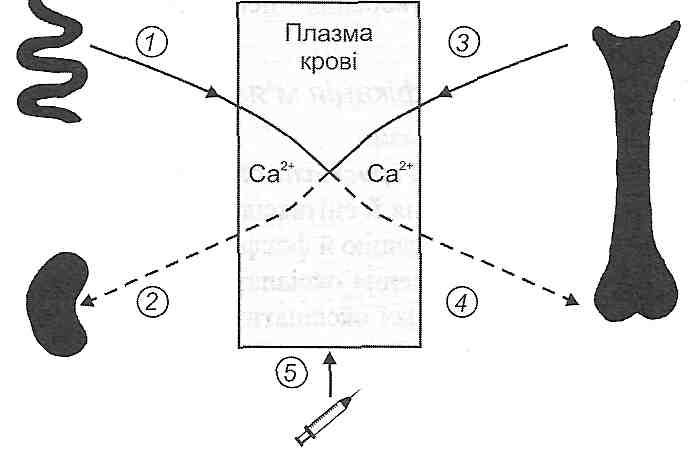
Рис. 88. Причини розвитку гіперкальціємічних станів
I. Посилене надходження кальцію з тонкої кишки в кров:
а) надмірний вміст кальцію в продуктах харчування;
б) посилене всмоктування кальцію в кишках, що буває найчастіше при гіпервітамінозі D.
II. Зменшення виведення кальцію з організму:
а) набуті порушення — хронічна ниркова недостатність;
б) спадкові порушення - сімейна гіпокальціурична гіперкальціємія.
III. Посилене надходження кальцію в кров з кісткової тканини:
а) гіперпаратиреоз;
б) злоякісні пухлини з метастазами в кісткову тканину;
в) множинні переломи кісток.
IV. Порушення відкладення кальцію в кісткову тканину, що спостерігається при гі-пофосфатемії.
24.17. Які захисно-компенсаторні реакції і власне патологічні зміни виникають при гіперкальціємії?
Захисно-компенсаторні реакції в цих умовах спрямовані на зменшення вмісту іонів кальцію в крові. До них відносять:
1) зменшення секреції паратирину;
2) зменшення утворення в нирках 1,25-(ОН)2-вітаміну D і збільшення утворення 24,25-(ОН)2-вітаміну D;
3) збільшення секреції кальцитоніну.
Власне патологічні зміни:
1) кальцієве ушкодження клітин (див. розд. 11);
2) кальцифікація м'яких тканин — звапніння;
3) зменшення збудливості збудливих тканин;
4) утворення кальцієвих каменів у нирках;
5) посилення шлункової секреції з утворенням пептичних виразок у шлунку;
6) розвиток артеріальної гіпертензії.
24.18. Як відбувається кальцифікація м'яких тканин?
У цьому процесі виділяють дві фази.
Перша фаза — ініціація утворення кристалів оксіапатиту. В її основі лежать два фізико-хімічних механізми: осадження й епітаксія. Осадження відбувається при локальному збільшенні концентрації кальцію й фосфату вище певного критичного значення. Епітаксія - це механізм утворення оксіапатиту на органічній матриці, кристалічна структура якої подібна до такої оксіапатиту. Функцію органічної матриці у тканинах можуть виконувати колаген і еластин.
Друга фаза —ріст кристалів оксіапатиту. Цей процес підпорядкований фізико-хімічним закономірностям. На його інтенсивність впливає рН середовища. У кислому середовищі ріст кристалів уповільнюється, у лужному - прискорюється.
24.19. Які причини можуть викликати розвиток гіпо- і гіперфосфатемії? Які зміни в організмі виникають при цьому?
Зменшення концентрації фосфатів у крові — гіпофосфшпемія - може бути обумовлене двома групами причин:
а) зменшенням надходження неорганічного фосфору в організм (голодування, синдром мальабсорбції, гіповітаміноз D);
б) посиленим виведенням фосфатів нирками (гіперпаратиреоз, фосфат-діабет, синдром Фанконі).
Зменшення неорганічного фосфору в організмі призводить до численних порушень, в основі яких— розлади окисного фосфорування в клітинах, зменшення утворення 2,3-дифосфогліцерату в еритроцитах, у результаті чого відбувається зсув кривої дисоціації оксигемоглобіну вліво й розвивається гіпоксія. При цьому виникають порушення в системі травлення (диспепсичні явища, анорексія), системі крові (гемолітична анемія, лейко- і тромбоцитопенія), нервовій системі (парестезії, атаксія, сплутаність свідомості, у важких випадках — кома), опорно-руховому апараті (остеомаляція, міопатія, фосфопенічний рахіт у дітей).
Причинами збільшення вмісту фосфатів у плазмі крові — гіперфосфатемії— можуть бути такі фактори:
а) посилене надходження фосфатів із клітин і тканин організму в кров (важке фізичне навантаження, гемоліз еритроцитів, лейкози, метастази й первинні злоякісні пухлини кісток);
б) порушення виведення фосфатів нирками (гіпопаратиреоз, недостатність нирок).
При гіперфосфатемії створюються сприятливі фізико-хімічні умови для утворення оксіапатиту в м'яких тканинах - розвивається їх кальцифікація.
25. Порушення кислотно-основного стану
25.1. Які буферні системи беруть участь у підтриманні кислотно-основного гомеостазу?
1. Гідрокарбонашна буферна система. її компонентами є вугільна кислота Н2СО. і гідрокарбонат натрію NaHC03 у співвідношенні 1:20. Підтримує сталість рН у плазмі крові й міжклітинній рідині.
2. Фосфатна буферна система. Представлена кислою (NaH2P04) і основною (Na,HP04) натрієвими солями фосфорної кислоти у співвідношенні 1:4. Бере участь у регуляції кислотно-основного стану в нирках та інших тканинах.
3. Гемоглобіновий буфер. Він представлений відновленим гемоглобіном і калієвою сіллю окисненого гемоглобіну. Діє в еритроцитах, попереджаючи зміни рН унаслідок постійного надходження із тканин вуглекислого газу.
4. Білковий буфер. Забезпечує підтримку сталості внутрішньоклітинного рН.
25.2. Яку участь бере система зовнішнього дихання в регуляції кислотно-основного стану?
Система зовнішнього дихання забезпечує сталість напруги вуглекислого газу в артеріальній крові.
При збільшенні рСО,, (гіперкапнія) стимулюється дихальний центр, розвивається гіпервентиляція й надлишок С02 виводиться з організму з видихуваним повітрям.
При зменшенні рС02 (гіпокапнія), навпаки, відбувається пригнічення дихального центру, розвивається гіповентиляція й CO, затримується в організмі.
25.3. Які процеси в нирках беруть участь у регуляції кислотно-основного стану?
1. Ацидогенез — утворення і секреція у просвіт ниркових канальців іонів водню. Відбувається у проксимальних і дистальних звивистих канальцях нефронів (рис. 89).
2. Амоніогепез - утворення і секреція в просвіт канальців аміаку. Цей процес попереджає зменшення рН сечі нижче 4,5 і пов'язане з цим ушкодження клітин ка-нальцевого епітелію (рис. 90).
3. Реабсорбція гідрокарбонату. Здійснюється в проксимальних звивистих канальцях нефронів при участі процесів ацидогснезу (рис. 91).
Рис. 89. Схема ацидогенезу в нирках
Рис. 90. Схема амоніогенезу в нирках

Рис. 91. Механізми реабсорбції гідрокарбонату в нирках
25.4. У чому полягає основна функціональна відмінність процесів ацидогенезу, що відбуваються в проксимальних і дистальних звивистих канальцях ниркових нефронів?
У проксимальних звивистих канальцях ацидогенез забезпечує реабсорбцію гідрокарбонату. Тут виявляється така залежність: що вищий рС02 артеріальної крові^ то вища інтенсивність ацидогенезу, то більша кількість гідрокарбонату може бути реабсорбована, і навпаки (рис. 92).
Рис. 92. Зв'язок міжрС02, ацидогенезом і реабсорбцією гідрокарбонату в нирках
Ацидогенез у дистальних звивистих канальцях забезпечує підкислення сечі, тобто її ацидифікацію. При цьому відбувається відтитровування гідрокарбонату плазми крові, завдяки чому зберігається її лужний резерв.
25.5. Назвіть основні форми порушень кислотно-основного стану.
Існує два типи порушень: ацидоз і алкалоз.
Ацидоз— це зміщення кислотно-основної рівноваги в кислий бік, алкалоз- у лужний.
Ацидози й алкалози можуть бути компенсованими, коли завдяки буферним системам і фізіологічним механізмам компенсації рН крові не виходить за межі фізіологічних коливань, і декомпенсованими, коли рН крові виходить за межі норми.
Залежно від механізмів розвитку ацидози й алкалози можуть бути газовими й негазовими. Газовими називають порушення кислотно-основного стану, обумовлені первинною зміною напруги вуглекислого газу в артеріальній крові. Не-газові порушення пов'язані з первинними змінами концентрації гідрокарбонату в крові.
25.6. Які показники використовують для характеристики порушень кислотно-основного стану?
Основні показники:
1) рН артеріальної крові (у нормі 7,37-7,43);
2) рС02 артеріальної крові (35-45 мм рт. ст.);
3) SB - стандартний гідрокарбонат (22-26 ммоль/л).
Додаткові показники:
1) ВВ - буферні основи (40-48 ммоль/л);
2) BE - зсув буферних основ (-2,5 - +2,5 ммоль/л).
25.7. Що таке газовий ацидоз?
Газовим називають ацидоз, основу якого становить збільшення напруги вуглекислого газу в крові (гіперкапнія).
25.8. Назвіть причини газового ацидозу.
1. Порушення виведення С02 з організму при всіх видах гіповентиляції (недостатність зовнішнього дихання) - ендогенний газовий ацидоз.
2. Вдихання повітря з високим вмістом CO, (тривале перебування в закритих приміщеннях з поганою вентиляцією, шахтах, підводних човнах) - екзогенний газовий ацидоз.
25.9. Які захисно-компенсаторні реакції розвиваються при газовому ацидозі?
Захисно-компенсаторні реакції при газовому ацидозі спрямовані на збільшення концентрації гідрокарбонату в плазмі крові і забезпечуються нирками. Тут посилюється ацидогенез у дистальних звивистих канальцях, що веде до утворення додаткової кількості гідрокарбонату. Посилення ацидогенезу в проксимальних звивистих канальцях забезпечує повну його реабсорбцію.
25.10. Які зміни показників кислотно-основного стану характерні для газового ацидозу?

25.11. Як здійснюють корекцію газового ацидозу?
Необхідно забезпечити виведення С02 з організму, для чого усувають причини недостатності зовнішнього дихання.
25.12. Що таке негазовий ацидоз?
Негазовий - це такий ацидоз, основу якого становить зменшення концентрації гідрокарбонату в плазмі крові.
25.13. Які види негазового ацидозу виділяють з урахуванням причин його виникнення?
1. Метаболічний ацидоз. 2. Екзогенний негазовий ацидоз. 3. Видільний ацидоз.
25.14. Що таке метаболічний негазовий ацидоз?
Метаболічний — це ацидоз, основу якого становить накопичення в організмі ендогенних кислот-метаболітів. Він розвивається при цукровому діабеті І типу (накопичуються кетонові тіла і молочна кислота), гіпоксії (молочна кислота), голодуванні (кетонові тіла), нирковій недостатності (фосфати, сульфати, сечова кислота), подагрі (сечова кислота).
25.15. Що таке екзогенний негазовий ацидоз?
Екзогенний негазовий - це ацидоз, що виникає внаслідок надходження або введення в організм екзогенних нелетких кислот. Може розвиватися при тривалому споживанні кислих продуктів, прийманні деяких ліків, зокрема, хлориду амонію; при отруєннях.
25.16. Що таке видільний ацидоз?
Видільний - це ацидоз, що виникає внаслідок втрати гідрокарбонату організмом при порушенні функціонування нирок і травної системи. Його різновидами є:
1) проксимальний нирковий канальцевий ацидоз. Розвивається в результаті первинних порушень реабсорбції гідрокарбонату в проксимальних звивистих канальцях;
2) дистальний нирковий канальцевий ацидоз. Розвивається внаслідок первинних порушень ацидогенезу в дистальних звивистих канальцях;
3) видільні ацидози, пов'язані з порушеннями діяльності травної системи:
а) втрата гідрокарбонату з підшлунковим соком при діареї;
б) гіперсалівація.
25.17. Які захисно-компенсаторні реакції розвиваються при негазовому ацидозі?
I. Дихальні механізми компенсації спрямовані на зменшення рС00. При збільшенні концентрації іонів водню збуджується дихальний центр, розвивається гіпервен-тиляція і збільшується виведення CO, з організму.
II. Ниркові механізми компенсації спрямовані на збереження гідрокарбонату в організмі. Зменшення рН крові викликає активацію ацидогенезу в дистальних звивистих канальцях. Завдяки цьому гідрокарбонат сечі відтитровується і зберігається в організмі, а рН сечі зменшується, тобто відбувається її ацидифікація.
25.18. Які зміни показників кислотно-основного стану характерні для негазового ацидозу?
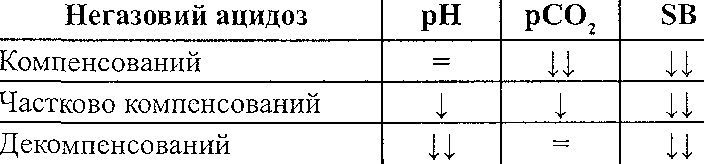
25.19. Що таке негазові ацидози зі збільшеною й нормальною аніонною різницею?
Аніонною різницею називають різницю концентрації іонів натрію і суми концентрацій аніонів гідрокарбонату і хлору в плазмі крові. У нормі вона становить 8-16 мекв/л (рис. 93).
Якщо зменшення концентрації гідрокарбонату при ацидозі покривається збільшенням концентрації інших аніонів, крім хлору, то аніонна різниця зростає. До таких ацидозів відносять усі метаболічні і екзогенні ацидози, крім тих, що обумовлені введенням хлоридів або попередників НС1.
При негазових ацидозах з нормальною аніонною різницею зменшення концентрації гідрокарбонату в плазмі крові покривається збільшенням концентрації аніонів хлору, тобто розвивається гіперхлоремія. До таких ацидозів відносять усі видільні ацидози й ацидози, що виникають при введенні хлоридів або попередників НС1.
Рис. 93. Вміст катіонів і аніонів у плазмі крові
25.20. Як здійснюють корекцію негазового ацидозу?
Корекція цього порушення має бути спрямована на нормалізацію концентрації гідрокарбонату в плазмі крові. Для цього внутрішньовенно вводять розчини гідрокарбонату натрію NaHCOr
25.21. Які власне патологічні зміни виникають в організмі при декомпенсованих ацидозах?
1. Ацидотичне ушкодження клітин (див. розд. 11).
2. Ацидотична кома (див. розд. 12).
3. Пригнічення скоротливої функції серця. Розвивається внаслідок блокади іонами водню кальцієвих каналів плазматичної мембрани й тропонінових центрів зв'язування кальцію в кардіоміоцитах.
25.22. Що таке газовий алкалоз?
Газовим називають алкалоз, основу якого становить зменшення напруги вуглекислого газу в крові (гіпокапнія).
25.23. Назвіть причини газового алкалозу.
Причиною газового алкалозу є гіпервентиляція легень. Вона буває при:
а) безпосередньому збудженні дихального центру (пухлини головного мозку, енцефаліти);
б) рефлекторному його збудженні при подразненні периферичних хеморецепторів (гірська хвороба, гарячка);
в) неправильно здійснюваному керованому штучному диханні.
25.24. Які захисно-компенсаторні реакції виникають при газовому алкалозі?
Компенсаторні реакції при газовому алкалозі спрямовані на зменшення концентрації гідрокарбонату в плазмі крові й забезпечуються нирками.
При зменшенні рС02, крові зменшується інтенсивність ацидогенезу в проксимальних звивистих канальцях нефронів, що веде до зменшення реабсорбції гідрокарбонату. Наслідком цього є збільшення рН сечі, з одного боку, і зменшення концентрації гідрокарбонату в плазмі крові, з другого.
25.25. Як здійснюють корекцію газового алкалозу?
Корекція цього порушення спрямована на збільшення напруги вуглекислого газу в крові, тобто на боротьбу з гіпокапнією. З цією метою для дихання використовують газові суміші, багаті С09, наприклад карбоген, що складається із 02 (95 %) і С02
(5%). " V *' •"'" :"
25.26. Які зміни показників кислотно- основного стану характерні для газового алкалозу?
25.27. Що таке негазовий алкалоз?
Пегозовим називають алкалоз, основу якого становить збільшення концентрації гідрокарбонату в плазмі крові.
25.28. Які види негазового алкалозу виділяють з урахуванням причин його виникнення?
I. Видільний негазовий алкалоз:
а) гіпохлоремічний;
б) гіпокаліємічний.
II. Екзогенний негазовий алкалоз (гіпернатріємічний).
25.29. Що таке гіпохлоремічний алкалоз?
Гіпохлоремічним називають алкалоз, пов'язаний із втратою аніонів хлору, у результаті чого розвивається гіпохлоремія. При цьому втрата аніонів хлору поповнюється збільшенням концентрації аніонів гідрокарбонату.
Найчастішою причиною розвитку такого алкалозу є нестримна блювота.
25.30. Що таке гіпокаліємічний алкалоз?
Гіпокаліємічним називають алкалоз, який розвивається внаслідок втрати організмом іонів калію.
Найчастішою причиною його виникнення є гіперальдостероиізм, при якому збільшується секреція іонів калію в сечу й розвивається гіпокаліємія.
25.31. Що таке екзогенний (гіпернатріємічний) алкалоз?
Екзогенний- це алкалоз, пов'язаний з надходженням в організм екзогенних основ. Найчастіше буває при введенні великої кількості гідрокарбонату натрію NaHC03, наприклад, при неправильній корекції негазового ацидозу.
25.32. Які зміни показників кислотно- основного стану характерні для негазового алкалозу?
25.33. Які захисно-компенсаторні реакції розвиваються при негазовому алкалозі?
Компенсаторні реакції в цих умовах спрямовані на збільшення напруги вуглекислого газу в крові й забезпечуються системою зовнішнього дихання. При збільшенні рН зменшується збудливість і пригнічується дихальний центр, що призводить до гі-повентиляції легень. Як наслідок, зменшується виведення С02, і рС02 крові зростає.
25.34. Як здійснюють корекцію негазового алкалозу?
При гіпохяоремічному алкалозі для відновлення концентрації аніонів хлору в плазмі крові вводять розчини хлориду амонію NH4C1.
При гіпокаліємічному алкалозі введенням в організм хлориду калію КСІ відновлюють концентрацію іонів калію.
В умовах екзогенного (гіпернатріємічного) алкалозу вводять діуретики - інгібітори карбоангідрази з метою пригнічення ацидогенезу в проксимальних звивистих канальцях нефронів і зменшення реабсорбції гідрокарбонату.
25.35. Які власне патологічні зміни виникають в організмі при декомпенсованих алкалозах?
1. Клітинний алкалоз. Призводить до:
а) порушення конформації й властивостей структурних, функціональних білків і білків-ферментів;
б) збільшення гідрофільності клітинних колоїдів, унаслідок чого розвивається набряк клітини;
в) порушення окисного фосфорування в мітохондріях.
2. Гіпокальціємія (див. розд. 24).
25.36. Який зв'язок існує між порушеннями кислотно-основного стану і порушеннями електролітного обміну?
Первинні порушення кислотно-основного стану закономірно ведуть до вторинних порушень обміну електролітів, і навпаки, первинні порушення обміну електролітів викликають розвиток вторинних порушень кислотно-основного стану.
Найтісніший зв'язок існує між порушеннями кислотно-основного стану й обміном іонів калію.
При декомпенсованих ацидозах розвивається гіперкаліємія, а при гіперкаліємії— ацидоз. Це багато в чому пояснюється змінами процесів секреції іонів калію й водню в дистальних звивистих канальцях ниркових нефронів.
При ацидозі посилення ацидогенезу в нирках супроводжується зменшенням секреції калію, у результаті чого його концентрація в плазмі крові зростає. Якщо ж первинно розвивається гіперкаліємія, то вона викликає посилення процесів секреції іонів калію в ниркові канальці й пригнічення ацидогенезу. Наслідком цього є збільшення концентрації іонів водню в плазмі крові, тобто розвивається ацидоз.
В умовах декомпенсованого алкалозу закономірно виникає гіпокаліємія, а в результаті первинної гіпокалїємії — вторинно алкалоз. При алкалозі в результаті пригнічення процесів ацидогенезу в нирках посилюється секреція іонів калію — розвивається гіпокаліємія. При первинній гіпокаліємії внаслідок зменшення секреції калію в ниркові канальці значно зростає інтенсивність ацидогенезу. У результаті збільшення секреції іонів водню в сечу їхня концентрація в плазмі крові зменшується, тобто розвивається алкалоз.
Зміни вмісту іонів натрію в плазмі крові також можуть супроводжуватися порушеннями кислотно-основного стану, якщо при цьому змінюється концентрація іонів гідрокарбонату (а не хлору). Так, при гіпернатріємії зі збільшенням концентрації гідрокарбонату розвивається алкалоз, і навпаки, гіпонатріємія, пов'язана зі зменшенням вмісту гідрокарбонатних аніонів у плазмі крові, є причиною ацидозу.
Збільшення концентрації іонів хлору в плазмі крові (гіперхлоремія) при нормальному рівні іонів натрію супроводжується розвитком ацидозу й, відповідно, зменшення вмісту іонів хлору (гіпохлоремія), якщо воно не пов'язане з гіпонатріємією, є причиною алкалозу.
Частина II. ПАТОЛОГІЧНА ФІЗІОЛОГІЯ ОРГАНІВ І СИСТЕМ
26. Патологічна фізіологія системи крові
26.1. Які функціональні, морфологічні й регуляторні особливості системи крові впливають на характер, етіологію й патогенез патологічних процесів у цій системі?
1. Генетична детермінованість розмноження, диференціювання, обміну речовин і структури клітин усіх кров'яних паростків, з якою можуть бути пов'язані геномні порушення або зміни генетичної регуляції, що часто виступають причиною захворювань у системі крові (лейкоз, спадкова нейтропенія, гемолітична анемія та ін.).
2. Висока мітотична активність гемопоетичної тканини, що обумовлює її підвищену вразливість при дії мутагенних факторів (іонізуюча радіація, віруси, хімічні мутагени), цитостатиків, при дефіциті пластичних матеріалів (заліза, ціанокоба-ламіну й фолієвої кислоти, білків). Це призводить до розвитку лейкозу, дефіцитної анемії, лейко- і тромбоцитопенії, виникнення мутантних клонів лімфоцитів, що продукують антитіла проти власних клітин крові (аутоімунні анемії, лейко- і тромбоцитопенія).
3. Регуляція утворення клітин крові за допомогою еритро-, лейко- і тромбоцитопо-етинів, при дефіциті яких виникають патологічні зміни крові.
4. Патогенні агенти (бактерії, віруси, ендо- і екзогенні хімічні речовини), що потрапляють у кров, можуть викликати лізис кров 'яних клітин (наприклад, еритроцитів при гемолітичній анемії) і порушення їхньої антигенної структури. Це, у свою чергу, може зумовлювати вторинний цитоліз унаслідок аутоімунних реакцій.
5. Ушкодження судин викликає крововтрату, при якій зменшується загальний об'єм крові й порушуються її функції.
26.2. Які ознаки свідчать про порушення в системі крові?
Такими ознаками є зміни:
1) загального об'єму крові;
2) кількості, структури й функції клітин крові - еритроцитів, лейкоцитів, тромбоцитів - унаслідок порушень кровотворення або руйнування формених елементів крові;
3) гемостазу;
4) біохімічних і фізико-хімічних властивостей крові.
Ці зміни виникають:
а) при дії патогенних факторів безпосередньо на систему крові;
б) у результаті порушень її нейрогуморальної регуляції;
в) при ураженні інших органів і систем. У цьому випадку кров реагує вторинно.
Висока частота вторинних змін у системі крові обумовлена її функціональними особливостями. Так, збільшення утворення еритроцитів, що забезпечують дихальну функцію крові (транспорт 02 і С02), є компенсаторною реакцією системи крові на гіпоксію. При інфекційних хворобах часто посилюється лейкопоез, у крові збільшується кількість лейкоцитів, що беруть участь у захисних, у тому числі імунних, реакціях організму. У випадку ушкодження судин насамперед активується система зсідання крові і система фібринолізу, що забезпечує зупинку кровотечі (гемостаз).
Патологія системи крові може виявляти себе як самостійними захворюваннями (наприклад, гемофілія, анемія Аддісона-Бірмера, гострий мієлобластний лейкоз), так і гематологічними синдромами, що супроводжують хвороби інших органів і систем (наприклад, при деяких уроджених вадах серця, нейтрофільний лейкоцитоз при захворюваннях, спричинених гнійною інфекцією).
26.3. Назвіть види порушень загального об'єму крові. Наведіть приклади.
Порушення об'єму крові виявляють себе гіповолемієїо або гіперволемією — зменшенням або збільшенням об'єму крові, якщо порівнювати з нормою (нормоволемією).
Гіпо- і гіперволемію поділяють на просту (зберігається нормальне співвідношення плазми і клітин крові), поліцитемічну (переважають клітини крові) і олігоци-темічну (переважає плазма).
Крім того, до порушень об'єму крові відносять зміни об'ємного співвідношення між клітинними елементами і плазмою при нормальному загальному об'ємі крові — оліго- і поліцитемічну нормоволемію (гемодилюція і гемоконцентрація). Показником об'ємного співвідношення є гематокрит, що визначає вміст клітинних елементів (переважно еритроцитів) у загальному об'ємі крові (у нормі 0,36-0,48, або 36-48 %).
Гіповолемія проста — зменшення об'єму крові без зміни гематокриту. Виникає відразу після гострої крововтрати і зберігається доти, доки рідина не перейде із тканини в кров.
Гіповолемія олігоцитемічна — зменшення об'єму крові з переважним зменшенням у ній клітин - еритроцитів. Спостерігається при гострій крововтраті в тих випадках, коли надходження крові й тканинної рідини в кровоносне русло не компенсує об'єм і особливо склад крові.
Гіповолемія поліцитемічна—зменшення об'єму крові внаслідок зменшення об'єму плазми при відносному збільшенні вмісту еритроцитів. Розвивається при зневодненні організму (пронос, блювота, посилене потовиділення, гіпервентиляція), шоку (вихід рідини в тканини внаслідок підвищення проникності стінок судин).
Гіперволемія проста— збільшення об'єму крові при збереженні нормального співвідношення між еритроцитами і плазмою. Виникає відразу ж після переливання великої кількості крові. Однак незабаром рідина виходить з кровоносного русла, а еритроцити залишаються, що веде до згущення крові. Проста гіперволемія при посиленій фізичній роботі обумовлена надходженням у загальний кровообіг крові з депо.
Гіперволемія олігоцитемічна — збільшення об'єму крові за рахунок плазми. Розвивається при затримці води в організмі у зв'язку із захворюваннями нирок, при вве-
денні кровозамінників. її можна моделювати в експерименті шляхом внутрішньовенного введення тваринам ізотонічного розчину натрію хлориду.
Гіперволемія поліцитемічна — збільшення об'єму крові за рахунок наростання кількості еритроцитів. Спостерігається при зниженні атмосферного тиску, а також при різних захворюваннях, пов'язаних з кисневим голодуванням (вади серця, емфізема), її розглядають як компенсаторне явище.
Однак при еритремії поліцитемічна гіперволемія є наслідком пухлинного розростання клітин еритроцитарного ряду кісткового мозку.
Олігоцитемічна нормоволемія виникає при анемії внаслідок крововтрати (об'єм крові нормалізувався за рахунок тканинної рідини, а кількість еритроцитів ще не відновилася), при гемолізі еритроцитів, порушеннях гемопоезу.
Поліцитемічна нормоволемія спостерігається при переливанні невеликих кількостей еритроцитарної маси.
26.4. Чим обумовлене патогенетичне значення гіпо- і гіперволемії?
Гіповолемія супроводжується порушенням дихальної й транспортної функцій еритроцитів; трофічної, екскреторної, захисної, регуляторної функцій крові. Це так чи інакше відбивається на сталості внутрішнього середовища організму (гомеостазі).
Гіперволемія обумовлює збільшення навантаження на серце. У випадку одночасного збільшення гематокриту (поліцитемічна гіперволемія) збільшується в'язкість крові, що може бути причиною порушень мікроциркуляції й провокувати утворення тромбів.
26.5. Що таке крововтрата? Які причини викликають крововтрату? Від чого залежать її перебіг і завершення?
Крововтрата - це патологічний процес, що виникає внаслідок кровотечі і характеризується складним комплексом патологічних порушень і компенсаторних реакцій, спрямованих проти зменшення об'єму циркулюючої крові й гіпоксії, обумовленої зниженням дихальної функції крові.
До етіологічних факторів, що викликають кровотечу, відносять:
1) порушення цілісності судин при пораненні або ураженні патологічним процесом (атеросклероз, пухлина, туберкульоз);
2) підвищення проникності судинної стінки (гостра променева хвороба);
3) зниження зсідання крові (геморагічний діатез).
Перебіг і результат крововтрати залежать від особливостей самої кровотечі (швидкості, величини, виду ушкодженої судини, механізму ушкодження); швидкості включення й вираженості компенсаторних реакцій організму; статі, віку, станів, що передують і супроводжують крововтрату (охолодження, травма, захворювання серця, глибокий наркоз). Серйозну небезпеку для життя людини являє втрата 50 % об'єму циркулюючої крові, смертельною є втрата крові понад 60 %.
26.6. Які стадії умовно виділяють у патогенезі гострої крововтрати?
І. Початкова стадія. Характеризується зменшенням об'єму циркулюючої крові -простою гіповолемією, зниженням артеріального тиску, гіпоксією переважно циркуляторного типу.
II. Компенсаторна стадія. Обумовлена здійсненням комплексу захисно-компенсаторних реакцій, спрямованих на ліквідацію наслідків втрати крові.
III. Термінальна стадія. Характеризується наростанням патологічних змін в організмі аж до настання смерті. Розвивається при недостатності компенсаторних реакцій, а також при інтенсивній і швидкій крововтраті, на тлі дії несприятливих факторів (охолодження, велика травма, серцево-судинні захворювання) і при відсутності лікувальних заходів.
26.7. Що є головною ланкою патогенезу гострої крововтрати?
У патогенезі гострої крововтрати центральне місце посідають порушення загальної гемодинаміки — зменшення об 'єму циркулюючої крові (гіповолемія) і обумовлене цим падіння артеріального тиску (гіпотензія) (рис. 94).
Зазначені порушення, з одного боку, є причиною розвитку власне патологічних змін в організмі (анемії, гіпоксії, ацидозу, інтоксикації), з другого- вмикають складний комплекс рефлекторних і гуморальних захисно-компенсаторних реакцій, спрямованих на відновлення й збереження гомеостазу.
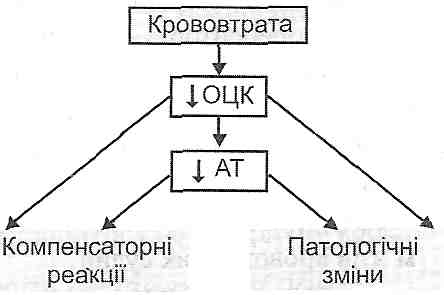
Рис. 94. Головна ланка патогенезу гострої крововтрати
26.8. Які захисно-компенсаторні реакції закономірно розвиваються при крововтраті?
Залежно від строків виникнення захисні реакції організму поділяють на термінові і нетермінові.
Залежно від спрямованості виділяють такі групи механізмів компенсації: Іо4 спрямовані на зменшення об'єму судинного русла;
2) спрямовані на збільшення об'єму циркулюючої крові;
3) спрямовані на відновлення складу крові;
4) спрямовані на захист від гіпоксії-(див. розд. 19).
26.9. Які компенсаторні реакції організму спрямовані на зменшення об'єму судинного русла в початковій стадії крововтрати?
Мета цієї групи реакцій — привести у відповідність об'єм судинного русла зменшеному об'єму циркулюючої крові (централізація кровообігу). Це дає можливість на деякий час підтримати необхідний тиск крові й забезпечити кровопостачання життєво важливих органів.
Зазначені реакції- термінові, у їхньому розвитку провідне значення мають рефлекси.
Зменшення об'єму судинного русла при крововтраті обумовлюється такими змінами (рис. 95):
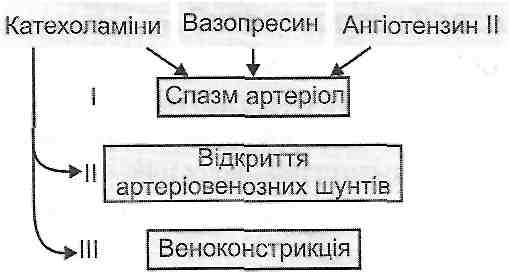
Рис. 95. Механізми централізації кровообігу при гострій крововтраті
1) спазм артеріол шкіри, м'язів, органів травної системи;
2) відкриття артеріовенозних анастомозів зазначених органів і тканин у результаті спазму прекапілярних сфінктерів;
3) веноконстрикція (скорочення гладких м'язів вен), що збільшує надходження крові до серця і зменшує ємність венозного відділу кровообігу.
В основі цих змін лежать такі механізми:
а) зниження артеріального тиску → збудження барорецепторів → активація симпа-тоадреналової системи → дія катехоламінів на а-адренорецептори гладких м'язів артерій, артеріол, прекапілярних сфінктерів і вен;
б) зменшення об'єму циркулюючої крові й артеріального тиску → збудження волю-мо- і барорецепторів → активація нейросекрсторних клітин гіпоталамуса, що продукують вазопресин → дія цього гормону на Vj-рецептори гладких м'язів судин з наступною вазоконстрикцією;
в) зменшення об'єму циркулюючої крові й активація симпатоадреналової системи → вивільнення клітинами юкстагломерулярного апарату нирок реніну → активація ренін-ангіотензинної системи з утворенням ангіотензину II → спазм гладких м'язів кровоносних судин.
26.10. У чому сутність компенсаторних реакцій організму, спрямованих на збільшення об'єму циркулюючої крові при крововтраті?
Реакції цієї групи здійснюються в період часу від кількох годин до кількох діб після кровотечі.
Збільшення об'єму циркулюючої крові в зазначений проміжок часу досягається за допомогою таких механізмів.
1. Перехід тканинної рідини в кровоносні судини. Основу цього процесу становить механізм, описаний Старлінгом. У результаті зменшення об'єму циркулюючої крові зменшується гідростатичний тиск у капілярах, що призводить до зменшення фільтрації води в артеріальній частині капілярів і збільшенню реабсорбції рідини у венозній. До зменшення гідростатичного тиску в капілярах призводить також розвиток спазму артеріол.
2. Посилення реабсорбції води та іонів натрію в нирках. Ця реакція, що запобігає втраті рідини із сечею, обумовлена такими механізмами:
а) дія вазопресину (антидіуретичного гормону) на У,-рецептори епітелію дистальних звивистих канальців і збірних трубок нирок, у результаті чого збільшується факультативна реабсорбція води;
б) активація ренін-ангіотензинної системи з наступним вивільненням альдостерону, що посилює реабсорбцію натрію в дистальних звивистих ка-нальцях нефронів;
в) активація симпатоадреналової системи, що призводить до перерозподілу течії крові між судинами кортикальних і юкстамедулярних нефронів, у результаті чого зростає площа й інтенсивність канальцевої реабсорбції води й натрію. 3. Вихід крові з депо в кровоносне русло. Основу цієї реакції становить активація симпатоадреналової системи й дія катехоламінів на судини печінки, селезінки, підшкірної жирової клітковини.
26.11. Які компенсаторні реакції забезпечують відновлення складу периферичної крові при крововтраті?
Відновлення складу периферичної крові забезпечується довгостроковими реакціями, які здійснюються в період часу від кількох діб до 1—2 тижнів після кровотечі. Вони виникають у відповідь на циркуляторну (зменшення об'єму циркулюючої крові) і гемічну (анемічну) гіпоксію. Основне значення надають кисневому голодуванню нирок, наслідком якого є утворення і надходження в кров великої кількості ниркових еритропоетинів. Останні діють на кровотворні клітини червоного кісткового мозку (еритропоетинчутливі клітини III класу) і стимулюють еритропоез - збільшується надходження молодих регенераторних форм еритроцитів у периферичну кров.
26.12. Які власне патологічні зміни можуть розвиватися при крововтраті?
1. Порушення системної' гемодинаміки (зменшення об 'єму циркулюючої крові, падіння артеріального тиску) і місцевого кровообігу (мікроциркуляції) аж до розвитку шоку.
2. Гостра постгеморагічна анемія (див. запит. 26.1.11).
3. Гіпоксія. Спочатку є циркуляторною, а потім гемічною (анемічною).
4. Негазовий ацидоз. Обумовлений гіпоксією й надходженням у кров молочної кислоти.
5. Порушення екскреторної функції нирок. При падінні артеріального тиску зменшується інтенсивність клубочкової фільтрації й розвиваються явища гострої ниркової недостатності: оліго- і анурія, інтоксикація (азотемія).
26.13. Що таке геморагічний шок? Назвіть головні патогенетичні ланки його розвитку.
Геморагічний шок — це шок, що виникає в результаті інтенсивної гострої крововтрати (рис. 96).
Провідним механізмом його розвитку є зменшення об 'єму циркулюючої крові, що викликає падіння
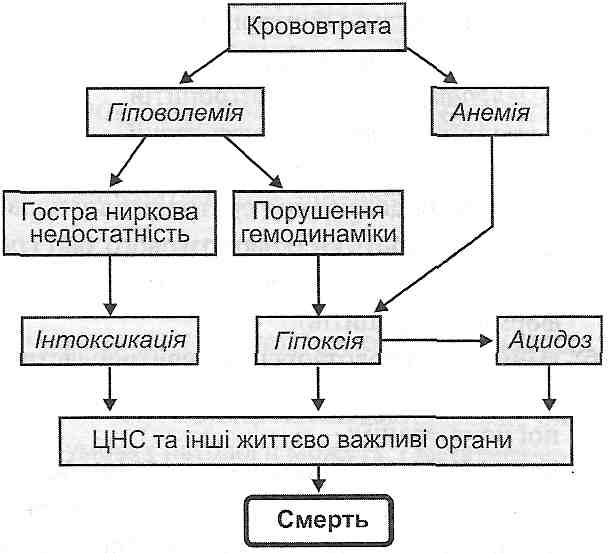
Рис. 96. Схема патогенеза геморагічного шоку
артеріального тиску, порушення мікроциркуляції, розлади кровопостачання життєво важливих органів (головного мозку, серця, нирок). Наслідком цього є розвиток гіпоксії, ацидозу й інтоксикації, що погіршує перебіг шоку, створює "зачаровані кола" у його патогенезі і в кінцевому підсумку веде до смерті.
26.1. Порушення системи еритроцитів
26.1.1. Якими кількісними змінами можуть виявляти себе патологічні процеси, пов 'язані з червоним паростком крові? Чим вони можуть бути обумовлені?
У нормі кількість еритроцитів у чоловіків дорівнює 4 -10і2-5 -1012, у жінок -3,5 -1012 — 4,5 -1012 в 1 л крові. Концентрація гемоглобіну у чоловіків становить 130-160, у жінок- 120-140 г/л.
В умовах патології можливі два види змін кількості еритроцитів і гемоглобіну в периферичній крові:
1) еритроцитоз - збільшення вмісту еритроцитів і гемоглобіну;
2) анемія — зменшення їх кількості.
Кількісні зміни еритроцитів можуть бути обумовлені:
а) порушенням співвідношення між їх утворенням (еритропоезом) і руйнуванням (еритродієрезом);
б) втратою еритроцитів при порушенні цілісності судин (крововтрата);
в) перерозподілом еритроцитів.
26.1.2. Які якісні зміни еритроцитів характерні для порушень червоного паростка крові? Чим вони можуть бути обумовлені?
До якісних змін еритроцитів відносять:
1) їхні регенераторні форми;
2) дегенеративні зміни еритроцитів;
3) клітини патологічної регенерації.
Причинами якісних змін еритроцитів можуть бути:
а) порушення дозрівання еритроцитів у червоному кістковому мозку або збільшення проникності кістковомозкового бар'єра, у результаті чого збільшується надходження в кров незрілих клітин з низьким вмістом гемоглобіну (регенераторні форми еритроцитів);
б) зміна типу кровотворення з еритробластичного на мегалобластичний, коли в кістковому мозку й крові з'являються мегалобласти і мегалоцити (клітини патологічної регенерації);
в) набуті і спадкові порушення обміну речовин, складу й структури еритроцитів, у тому числі синтезу гемоглобіну (зменшення утворення або синтез аномальних гемоглобінів), що веде до появи в крові дегенеративних форм еритроцитів.
26.1.3. Які клітини належать до регенераторних форм еритроцитів? Дайте їх коротку характеристику.
Регенераторні форми еритроцитів (клітини фізіологічної регенерації) — це молоді незрілі клітини червоного паростка крові, надходження яких у периферичну кров свідчить про посилення регенерації клітин еритроїдного ряду в червоному кістковому мозку або збільшення проникності кістковомозкового бар'єра.
До регенераторних форм відносять:
а) ретикулоцити (рис. 97; див. форзац). їх виявляють у мазку крові при суправі-тальному фарбуванні (барвник— бриліанткрезилблау). Являють собою без'ядерні клітини брудно-зеленого забарвлення (кольори "болотної зелені") із чорними включеннями у вигляді гранул (substantia granulofilamentosa). У нормі їхній вміст у крові становить 0,2-2 %. При посиленій регенерації клітин червоного паростка крові їх кількість може зростати до 50 %;
б) поліхроматофіли (рис. 98; див. форзац). їх виявляють у мазку крові при фарбуванні за Романовським-Гімза. Є без'ядерними клітинами, цитоплазма яких виявляє властивість поліхроматофілії, тобто сприймає як кислотні, так і основні барвники. Тому поліхроматофіли відрізняються від зрілих еритроцитів ціанотичним відтінком свого забарвлення. Власне кажучи, ретикулоцити і поліхроматофіли є клітинами однакового ступеня зрілості - безпосередніми попередниками еритроцитів. Різні назви пов'язані з різними їхніми властивостями, які виявляються при різних способах фарбування;
в) нормобласти (ацидофільні, поліхроматофільні, базофільні). Це ядерні попередники еритроцитів. У нормі в периферичній крові їх нема, вони містяться тільки в червоному кістковому мозку. При посиленій регенерації клітин еритроїдного ряду можуть з'являтися в крові ацидофільні і поліхроматофільні, рідше базофільні нормобласти. Іноді, при гіперрегенеративних анеміях, у крові можна виявити еритробласти (попередники нормобластів).
26.1.4. Які дегенеративні зміни можуть бути характерні для еритроцитів в умовах патологічних процесів, пов'язаних з червоним паростком крові?
Дегенеративними називають якісні зміни еритроцитів, що свідчать про неповноцінність цих клітин.
Такі зміни характеризуються такими явищами (рис. 99, 100; див. форзац):
а) анізоцитоз — зміна величини еритроцитів. Можлива поява макроцитів — еритроцитів з діаметром понад 8 мкм і мікроцитів — клітин, діаметр яких менший 6,5 мкм (середній діаметр нормального еритроцита близько 7,2 мкм);
б) пойкілоцитоз — зміна форми еритроцитів. У нормі еритроцити мають форму дисків, увігнутих всередину з обох боків. В умовах патології можуть з'являтися грушоподібні, витягнуті, серпоподібні, овальні еритроцити, а також еритроцити сферичної форми (сфероцити);
в) зміна забарвлення еритроцитів, що залежить від вмісту в них гемоглобіну. Еритроцити, інтенсивно забарвлені, називають гіперхромними, із блідим забарвлен-
ням - гіпохромними. Еритроцити, у яких забарвлена у вигляді кільця тільки периферична частина, де міститься гемоглобін, а в центрі - просвітління, мають назву анупоцитів. У випадку, коли еритроцити виявляють різну інтенсивність забарвлення ведуть мову про анізохромію; г) наявність патологічних включень. До них, зокрема, відносять тільця Жоллі-утворення розміром 1-2 мкм, які є залишками ядерної субстанції; кільгія Кебо-та - залишки ядерної оболонки, що мають форму кільця або вісімки; базофільна зернистість — залишки базофільної речовини цитоплазми, що свідчать про токсичне ураження червоного кісткового мозку.
26.1.5. Які клітини еритроцитарного ряду відносять до клітин патологічної регенерації?
При зміні типу кровотворення з еритробластичного на мегалобластичний у крові з'являються так звані клітини патологічної регенерації (рис. 101; див. форзац):
а) мегалобласти - дуже великі клітини (діаметром 12-15 мкм) з базофільною, по-ліхроматофільною або ацидофільною цитоплазмою, що містять велике, зазвичай ексцентрично розташоване ядро з ніжною хроматиновою ґраткою;
б) мегалоцити — без'ядерні клітини, які утворюються при дозріванні мегалобластів. Мають діаметр 10-12 мкм і більше, зазвичай інтенсивно забарвлені, злегка овальної форми, без властивого еритроцитам просвітління в центральній частині.
Поява зазначених клітин у червоному кістковому мозку й крові характерна для так званих мегалобластичних анемій, зокрема, Вр-фолієводефіцитної анемії.
26.1.6. Що таке єритроцитоз? Які причини й механізми його розвитку?
Еритроцитоз — це збільшення в крові кількості еритроцитів понад б • 1012 віл і концентрації гемоглобіну понад 170 г/л.
Еритроцитоз поділяють на абсолютний і відносний.
Абсолютний еритроцитоз - це підвищення вмісту еритроцитів і гемоглобіну в одиниці об'єму крові внаслідок посилення еритропоезу. За етіологією виділяють набутий і спадковий абсолютний еритроцитоз.
Набутий абсолютний еритроцитоз виникає в результаті збільшення продукції еритропоетину переважно в нирках. Це може бути викликано такими причинами:
1) порушеннями нейрогуморальної регуляції- збудженням симпатичної частини нервової системи, гіперфункцією деяких ендокринних залоз. Катехоламіни, АКТГ, тиреоїдні гормони, глюкокортикоїди посилюють утилізацію кисню, що сприяє розвитку гіпоксії й утворенню еритропоетинів у нирках;
2) гіпоксичною, дихальною, циркуляторною гіпоксією - у випадку гірської хвороби, недостатності зовнішнього дихання й кровообігу (див. розд. 19);
3) місцевою гіпоксією нирок унаслідок їх ішемії (гідронефроз, стеноз ниркових артерій);
4) гіперпродукцією еритропоетинів деякими пухлинами (гіпернефрома, рак печінки та ін.).
Крім того, абсолютний еритроцитоз розвивається при істинній поліцитемії (еритремія, або хвороба Вакеза), що є різновидом хронічного лейкозу.
Причиною виникнення спадкового абсолютного еритроцитозу може бути генетично обумовлений дефект глобіну в молекулі гемоглобіну або дефіцит в еритроцитах 2,3-Дифосфогліцерату, що є регулятором оксигенації й дезоксигенації гемоглобіну. При цьому підвищується спорідненість гемоглобіну до кисню і зменшується віддача останнього тканинам (крива дисоціації оксигемоглобіну зміщена вліво). Розвивається гіпоксія, стимулюється продукція еритропоетинів, під впливом яких посилюється еритропоез.
Гіпоксичне, або дисрегуляторне посилення утворення еритропоетинів супроводжується підвищенням функціональної активності еритроцитарного паростка кісткового мозку, що виявляється збільшенням у крові вмісту еритроцитів, гемоглобіну, гематокриту. При цьому може збільшитися об'єм циркулюючої крові (поліцитемічна гіперволемія), її в'язкість, сповільнюється швидкість течії крові, порушується серцева діяльність. Артеріальний тиск підвищується, відзначається повнокрів'я внутрішніх органів, гіперемія шкіри й слизових оболонок, спочатку посилюється тромбоутворення, а потім виникають кровотечі (розвивається ДВЗ-синдром, див. розд. 26.3).
Зміни в крові при набутих абсолютних еритроцитозах часто носять компенсаторний характер, сприяють поліпшенню кисневого постачання тканин в умовах гіпоксії. Із припиненням дії етіологічного фактора відбувається нормалізація кількості еритроцитів і гемоглобіну. Саме в цьому полягає корінна відмінність абсолютного еритроцитозу, що характеризується підвищенням фізіологічної регенерації еритроцитарного паростка кісткового мозку, від еритремії (хвороба Вакеза),- при якій первинне й необоротне зростання кількості еритроцитів обумовлене гіперплазією еритроцитарного паростка пухлинної природи.
Відносний еритроцитоз — це збільшення вмісту еритроцитів і гемоглобіну в одиниці об'єму крові внаслідок зменшення об'єму плазми. Його розвиток пов'язаний з дією факторів, які обумовлюють зневоднення організму (див. розд. 23) або перерозподіл крові, що викликає поліцитемічну гіповолемію (наприклад, шок, опіки).
26.1.7. Дайте визначення поняття "анемія".
Анемія — це гематологічний синдром або самостійне захворювання, що характеризується зменшенням кількості еритроцитів і (або) вмісту гемоглобіну в одиниці об'єму крові, а також якісними змінами еритроцитів.
26.1.8. Якими гематологічними й загальними клінічними ознаками може виявляти себе анемія?
Гематологічні ознаки анемій поділяють на кількісні і якісні. До кількісних відносять:
1) зменшення вмісту еритроцитів в одиниці об'єму крові (у чоловіків нижче 4 • 1012, у жінок нижче 3,5 -1012 в 1 л крові);
2) зменшення концентрації гемоглобіну (у чоловіків нижче 130 г/л, у жінок нижче 120 г/л);
3) зменшення гематокриту (у чоловіків нижче 43 %, у жінок нижче 40 %);
4) зміни колірного показника (норма 0,85-1).
Якісними ознаками анемій є поява:
1) регенераторних форм еритроцитів (див. запит. 26.1.3);
2) дегенеративних змін у клітинах еритроцитарного ряду (див. запит. 26.1.4);
3) клітин патологічноїрегенерації(див. запит. 26.1.5).
Загальні клінічні прояви анемій:
1) гіпоксія - синдром, що виникає при будь-якому виді анемії (див. розд. 19);
2) синдроми, обумовлені особливістю патогенезу кожного окремого виду анемії (наприклад, при В12-фолієводефіцитній анемії- неврологічні розлади й ураження травної системи, при гемолітичній анемії- жовтяниця).
26.1.9. Як класифікують анемії? Наведіть приклади кожного виду анемій.
I. Патогенетична класифікація:
1) постгеморагічні анемії (наприклад, анемія після гострої крововтрати);
2) гемолітичні анемії (наприклад, серпоподібноклітинна);
3) анемії, обумовлені порушеннями еритропоезу (наприклад, залізодефіцитна).
II. За етіологією:
1) спадкові (наприклад, талассмія);
2) набуті (наприклад, хронічна постгеморагічна анемія).
III. За регенеративною здатністю червоного кісткового мозку:
1) регенераторні (наприклад, гостра постгеморагічна анемія);
2) гіперрегенераторні (наприклад, набута гемолітична анемія);
3) гіпорегенераторні (наприклад, залізодефіцитна анемія);
4) арегенераторні (наприклад, апластична анемія).
IV. За колірним показником (КП):
1) нормохромні (КП = 0,85-1; наприклад, гостра постгеморагічна анемія в перші кілька діб після крововтрати);
2) гіпохромні (КП < 0,85; наприклад, залізодефіцитна анемія);
3) гіперхромні (КП > 1; наприклад, ВІ2-фолієводефіцитна анемія).
V. За типом кровотворення:
1) анемії з еритробластичним типом кровотворення (наприклад, залізодефіцитна анемія);
2) анемії з мегалобластичним типом кровотворення (наприклад, В|2-фолієво-дефіцитна анемія).
VI. За клінічним перебігом:
1) гострі (наприклад, анемія після гемотрансфузійного шоку);
2) хронічні (наприклад, гіпопластична анемія).
26.1.10. Які ознаки свідчать про регенераторний характер анемії? »
Ознаками посиленої регенерації клітин еритроїдного ряду є:
1) з боку периферичної крові - збільшення вмісту ретикулоцитів і поліхроматофілів та поява нормобластів (регенераторні форми еритроцитів);
2) з боку червоного кісткового мозку -зміщення лейкоеритроїдного співвідношення від 3:1 до 1:1 і навіть до 1:2 і 1:3.
26.1.11. Що таке постгеморагічні анемії? Як їх класифікують?
Постгеморагічна анемія - це анемія, що розвивається в результаті крововтрати.
Залежно від характеру крововтрати виділяють два види анемій цієї групи: 1) гостру постгеморагічну і 2) хронічну постгеморагічну анемію.
Гостра постгеморагічна анемія виникає після швидкої масивної крововтрати при пораненні судин або їх ушкодженні патологічним процесом.
Хронічна постгеморагічна анемія розвивається внаслідок повторних, часто невеликих крововтрат, викликаних ураженням кровоносних судин при деяких хворобах (дисменорея, виразкова хвороба шлунка, геморой та ін.) і порушенням судин-но-тромбоцитарного і коагуляційного гемостазу (геморагічний діатез). Втрата заліза при частих кровотечах надає цій анемії залізодефіцитний характер.
26.1.12. Опишіть картину крові при гострій постгеморагічній анемії.
Картина крові при гострій постгеморагічній анемії зазнає змін залежно від часу, що пройшов після крововтрати. З урахуванням цього можна виділити три періоди, кожний з яких характеризується певною картиною периферичної крові (рис. 102).
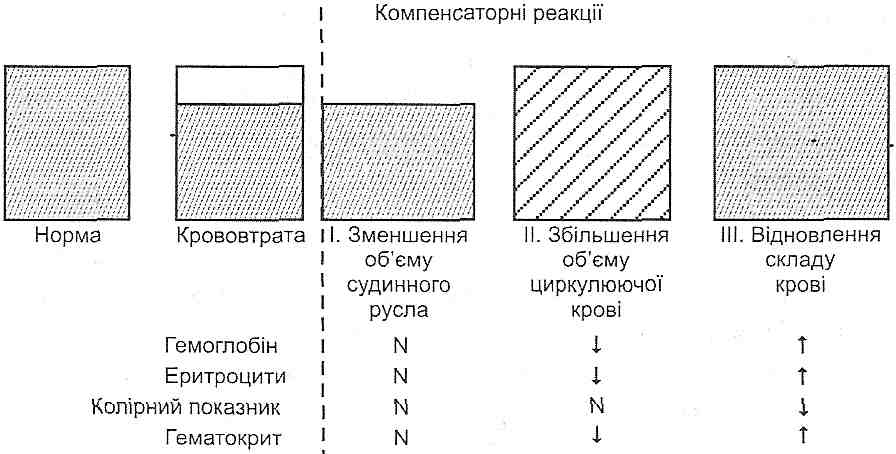
Рис. 102. Картина периферичної крові в різні періоди після гострої крововтрати (N- норма)
I. Перші кілька годин після гострої крововтрати. У цей період часу зменшується загальний об'єм крові, а також загальна кількість еритроцитів в організмі. Однак в одиниці об'єму крові вміст еритроцитів і концентрація гемоглобіну не міняються. Це пояснюється тим, що відразу ж після крововтрати спрацьовують термінові компенсаторні реакції, спрямовані на зменшення об'єму судинного русла, і ще недостатньо виражені реакції, спрямовані на поповнення об'єму циркулюючої крові (перехід рідини із тканин у кров).
II. Період часу від кількох годин до кількох діб після гострої крововтрати. У результаті переходу рідини з інтерстиціального простору в кровоносні судини відбуваєть-
ся розведення крові (гемодшюі/ія). Як результат, зменшується кількість еритроцитів і гемоглобіну в одиниці об'єму крові, падає гематокрит. Колірний показник залишається без змін (нормохромна анемія). Якісні зміни еритроцитів у мазку крові ще не виявляються. III. Період часу від кількох діб до 1—2 тижнів після гострої крововтрати. Найбільш характерною рисою картини крові в цей період є поява великої кількості регенераторних форм еритроцитів (див. запит. 26.1.3), що пов'язане з посиленням еритропоезу в червоному кістковому мозку. Оскільки молоді незрілі еритроцити містять гемоглобіну менше в порівнянні зі зрілими клітинами, колірний показник зменшується й анемія стає гіпохромною.
26.1.13. Визначте місце гострої постгеморагічної анемії в різних класифікаціях анемій.
|
Класифікація |
Анемія після гострої крововтрати |
І. |
За патогенезом |
Постгеморагічна |
II. |
За етіологією |
Набута |
III. |
За регенераторною здатністю червоного кісткового мозку |
Регенераторна |
IV. |
За колірним показником |
Спочатку нормохромна, потім гіпохромна |
V. |
За типом кровотворення |
3 еритробластичним типом кровотворення |
VI. |
За клінічним перебігом |
Гостра |
26.1.14. Опишіть картину крові при хронічній постгеморагічній анемії.
У зв'язку із втратою заліза при частих кровотечах розвиваються гематологічні ознаки залізодефіцитної анемії: зменшується концентрація гемоглобіну й колірний показник, у мазку крові з'являються дегенеративні форми еритроцитів (мікро- і по-йкілоцитоз, гіпохромія). Кількість еритроцитів і гематокрит можуть залишатися без змін.
26.1.15. Визначте місце хронічної постгеморагічної анемії в різних класифікаціях анемій.
|
Класифікація |
Анемія після повторних крововтрат |
І. |
За патогенезом |
Постгеморагічна |
II. |
За етіологією |
Набута |
III. |
За регенераторною здатністю червоного кісткового мозку |
Гіпорегенераторна |
IV. |
За колірним показником |
Гіпохромна |
V |
За типом кровотворення |
3 еритробластичним типом кровотворення |
VI. |
За клінічним перебігом |
Хронічна |
26.1.16. Що таке гемолітичні анемії? Як їх класифікують?
Гемолітичними називають анемії, що виникають унаслідок руйнування (гемолізу) еритроцитів.
Класифікація. І. За походженням:
1) набуті;
2) спадково обумовлені. її. За причинами гемолізу:
1) анемії, обумовлені екзоеритроцитарними факторами (екстракорпускулярні);
2) анемії, обумовлені ендоеритроцитарними факторами (корпускулярні). НІ. За механізмами гемолізу:
1) анемії із внутрішньосудинним гемолізом;
2) анемії із внутрішньоклітинним гемолізом. IV. За клінічним перебігом:
1) гострі;
2) хронічні.
26.1.17. Які ендоеритроцитарні фактори можуть бути причиною розвитку гемолітичної анемії?
Розвиток гемолітичної анемії може бути пов'язаний з трьома групами дефектів еритроцитів:
1) дефекти мембрани (мембранопаті'і);
2) порушення ферментів (ферменто-, або ензимопатії);
3) зміни структури гемоглобіну (гемоглобінопатії).
26.1.18. Як визначити, які фактори (ендо- чи екзоеритроцитарні) є причиною гемолізу еритроцитів?
Із цією метою використовують пробу Моллісона у двох варіантах її постановки.
I. Еритроцити хворого з гемолітичною анемією вводять здоровій людині. Можливі результати:
а) якщо відбувся гемоліз цих еритроцитів, то анемія ендоеритроцитарна;
б) якщо гемолізу немає, то анемія екзоеритроцитарна.
II. Еритроцити здорової людини вводять хворому з гемолітичною анемією. Можливі результати:
а) якщо відбувається гемоліз цих еритроцитів, то анемія екзоеритроцитарна;
б) якщо гемолізу немає, то анемія ендоеритроцитарна.
26.1.19. Назвіть можливі причини й основні механізми внутрішньосудинного гемолізу еритроцитів.
Внутрішньосудиннш гемоліз виникає в кровоносних судинах унаслідок дії факторів, що ушкоджують еритроцити. Ці фактори отримали назву гемолітичних. До них відносять:
а) фактори фізичної природи (механічна травма, іонізуюча радіація, ультразвук, температура);
б) хімічні агенти (гемолітичні отрути);
в) біологічні фактори (збудники інфекційних захворювань, токсини, ферменти);
г) імунні фактори (антитіла).
Механізми внутрішньосудинного гемолізу.
I. Механічний гемоліз. Виникає внаслідок механічного руйнування мембран еритроцитів, наприклад, при роздавлюванні еритроцитів у судинах стопи (маршовий гемоліз).
II. Осмотичний гемоліз. Виникає тоді, коли осмотичний тиск усередині еритроцита більший, ніж осмотичний тиск плазми крові. У цьому випадку вода за законами осмосу надходить в еритроцит, об'єм його зростає, і в кінцевому підсумку відбувається розрив мембрани. Причиною осмотичного гемолізу може бути або зменшення осмотичного тиску середовища, у якому перебувають еритроцити (гіпотонічні розчини), або збільшення осмотичного тиску в самих еритроцитах. Останнє, як правило, пов'язане зі збільшенням концентрації іонів натрію усередині еритроцитів у результаті підвищення проникності їх мембрани або внаслідок порушення роботи Na-K-насосів.
III. Окисний гемоліз. Розвивається внаслідок вільнорадикального окиснення ліпідів і білків плазматичної мембрани еритроцитів. Результатом цього є збільшення проникності еритроцитарної мембрани, що потім веде до реалізації осмотичного механізму гемолізу.
IV. Детергентний гемоліз. Пов'язаний з розчиненням ліпідних компонентів мембрани еритроцитів речовинами-детергентами. Цей вид гемолізу викликають жовчні кислоти (холемічний синдром), жиророзчинні хімічні агенти, деякі токсини бактерій (лецитинази).
V. Комплементзалежний гемоліз. Обумовлений руйнуванням (перфорацією) мембрани еритроцитів активним комплементом. Цей механізм лежить в основі імунного гемолізу.
26.1.20. Які фактори можуть спричиняти окисний гемоліз еритроцитів?
Основу окисного гемолізу становлять реакції вільнорадикального окиснення, і зокрема, процеси пероксидного окиснення ліпідів еритроцитарної мембрани (див. розд. 11). Існує два механізми активації окисного гемолізу еритроцитів.
I. Посилене утворення вільних радикалів. Це буває при:
а) дії екзогенних речовин-окислювачів (деякі лікарські препарати, гемолітичні отрути, токсичні дози вітаміну D; продукти, що містяться в бобах (Vicia fava);
б) дії іонізуючої радіації;
в) гіпероксії.
II. Порушення діяльності антиоксидантних систем еритроцитів. Це може бути обумовлено:
а) спадковими або набутими порушеннями активності ферментів глюшатіоно-вої антиоксидантної системи (глютатіонпероксидази і глютатіонредуктази);
б) дефіцитом селену — мікроелемента, необхідного для функціонування глю-татіонпероксидази;
в) пригніченням реакцій пентозного циклу (наприклад, дефіцит глюкозо-6-фосфатдегідрогенази).
26.1.21. Які порушення розвиваються в організмі в результаті внутрішньосудинного гемолізу еритроцитів?
В нутрішньосу динний гемоліз супроводжується виходом гемоглобіну з клітин у плазму крові, де він частково з'єднується з білком гаптоглобіном (рис. 103). При цьому відбуваються такі процеси.
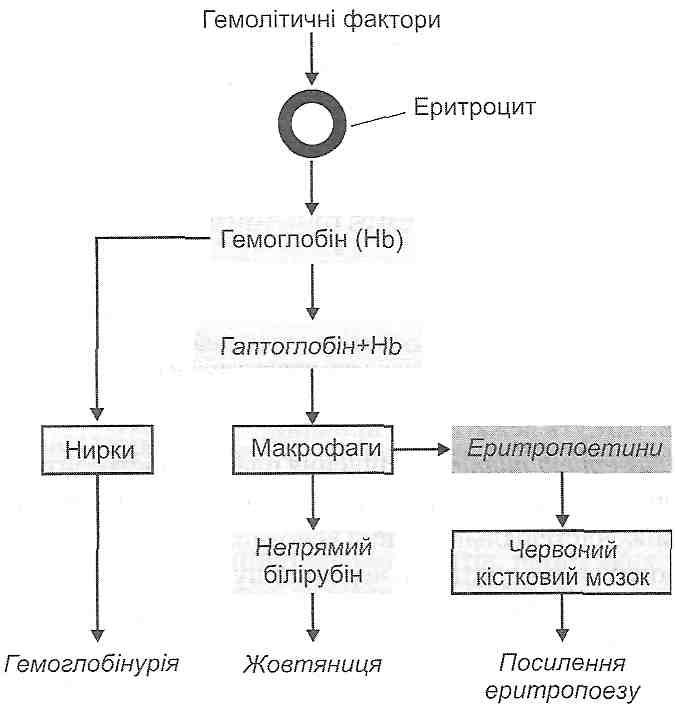
Рис. 103. Схема патогенезу порушень при енутрігиньосудинному гемолізі
1. Комплекс гемоглобін-гаптоглобін поглинається макрофагами і викликає утворення й вивільнення останніми макрофагальних еритропоетинів. Еритропоетини, впливаючи на червоний кістковий мозок, стимулюють еритропоез. У результаті в червоному кістковому мозку й периферичній крові з'являються ознаки посиленої регенерації клітин еритроїдного ряду.
2. Поглинений макрофагами гемоглобін зазнає біохімічних перетворень, у результаті яких білкова частина молекули розщеплюється до амінокислот, а з гема утворюється білірубін. Останній зв'язується з білками й надходить у кров (непрямий білірубін). У результаті розвивається синдром, відомий під назвою гемолітична жовтяниця (див. розд. 31).
З. Частина не зв 'язаного з гаптоглобіном гемоглобіну фільтрується в нирках. Це призводить, з одного боку, до появи гемоглобіну в сечі (гемоглобінурія), з другого - до "забивання" nop ниркового фільтра, що може бути причиною появи ознак гострої ниркової недостатності.
26.1.22. Що таке внутрішньоклітинний гемоліз еритроцитів? Чим він може бути обумовлений?
Внутрішньоклітинний гемоліз розвивається внаслідок поглинання і перетравлювання еритроцитів макрофагами (рис. 104).
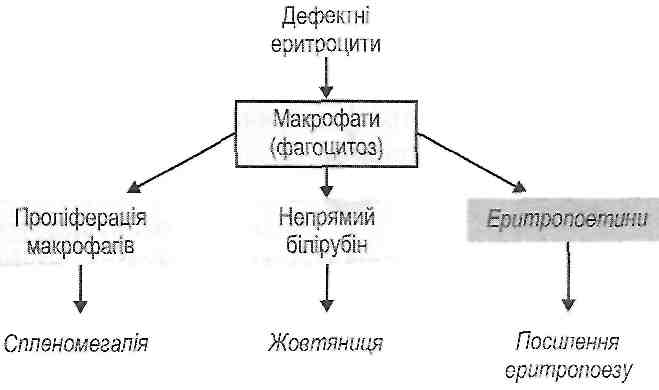
Рис. 104. Схема патогенезу порушень при внутрішньоклітинному гемолізі
У його основі можуть лежати такі причини:
а) поява дефектних еритроцитів. Зменшення пластичності еритроцитів, їхньої здатності до деформації, набряк призводять до того, що вони не можуть вільно проходити через міжендотеліальні щілини венозних синусів селезінки ("селезінковий фільтр") і надовго затримуються в червоній пульпі, контактуючи з макрофагами. Остання обставина і викликає поглинання дефектних еритроцитів макрофагами;
б) поява на поверхні еритроцитів хімічних груп, здатних специфічно взаємодіяти з рецепторами макрофагів. Такі групи виявляються при старінні еритроцитів (оголюються структури сіалових кислот еритроцитарної мембрани), а також при фіксації на їхній поверхні антитіл (з'являються Рс-фрагменти імуноглобулінів). В останньому випадку активується антитілозалежний фагоцитоз еритроцитів;
в) гіперспленізм - збільшення фагоцитарної активності макрофагів селезінки.
26.1.23. Які порушення розвиваються в організмі в результаті внутрішньоклітинного гемолізу еритроцитів?
Посилений фагоцитоз еритроцитів викликає такі зміни:
а) утворення й вивільнення макрофагами epumponoemimie, у результаті чого посилюється еритропоез у червоному кістковому мозку і з'являється велика кількість регенераторних форм еритроцитів у периферичній крові;
б) утворення великої кількості білірубіну, що обумовлює розвиток жовтяниці;
в) проліферацію макрофагів, що призводить до збільшення селезінки (сплеиомегалїі).
26.1.24. Які причини можуть викликати розвиток набутої гемолітичної анемії?
Залежно від причин розвитку виділяють такі види набутої гемолітичної анемії.
I. Анемії, обумовлені механічним ушкодженням еритроцитів.
II. Імунні гемолітичні анемії.
III. Токсичні гемолітичні анемії.
IV. Інфекційні гемолітичні анемії.
V. Набуті мембранопатії.
26.1.25. Наведіть приклади анемій, обумовлених механічним ушкодженням еритроцитів.
1. Механічний гемоліз при протезуванні судин або клапанів серця.
2. "Маршова " гемоглобінурія — травматизація еритроцитів у капілярах стоп під час тривалого маршу.
3. Мікроангіопатична гемолітична анемія (хвороба Мошковича) — травматизація еритроцитів при зіткненні їх з нитками фібрину. Буває при ДВЗ-синдромі.
26.1.26. Що таке імунні гемолітичні анемії? Назвіть можливі причини.
Імунні гемолітичні анемії—це анемії, що виникають за участі специфічних імунних механізмів. Вони обумовлені взаємодією гуморальних антитіл з антигенами, фіксованими на поверхні еритроцитів, і тому є проявом II типу алергічних реакцій за класифікацією Кумбса і Джелла (див. розд. 10).
Залежно від причин розвитку виділяють такі види імунних гемолітичних анемій:
1. Алоімунні (ізоімунні) гемолітичні анемії. їх причиною можуть бути: а) надходження ззовні антитіл проти власних еритроцитів (гемолітична хвороба новонароджених) і б) надходження в організм еритроцитів, проти яких у плазмі є антитіла (переливання крові, не сумісної за групами АВО або Rh).
2. Аутоімунні гемолітичні анемії. Обумовлені утворенням в організмі антитіл проти власних еритроцитів. Це може бути пов'язано або з первинними змінами самих еритроцитів (поява аутоантигенів), або зі змінами в імунній системі (скасування імунологічної толерантності, поява "заборонених" клонів лімфоцитів).
3. Гетероімунні (гаптенові) гемолітичні анемії. Виникають при фіксації на поверхні еритроцитів чужорідних антигенів (гаптенів), зокрема, лікарських препаратів (пеніцилін, сульфаніламіди), вірусів.
26.1.27. Що таке гемолітична хвороба новонароджених?
Гемолітична хвороба новонароджених - це хвороба, що виникає в результаті гемолізу еритроцитів плода й новонародженого, викликаного антитілами матері.
Найчастіше бувають два варіанти гемолітичної хвороби новонароджених: резус-конфлікт і АВО-конфлікт.
Резус-конфлікт. Розвивається у випадку вагітності Шг-матері Ші+-плодом (найчастіше при повторній вагітності). Спочатку відбувається імунізація матері Rh+-epn-троцитами плода, які можуть потрапляти в організм матері під час пологів або при
дефектах плаценти. Найбільш імовірною є імунізація під час пологів, тому резус-конфлікт виникає найчастіше в умовах повторної вагітності Кп+-плодом.
У відповідь на надходження Ші+-еритроцитів в організмі матері синтезуються антитіла проти D-антигену. Ці антитіла (Ig G) здатні проникати через плаценту в організм плода й викликати гемоліз його еритроцитів.
АВО-конфлікт. Найчастіше виникає в ситуаціях, коли мати має групу крові 0(1), а плід - А(ІІ) або В(III). Нормальні ізоаглютиніни в системі АВО належать до класу IgM. Ці антитіла не проникають через плаценту й тому не можуть бути причиною АВ0-конфлікту. Однак у 10 % здорових людей, що мають групу крові 0(1), є антитіла проти аглютиногенів А і В, представлені IgG. Наявність цих антитіл не залежить від попередньої імунізації. Аглютиніни IgG проникають через плаценту і можуть викликати гемоліз еритроцитів плода з групами крові А(П), В(Ш). Серед дітей-первістків гемолітична анемія як результат АВО-конфлікту буває з такою ж частотою, як і у дітей, народжених після других, третіх і наступних пологів, на відміну від резус-конфлікту, при якому частота гемолітичної анемії збільшується зі збільшенням кількості пологів.
26.1.28. Які типи антитіл можуть викликати гемоліз еритроцитів? Який його механізм?
Імунний гемоліз еритроцитів може бути зумовлений такими видами антитіл.
1. Гемаглютиніни. Належать до імуноглобулінів класів IgM і IgG. Викликають аглютинацію (склеювання) еритроцитів. Відомі теплові й холодові гемаглютиніни з оптимумом реакції зв'язування антигенів при звичайній температурі тіла або при зниженні температури до 32 °С (наприклад, у кінцівках за холодної погоди). У цих умовах механізм гемолізу еритроцитів - внутрішньоклітинний: агрегати еритроцитів при проходженні через селезінку зазнають фагоцитозу, що його здійснюють макрофаги.
2. Гемолізини. Є антитілами переважно класу IgG. Здатні фіксувати комплемент. Відомі гемолізини з оптимумом реакції зв'язування антигенів при звичайній температурі тіла (теплові) і при зниженні температури крові (холодові).
Гемоліз еритроцитів під дією гемолізинів має два механізми:
а) енутрішньосудинний — комплементзалежний гемоліз. У результаті фіксації, а потім і активації комплементу відбувається руйнування мембрани еритроцитів;
б) внутрішньоклітинний- антитілоопосередкований фагоцитоз. Макрофаги (Гс-рецептори) взаємодіють із F -фрагментами гемолізинів, фіксованих на еритроцитах. При цьому або відбувається повне поглинання еритроцитів макрофагами, або макрофаг "відкушує" частину еритроцита, перетворюючи його на мікросфероцит.
26.1.29. Що може бути причиною розвитку токсичної гемолітичної анемії?
Токсичну гемолітичну анемію можуть викликати: а) екзогенні хімічні агенти: миш'яковистий водень, свинець, солі міді, фенілгідра-зин, резорцин та ін.;
б) ендогенні хімічні фактори: жовчні кислоти, продукти, що утворюються при сш вій хворобі, уремії;
в) отрути біологічного походження: зміїна, бджолина, отрута деяких видів паву.
26.1.30. Назвіть можливі причини розвитку інфекційних гемолітичних анемій.
Гемолітичну анемію викликає цілий ряд інфекційних агентів, зокрема, гем тичнйй стрептокок, малярійний плазмодій, токсоплазма, лейшманії.
Причиною гемолізу при інфекційних захворюваннях можуть бути або розм ження збудників в еритроцитах (малярійний плазмодій), або дія токсинів-гемолїзж (гемолітичний стрептокок).
26.1.31. Що таке набутімембранопатії? Наведіть приклади.
Набуті мембранопатії— це гемолітичні анемії, які виникають унаслідок і тих у процесі індивідуального розвитку дефектів мембран еритроцитів^
Прикладом може бути пароксизмальна нічна гемоглобінурія (хвороба Марк еи—Мікелі). Це захворювання виникає в результаті соматичної мутації кровотвор клітин, унаслідок якої з'являються аномальні популяції еритроцитів, лейкопж тромбоцитів з дефектами мембрани. Вважається, що порушення мембран заза них клітин пов'язані зі зміною співвідношення жирових кислот, що входять лося ду їхніх фосфоліпідів (зменшується вміст ненасичених і збільшується — насилав жирових кислот). Еритроцити аномальної популяції набувають здатності фіксуа комплемент, що є передумовою комплементзалежного гемолізу. Зменшення рН і редовища є чинником, який провокує внутрішньосудинний гемоліз. Цим поясні ся той факт, що руйнування еритроцитів розвивається найчастіше вночі (у нічне рН крові трохи зменшується).
Таким чином, пароксизмальна нічна гемоглобінурія є набутою ендоеритрс тарною гемолітичною анемією із внутрішньосудинним гемолізом еритроцитів.
26.1.32. Які зміни характерні для картини периферичної крові і червоного кісткового мозку при набутій гемолітичній анемії?
Периферична кров. Зменшується кількість еритроцитів і концентрація гв глобіну, хоча при внутрішньосудинному гемолізі вміст останнього може не змеа ватися за рахунок гемоглобіну, що перебуває в плазмі крові. Колірний показни правило, у нормі, однак може бути й більший за одиницю, що пов'язано із пая троцитарним гемоглобіном. У мазку крові виявляється значна кількість регенераи них форм еритроцитів: ретикулоцитів, поліхроматофілів, нормобластів, що свідчі про регенеративний, а іноді й гіперрегенеративний характер анемії.
Червоний кістковий мозок. Лейкоеритроцитарний індекс становить не 3^ у нормі, а 2:1,1:1, 1:2. Такі зміни є ознакою посиленої регенерації клітин червоа паростка крові. Показано, що здоровий червоний кістковий мозок може компез вати 6—8-кратне збільшення темпів руйнування еритроцитів без розвитку помі анемії.
26.1.33. Що може лежати в основі розвитку спадкових гемолітичних анемій?
Залежно від механізмів розвитку всі спадково обумовлені гемолітичні анемії поділяють на три групи.
I. Мембранопатії. Основу цієї групи анемій становлять дефекти мембран еритроцитів.
II. Ферментопатії (ензимопатії). Анемії цієї групи обумовлені спадковими порушеннями ферментів еритроцитів.
III. Гемоглобінопатії - анемії, що виникають унаслідок якісних змін гемоглобіну.
26.1.34. Чим може бути обумовлений розвиток спадкових мембранопатій?
Спадкові мембранопатії можуть бути обумовлені двома групами дефектів ери-троцитарної мембрани. З урахуванням цього виділяють дві групи мембранопатій.
1. Мембранопатії, обумовлені порушеннями мембранних білків. До них, зокрема, відносять:
а) мікросфероцитарну анемію Мінковського-Шоффара;
б) овалоклітинну гемолітичну анемію (еліптоцитоз, овалоцитоз);
в) стоматоцитоз.
2. Мембранопатії, пов'язані з порушеннями мембранних ліпідів. У цю групу входять:
а) акантоцитоз (є проявом абеталіпопротеїнемії);
б) анемія, обумовлена дефіцитом ЛХ4Г(лецитин-холестерол-ацилтрансферази).
26.1.35. Дайте характеристику мікросфероцитарної анемії Мінковського-Шоффара.
Анемія Мінковського-Шоффара є спадковою, ендоеритроцитарною (мембра-нопатія) гемолітичною анемією з внутрішньоклітинним гемолізом. Тип спадкування - аутосомно-домінантний.
Спадковий дефект виникає в мембранному білку еритроцитів - спектрі/ні. Унаслідок цього значно збільшується проникність еритроцитарної мембрани для іонів натрію. Натрій і вода переходять з плазми всередину еритроцитів. Це врікликає активацію Na-K-насосів і гліколізу, завдяки чому еритроцити підтримують у цих умовах свій об'єм. Якщо ж зазначені механізми виявляються функціонально недостатніми, то об'єм еритроцитів збільшується й вони набувають сферичної форми, тобто перетворюються на сфероціїти (кульки).
Сфсроцити втрачають свою пластичність, а отже, і здатність до деформації. Через це вони не можуть проходити через вузькі міжендотеліальні щілини венозних синусів селезінки й на тривалий час затримуються в ній. Макрофаги селезінки "відкушують" частину мембрани еритроцитів і перетворюють останніх нз. мікросфероцити.
При наступних проходженнях мікросфероцитів через селезінку макрофаги повністю фагоцитують змінені еритроцити -відбувається внутрішньоклітинний гемоліз. Таким чином, тривалість життя еритроцитів зменшується до 8-12 діб замість 120.
З урахуванням патогенезу зазначеної анемії непоганий лікувальний ефект має видалення селезінки — спленектомія.
26.1.36. Чим може бути обумовлений розвиток спадкових ферментопатій ?
Причиною розвитку спадкових ферментопатій можуть бути порушення таких ферментних систем еритроцитів.
1. Дефіцит ферментів пентозного циклу. Найпоширенішою ферментопатією є глюкозо-6-фосфатдегідрогеназодефіцитна анемія, обумовлена повною відсутністю або значним зменшенням активності глюкозо-6-фосфатдегідрогенази. Відомо близько 250 мутантних форм цього ферменту (див. запит. 26.1.37).
2. Дефіцит ферментів гліколізу. Описано спадкові дефекти 11 з 13 ферментів гліколізу. Найбільш поширеним є дефіцит піруеаткінази.
Основою патогенезу анемій, що виникають у результаті порушень реакцій гліколізу, є дефіцит АТФ, що закономірно призводить до порушення енергозабезпечення Na-K-насосів плазматичних мембран. Наслідком цього є збільшення концентрації іонів натрію і, як результат, осмотичного тиску всередині еритроцитів, що, у свою чергу, викликає надходження води в клітини. При цьому еритроцити набухають і перетворюються на сфероцити, які зазнають фагоцитозу з боку макрофагів.
3. Дефіцит ферментів циклу глютатіону (глютатіонсинтетази, глютатіонредукта-зи, глютатіонпероксидази).
Порушення циклу глютатіону призводять до пригнічення роботи глютатіонпе-роксидазної антиоксидантної системи, наслідком чого є активація процесів пе-роксидного окиснення ліпідів в еритроцитах. Реакції вільнорадикального окис-нення ліпідів мембран порушують їх бар'єрні властивості й значно збільшують проникність еритроцитарної мембрани до іонів, зокрема, іонів натрію. Останні за градієнтом концентрації надходять у клітини, збільшуючи осмотичний тиск усередині еритроцитів. Подальші процеси аналогічні тим, які розвиваються при інших описаних тут ферментопатіях.
4. Дефіцит ферментів утилізації АТФ. Прикладом цієї групи ферментопатій є дефіцит білкових компонентів Na-K-АТФ-ази еритроцитарної мембрани. Первинні порушення роботи Na-K-насосів еритроцитів призводять до збільшення концентрації іонів натрію в клітинах з усіма наслідками, що звідси випливають.
26.1.37. Дайте характеристику глюкозо-6-фосфатдегідрогеназо-дефіцитної анемії.
Глюкозо-6-фосфашдегідрогеназодефіцитна анемія є спадково обумовленою гемолітичною анемією, ендоеритроцитарною (ферментопатія), з внутрішньосудинним гемолізом. Тип спадкування - зчеплений з Х-хромосомою.
У патогенезі цієї анемії центральне місце посідають такі процеси. Дефіцит глю-козо-6-фосфатдегідрогенази є причиною порушень реакцій пентозного циклу, внаслідок чого зменшується утворення НАДФН і відновлення (регенерація) глютатіону в еритроцитах. Це, у свою чергу, призводить до зниження активності антиоксидантної глютатіонпероксидазної системи і збільшення небезпеки активації вільнорадикаль-
ного окиснення. Цим, зокрема, пояснюється збільшення чутливості еритроцитів до дії екзогенних окислювачів (лікарських препаратів, токсичних продуктів рослинного походження) і активація пероксидного окиснення ліпідів мембран. Останнє призводить до збільшення проникності еритроцитарних мембран і надходження в клітини іонів натрію й води. Розвивається набухання й набряк еритроцитів, а потім - їх внутрішньо судинний гемоліз.
Однією з клінічних форм глюкозо-6-фосфатдегідрогеназодефіцитної анемії є фа-візм, що виникає при вживанні бобів Vicia fava, які містять токсичні продукти з вираженими окисними властивостями.
26.1.38. Чим може бути обумовлений розвиток спадкових гемоглобінопатій ?
В основі розвитку спадкових гемоглобінопатій можуть бути дві групи порушень: якісні і кількісні. Тому й розрізняють якісні і кількісні гемоглобінопатії.
Сутність якісних гемоглобінопатій становлять порушення первинної структури ланцюгів гемоглобіну, наприклад, заміна амінокислот, подовження або вкорочення ланцюгів молекул "гемоглобіну Найпоширенішою клінічною формою цієї групи гемоглобінопатій є серпоподібноклітинна анемія.
Кількісні гемоглобінопатії характеризуються порушеннями синтезу ланцюгів гемоглобіну. Прикладом анемій цієї групи є а- і р-таласемії.
26.1.39. Дайте характеристику серпоподібноклітинної анемії.
Серпоподібноклітинна анемія є спадково обумовленою гемолітичною анемією, ендоеритроцитарною (гемоглобінопатія), з внутрішньоклітинним гемолізом. Тип спадкування - неповне домінування.
Сутність дефекту полягає в тому, що в Р-ланцюгу молекули гемоглобіну в 6-му положенні від N-кінця глютамінову кислоту заміщено на валін. Це призводить до появи патологічної форми гемоглобіну, яку позначають HbS. Основна функціональна відмінність HbS від звичайних форм гемоглобіну полягає в тому, що у відновленому стані розчинність HbS зменшується майже в 100 разів. Це призводить до того, що HbS випадає в осад - утворюються кристали, які деформують еритроцити. Як наслідок, клітини червоної крові набувають серпоподібної форми, важко проходять через вузькі капіляри і міжендотеліальні простори венозних синусів селезінки. Цим пояснюється інтенсивний фагоцитоз серповидних еритроцитів макрофагами (внутрішньоклітинний гемоліз) і виражені трофічні зміни в тканинах, аж до мікротромбозів і некрозів.
Серпоподібність еритроцитів значно зростає при проходженні їх через тканини, які характеризуються низькими значеннями р02 (жирова тканина, селезінка), а також в умовах гіпоксії.
26.1.40. У чому сутність таласемій?
Толасеміїе спадково обумовленими гемолітичними анеміями, ендоеритроцитар-ними, з внутрішньоклітинним гемолізом. їх відносять до кількісних гемоглобінопатій, оскільки порушується синтез ланцюгів молекул гемоглобіну.
У людини в нормі гемоглобін представлено такими формами: НЬА] (аарр) - 95-96 %, НЬА2 (ааДД), у новонароджених — HbF (aayy).
Якщо порушується синтез а-ланцюгів, то розвивається а-таласемія. При цьому не утворюється НЬА,, НЬА і HbF. Замість а-ланцюгів клітини червоної крові синтезують р- і у-ланцюги. Тому при а-таласемії в еритроцитах з'являються патологічні форми гемоглобіну: у дорослих - НЬН (РРРР), а у новонароджених HbBart (уууу). НЬН і HbBart нестабільні, тому легко випадають в осад, унаслідок чого еритроцити набувають форми мішеней (звідси ще одна назва а-таласемії - мішенеклітинна анемія). Змінені еритроцити фагоцитуються макрофагами — розвивається внутрішньоклітинний гемоліз.
При р-таласемії (хвороба Кулі) порушений синтез р-ланцюгів молекул гемоглобіну. Тому нема HbAj, а компенсаторно збільшується утворення НЬА2. У новонароджених синтез HbF не порушений.
26.1.41. Якими клінічними синдромами можуть виявляти себе гемолітичні анемії?
Крім гематологічних ознак (змін периферичної крові і червоного кісткового мозку), для гемолітичних анемій характерні такі клінічні ознаки й синдроми.
1. Гіпоксія. Обумовлена анемією й виявляється різкою слабкістю, неприємними відчуттями у ділянці серця, серцебиттям, задишкою.
2. Гемолітична жовтяниця (див. розд. 31).
3. Посилене утворення жовчних каменів, особливо білірубінових. Пояснюється значним збільшенням вмісту білірубіну в жовчі та збільшенням її в'язкості.
4. Гемоглобінурія. Розвивається при внутрішньосудинному гемолізі. Гемоглобін, що вивільняється зі зруйнованих еритроцитів, зв'язується з білком плазми крові гаптоглобіном. 100 мл плазми крові містить стільки гаптоглобіну, що він може зв'язати 125 мг гемоглобіну. Якщо ж концентрація гемоглобіну в плазмі крові перевищує 125 мг %, то незв'язаний гемоглобін проходить через нирковий фільтр і з'являється в сечі.
5. Спленомегалія — збільшення селезінки. Є характерною для внутрішньоклітинного механізму гемолізу еритроцитів. В основі цього явища лежить підвищення функціональної активності макрофагів, що викликає інтенсивну їх проліферацію. Спленомегалія часто супроводжується збільшенням печінки (проліферація печінкових макрофагів).
6. Гемосидероз— відкладення гемосидерину в макрофагах. Гемосидерин— це частково денатурований і депротеїнізований феритин, тобто білок, що містить багато заліза в негемовій формі (вміст Fe у гемосидерині — 25-30 %).
7. Порушення мікроциркуляції. Часто виникають при інтенсивному внутрішньосудинному гемолізі й обумовлені розвитком ДВЗ-синдрому (див. розд. 26.3).
8. Гарячка. Розвивається в результаті різкої активдції фагоцитарної функції макрофагів, унаслідок чого вони вивільняють інтерлейкін-1 (див. розд. 15).
26.1.42. Як класифікують групу анемій, пов'язаних з порушеннями еритропоезу?
I. За походженням анемії, пов'язані з порушеннями еритропоезу, можуть бути набутими і спадково обумовленими.
II. За причинами виникнення виділяють такі групи анемій:
а) мієлотоксичні. Розвиваються внаслідок ушкодження кровотворних клітин під дією екзогенних (радіація, хімічні агенти, віруси) і ендогенних факторів (імунні фактори, токсичні продукти обміну речовин);
б) дефіцитні. Причиною їх виникнення є дефіцит факторів, необхідних для кровотворення, - недостатність заліза, фолієвої кислоти, білків, вітамінів В6іВ,2;
в) дисрегуляторні. Виникають як наслідок розладів регуляції еритропоезу (порушення співвідношення між еритропоетинами й інгібіторами еритропоезу при недостатності нирок, ушкодження елементів строми, що є мікрооточен-ням для кровотворних клітин);
г) пов 'язані зі зменшенням плацдарму еритропоезу. Є наслідком заміщення кровотворної тканини лейкозними клітинами, сполучною тканиною (фіброз), метастазами пухлин.
III. Залежно від сутності процесів, що лежать в основі розвитку анемій, виділяють:
а) порушення утворення еритроцитів: дефіцит кровотворних клітин унаслідок їх ушкодження або заміщення, порушення розмноження клітин кровотворення (порушення ресинтезу ДНК), дефекти дозрівання еритроцитів і виходу їх у кровоносні судини (неефективний еритропоезу,
б) порушення синтезу гемоглобіну: дефіцит заліза, порушення синтезу порфіринів (спадкові порушення ферментів, отруєння свинцем, дефіцит вітаміну В6, розлади синтезу білкових ланцюгів молекул гемоглобіну).
26.1.43. Що таке гіпопластична анемія? Які її етіологія й патогенез?
Гіпопластична (апластична) анемія - це захворювання системи крові, що характеризується пригніченням кровотворної функції червоного кісткового мозку і виявляється недостатнім утворенням еритроцитів, гранулоцитів і тромбоцитів (пан-цитопенією) або одних тільки еритроцитів (парціальна гіпопластична анемія, ери-тробластофтиз).
Розрізняють набуті і спадково обумовлені форми гіпопластичної анемії.
Набуті форми можуть мати такі причини:
1) фізичні фактори (іонізуюча радіація);
2) хімічні агенти (бензол, свинець, пари ртуті, лікарські препарати: цитостатичні засоби, левоміцетин, сульфаніламіди);
3) біологічні фактори (вірус гепатиту).
Крім того, до набутих відносять так звану ідіопатичну форму, причину якої встановити не вдається. На цю форму припадає 50-75 % всіх випадків гіпопластичної анемії.
Усі наведені етіологічні фактори поділяють на дві групи.
I. Фактори з облігатним (обов 'язковим) мієлотоксичним ефектом. До них відносять іонізуючу радіацію, бензол та його похідні, протипухлинні препарати (цитостатики), неорганічні сполуки миш'яку та ін.
II. Фактори з факультативним мієлотоксичним ефектом, що може виявлятися лише в окремих поодиноких випадках (наприклад, апластична постмедикаментозна анемія). У розвитку анемії після прийому лікарських препаратів велике, якщо не вирішальне, значення мають індивідуальні особливості організму. Встановлено, що не існує прямого зв'язку між розвитком гіпопластичної анемії, з одного боку, і дозою, а також тривалістю застосування лікарських препаратів, з другого. Розвиток гіпопластичної анемії може бути пов'язаний з прийманням деяких антибіотиків (левоміцетин), протисудомних, антитиреоїдних, антигістамінних засобів, транквілізаторів.
Прикладом спадково обумовленої форми гіпопластичної анемії є анемія Фан-коні — захворювання з аутосомно-рецесивним типом спадкування. Сутність дефекту полягає в порушеннях систем репарації ДНК. Унаслідок цього легко виникають невідновлювані ушкодження ДНК під впливом ультрафіолетового випромінювання й хімічних агентів. Порушується кровотворення, часто розвиваються гострі лейкози.
У патогенезі гіпопластичної анемії провідне місце посідають два механізми.
1. Ушкодження стовбурових клітин. Доказом цього є панцитопенія, тобто порушення утворення всіх формених елементів крові, що мають загального попередника (еритроцитів, гранулоцитів, тромбоцитів).
2. Ушкодження клітин мікрооточення - порушення стромальних клітин, які впливають на процеси розмноження й дозрівання клітин крові. Можливість реалізації даного патогенетичного механізму доводиться існуванням чистих ліній мишей ("сталеві" миші) з первинними спадково обумовленими дефектами стромальних клітин. Для всіх представників "сталевих" мишей характерний розвиток гіпопластичної анемії.
26.1.44. Дайте характеристику картини периферичної крові і червоного кісткового мозку при гіпопластичній анемії.
Для картини периферичної крові характерне зменшення вмісту еритроцитів і концентрації гемоглобіну, при цьому колірний показник у межах норми. У мазку крові, як правило, не виявляють регенераторних форм еритроцитів (ретикулоцитів, поліхроматофІлів). Зменшується вміст гранулоцитів (особливо нейтрофілів) і тромбоцитів. Кількість лімфоцитів може залишатися без змін.
У червоному кістковому мозку зменшується кількість кровотворних клітин зі збільшенням вмісту жирової тканини (картина спустошення червоного кісткового мозку). У зв'язку з тим, що залізо не використовується для кровотворення, збільшується його вміст в еритробластах і позаклітинно.
26.1.45. Визначте місце гіпопластичної анемії в різних класифікаціях анемій.
|
Класифікація |
Гіпопластична анемія |
І. |
За патогенезом |
Анемія, пов'язана з порушеннями еритропоезу |
II. |
За етіологією |
Набута, є спадкові форми |
III. |
За регенераторною здатністю червоного кісткового мозку |
Гіпо- або арегенераторна |
IV. |
За колірним показником |
Нормохромна |
V. |
За типом кровотворення |
3 еритробластичним типом кровотворення |
VI. |
За клінічним перебігом |
Хронічна |
26.1.46. Якими синдромами виявляє себе гіпопластична анемія?
Прояви гіпопластичної анемії пов'язані зі зменшенням утворення трьох видів формених елементів крові: еритроцитів, гранулоцитів і тромбоцитів. Це призводить до розвитку таких клінічних синдромів.
I. Анемія і пов'язаний з нею гіпоксачниїї синдром.
II. Геморагічний синдром (див. розд". 26.3).
III. Запальні процеси, обумовлені інфекційними агентами (пневмонія, отит, пієліт та ін.).
26.1.47. Що таке металобластичні анемії? Наведіть приклади.
Мегалобласмичні анемії-це група анемій, в основі яких порушення синтезу нуклеїнових кислот у клітинах і, як наслідок, порушення розмноження останніх. До мегалобластичних відносять:
1) анемію, обумовлену дефіцитом вітаміну В і фолієвої кислоти, - Вп-фолієводефі-цитну анемію;
2) перніціозну анемію Аддісона-Бірмера (етіологію не встановлено);
3) анемію, пов'язану з порушеннями ферментів синтезу пуринових і піримідинових основ, — В 12-рефрактерну анемію (стійку до лікування вітаміном В|2).
За походженням мегалобластичні анемії можуть бути набутими (виявляються звичайно у дорослих і осіб літнього віку) і спадково обумовленими. В останньому випадку порушення можуть мати стосунок до системи транспорту речовин (усмоктування в травному каналі) і реакцій метаболізму (ферментопатії).
26.1.48. Яка роль вітаміну В12 і фолієвої кислоти в забезпеченні кровотворення?
Вітамін В (ціанокобаламін) надходить в організм у складі харчових продуктів (м'ясо, яйця, сир, печінка) у зв'язаному з білками вигляді. У результаті гідролізу білків вітамін В]2 вивільняється в шлунку, де утворюється комплекс вітаміну В,2 з гастромукопротеїном (внутрішнім фактором Кастла). 1 мг гастромукопротеїну зв'язує 25 мг вітаміну В]2. Комплекс, що утворився, всмоктується в середньому й нижньому відділах клубової кишки.
Транспорт вітаміну В12 кров'ю здійснюється за допомогою специфічних транспортних білків - транскобаламінів. Печінка є важливим депо вітаміну В12. Запаси його тут настільки великі, що потрібно 3-6 років, аби при порушеннях надходження вітаміну В в організмі виникли ознаки його дефіциту.
У клітинах з вітаміну В12 утворюється дві його коферментні форми: метилкоба-ламін і 5-дезоксиаденозилкобаламін.
Метилкобаламін необхідний для синтезу ДНК у клітинах, тому він істотно впливає на кровотворення (рис. 105).
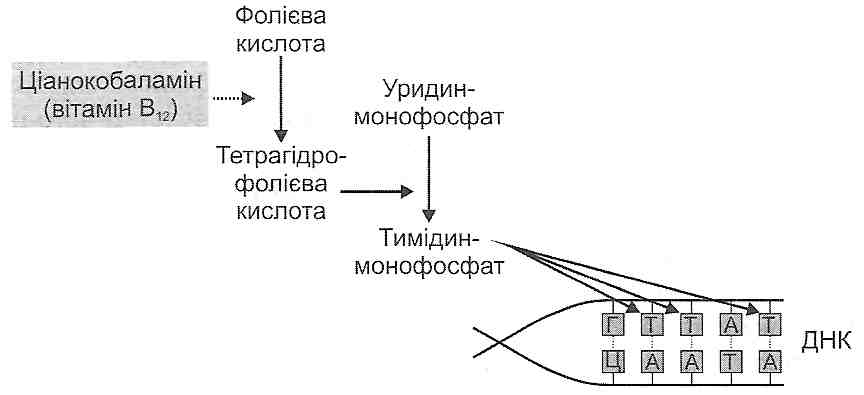
Рис. 105. Вплив ціанокобаламіну на кровотворення
Цей кофермент входить до складу ферментного комплексу, що перетворює фолієву кислоту на її коферментну форму — тетрагідрофолієву кислоту. Тетрагідро-фолієва кислота, у свою чергу, є коферментом ферментного комплексу, що каталізує реакцію перетворення уридинмонофосфату на тимідинмонофосфат, що йде на побудову ланцюгів ДНК.
Друга коферментна форма вітаміну В]2 - 5-дезоксіаденозилкобаламін — необхідна для нормального обміну жирових кислот. Вона бере участь у реакціях перетворення токсичного продукту обміну - метилмалонової кислоти на сукцинат, котрий потім надходить у цикл Кребса.
26.1.49. Назвіть основні причини недостатності вітаміну В12в організмі.
1. Екзогенна (аліментарна) недостатність — недостатнє надходження вітаміну В12 в організм з продуктами харчування. Може розвиватися у маленьких дітей при вигодовуванні їх козяч им молоком або сухими молочними сумішами.
2. Порушення всмоктування вітаміну BJ2:
а) порушення утворення й секреції гастромукопротеїну (внутрішнього фактора Кастла). Буває при спадково обумовлених порушеннях, атрофії слизової оболонки шлунка, аутоімунних ушкодженнях парієтальних клітин слизової шлунка, після гастректомії або видалення понад 2/3 шлунка;
б) порушення функції тонкої кишки: хронічні проноси (целіакія, спру), резекція великих ділянок кишки;
в) конкурентне використання вітаміну В12 гельмінтами й мікрофлорою кишок (дифілоботріоз, синдром "сліпої петлі").
3. Порушення утворення транскобаламінів у печінці.
4. Порушення депонування вітаміну Впу печінці (наприклад, при цирозі).
5. Посилене використання вітаміну Вп (наприклад, при вагітності).
26.1.50. Який патогенез порушень, що розвиваються в організмі при дефіциті вітаміну В12?
Дефіцит вітаміну В12 призводить до розвитку розладів, пов'язаних з порушенням утворення двох коферментних форм цього вітаміну: метилкобаламіну та 5-дезоксіа-денозилкобаламіну.
Недостатнє утворення метилкобаламіну, як і дефіцит фолієвої кислоти, викликає порушення утворення тетрагідрофолієвої кислоти, у результаті чого не відбувається перетворення уридинмонофосфату в тимідинмонофосфат. Унаслідок дефіциту пуринових основ порушуються синтез ДНК і розмноження клітин, у першу чергу кровотворних і епітелію травного каналу.
У червоному кістковому мозку еритробластичний тип кровотворення змінюється на мегалобластичний, зростає неефективний еритропоез, стає коротшою тривалість життя еритроцитів. Розвивається анемія, при якій клітини патологічної регенерації й еритроцити з вираженими дегенеративними порушеннями з'являються не тільки в кістковому мозку, але й у крові.
Зміни в клітинах мієлоїдного й мегакаріоцитарного ряду виявляються зменшенням кількості лейкоцитів і тромбоцитів, вираженою атипією клітин (гігантські нейтрофіли, мегакаріоцити з дегенеративними змінами в ядрі).
Виникнення при дефіциті вітаміну В]2 і фолієвої кислоти атипового мітозу та поява гігантських клітин епітелію травного каналу призводять до розвитку запально-атрофічних процесів у слизовій оболонці його відділів (глосит, стоматит, езофагіт, ахі-лічний гастрит, ентерит). Це посилює первинні порушення секреції або всмоктування гастромукопротеїну, а отже, посилює дефіцит вітаміну Вр ("зачароване коло").
У результаті недостатності другої коферментної форми вітаміну В — 5-дезоксіа-денозилкобаламіну в організмі накопичуються пропіонова і метилмалонова кислоти, токсичні для нервових клітин. Крім того, у нервових волокнах синтезуються жирові кислоти зі зміненою структурою, що призводить до порушення утворення мієліну і ушкодження аксонів. Розвивається дегенерація задніх і бічних стовпів спинного мозку (фунікулярний мієлоз), уражаються черепні й периферичні нерви.
26.1.51. Дайте характеристику картини периферичної крові і червоного кісткового мозку при В 12-фолієводефіцитній анемії.
Найхарактернішою рисою цієї анемії є поява в крові й червоному кістковому мозку клітин патологічної регенерації — мегалобластів та їхніх без'ядерних форм-мегалоцитів.
На тлі істотного зменшення вмісту еритроцитів і концентрації гемоглобіну колірний показник збільшений, що пояснюється більшим діаметром еритроцитів. Характерне явище дегенерації еритроцитів: анізоцитоз (макроцитоз), пойкілоцитоз (поява клітин овальної форми), патологічні включення (тільця Жоллі, тільця Кебота).
У крові зменшений вміст гранулоцитів (особливо нейтрофілів) і тромбоцитів. Виявляються гігантські нейтрофіли з гіперсегментованими ядрами.
26.1.52. Визначте місце В 12-фолієводефіцитної анемії в різних класифікаціях анемій.
|
Класифікація |
В^-фолієводефіцитна анемія |
І. |
За патогенезом |
Анемія, пов'язана з порушеннями еритропоезу |
II. |
За етіологією |
Набута, є спадкові форми |
III. |
За регенераторною здатністю червоного кісткового мозку |
Гіпорегенераторна |
IV. |
За колірним показником |
Гіперхромна |
V. |
За типом кровотворення |
3 мегалобластичним типом кровотворення |
VI. |
За клінічним перебігом |
Хронічна |
26.1.53. Якими синдромами виявляє себе В 12-фолієводефіцитна анемія?
При В -фолієводефіцитній анемії спостерігають:
1. Гематологічний синдром:
а) анемія і пов'язана з нею гіпоксія;
б) лейкопенія (нейтропенія), що призводить до зниження резистентності організму до інфекцій;
в) тромбоцитопенія, що викликає розвиток геморагічного синдрому.
2. Ураження травного каналу, що виявляються розвитком запально-атрофічних змін у слизовій оболонці: глосит, стоматит, езофагіт, ахілічний гастрит, ентерит.
3. Ураження центральної й периферичної нервової системи: фунікулярний мієлоз, дегенерація периферичних нервів.
26.1.54. У чому полягає фізіологічне значення заліза?
В організмі дорослої людини масою 70 кг міститься 4,5 г заліза. Воно в гемовій і негемовій формах входить до складу цілого ряду білків.
I. Білки, які містять залізо в гемовій формі:
а) гемоглобін;
б) міоглобін;
в) цитохроми;
г) каталаза;
ґ) лактоферин.
II. Білки, які містять залізо в негемовій формі:
а) феритин;
б) гемосидерин;
в) трансферин;
г) ферменти: аконітаза, ксантиноксидаза, НАДН-дегідрогеназа.
26.1.55. Як відбувається обмін заліза в організмі?
Залізо надходить в організм у складі харчових продуктів (м'ясо, печінка, риба, рис, горох, курага). Найкраще всмоктується залізо, що входить до складу харчових білків у формі гема. Стінки кишок містять фермент гемоксигеназу, що розщеплює гем харчових продуктів на білірубін, оксид вуглецю (II) та іони заліза, останні і всмоктуються в тонкій кишці. Цьому процесу сприяють аскорбінова кислота, фруктоза, піровиноградна кислота. Системи транспорту заліза в кишках можуть забезпечити всмоктування максимум 2,5 мг заліза за добу, незалежно від потреби організму в цьому елементі.
Транспорт заліза в організмі здійснюється за допомогою білка трансферину, а депонування відбувається у формі іншого білка - феритину. Надходячи в клітини, залізо зв'язується з внутрішньоклітинним білком сидерохіліном, що транспортує залізо в мітохондрії, де синтезується гем.
Фізіологічні втрати заліза невеликі. У чоловіків вони становлять менш ніж 1 мг/ добу (втрати із сечею, потом, злущеним епітелієм шкіри). У жінок вони набагато більші і обумовлені менструаціями, вагітністю, пологами, лактацією.
26.1.56. Назвіть можливі причини розвитку залізодефіцитної анемії.
1. Недостатнє надходження заліза в організм:
а) аліментарна анемія в грудних дітей (вигодовування коров'ячим або козячим молоком);
б) порушення всмоктування заліза (резекція шлунка, кишок, гастрити, ентерити).
2. Крововтрати. Це найпоширеніша причина дефіциту заліза в організмі. Найчастіше буває при невеликих, але повторних кровотечах (хронічна постгеморагічна анемія).
3. Посилене використання заліза - вагітність, лактація.
26.1.57. Який патогенез порушень, що розвиваються в організмі у зв 'язку з дефіцитом заліза ?
Недостатність заліза в організмі призводить до порушення синтезу залізовмісних білків, а отже, до розладів функцій, у виконанні яких беруть участь ці білки. Найбільше значення мають такі лінії патогенезу:
1) дефіцит заліза → порушення синтезу гема і гемоглобіну → анемія;
2) дефіцит заліза → порушення синтезу гема → порушення утворення цитохро-мів → розлади клітинного дихання (порушення утилізації кисню) → тканинна гіпоксія;
3) дефіцит заліза → порушення синтезу гема → зменшення активності каталази → порушення функції антиоксидантних систем → активація вільнорадикального окиснення → ушкодження клітин → гемоліз еритроцитів і розвиток дистрофічних змін у клітинах;
4) дефіцит заліза → порушення синтезу гема → зменшення синтезу міоглобіну → зниження пристосувальних можливостей клітин щодо гіпоксії.
26.1.58. Дайте характеристику картини периферичної крові і червоного кісткового мозку при залізодефіцитній анемії.
Для залізодефіцитної анемії характерне зниження концентрації гемоглобіну в периферичній крові і зменшення колірного показника, що свідчить про зменшення насичення кожного окремого еритроцита гемоглобіном. Кількість еритроцитів в одиниці об'єму крові при цьому або трохи зменшується, або залишається без змін.
При біохімічних дослідженнях виявляється зниження вмісту заліза в сироватці крові, а також ступеня насичення залізом трансферину.
У мазку крові зменшується кількість регенераторних форм еритроцитів (ретику-лоцитів, поліхроматофілів) і з'являються дегенеративні форми. Характерні гіпохромія (з'являються анулоцити), анізоцитоз (мікроцитоз), пойкілоцитоз.
У червоному кістковому мозку зменшується вміст сидеробластів і сидероцитів (еритробластів і нормоцитів, що містять гранули заліза), частка яких у нормі становить 20-40 %. У той же час збільшується вміст базофільних і поліхроматофільних форм клітин еритроїдного ряду при одночасному зменшенні оксифільних. Зазначений феномен отримав назву "синього кісткового мозку ".
26.1.59. Визначте місце залізодефіцитної анемії в різних класифікаціях анемій.
|
Класифікація |
Залізодефіцитна анемія |
І. |
За патогенезом |
Анемія, пов'язана з порушеннями еритропоезу |
II. |
За етіологією |
Набута |
III. |
За регенераторною здатністю червоного кісткового мозку |
Гіпорегенераторна |
IV. |
За колірним показником |
Гіпохромна |
V. |
За типом кровотворення |
3 еритробластичним типом кровотворення |
VI. |
За клінічним перебігом |
Хронічна |
26.1.60. Якими синдромами виявляє себе залізодефіцитна анемія?
1. Гематологічний синдром. Охоплює порушення з боку периферичної крові і червоного кісткового мозку.
2. Гіпоксія. Виявляється загальною слабкістю, запамороченнями, серцебиттям, задишкою, непритомностями. Кисневе голодування в умовах дефіциту заліза має два механізми: кров'яний (зменшення кисневої ємності крові) і тканинний (порушення клітинного дихання й утилізації кисню).
3. Синдром трофічних порушень. Виявляється такими ознаками, як сухість і тріщини шкіри, тріщини в кутах рота, ангулярний стоматит, ураження нігтів (зміни форми, стоншення), атрофія сосочків язика (атрофічний глосит), атрофічний гастрит. Вважають, що розвиток зазначених порушень, з одного боку, пов'язаний з гіпоксичним і вільнорадикальним ушкодженням клітин, з другого — з розладами вторинних метаболічних шляхів, у здійсненні яких беруть участь ферменти, що містять залізо.
4. Сидеропенічний синдром. Це специфічний синдром, що виникає при дефіциті заліза. Він виявляється спотвореннями смаку й нюху. Хворі часто їдять крейду, зубний порошок, вугілля, глину, пісок, лід, сирі крупи, тісто, сирий м'ясний фарш. Мають пристрасть до запахів гасу, бензину, ацетону, вихлопних газів автомобілів. Патогенез зазначених порушень досі невідомий.
5. Синдром м'язової слабкості. М'язова слабість, що виникає при дефіциті заліза, завжди більш значна, ніж та, котрої варто було б очікувати, виходячи зі ступеня анемії. Вона виявляється слабкістю і стомлюваністю скелетних м'язів, слабкістю міокарда (міокардіопатія), порушеннями ковтання (дисфагія), порушеннями сечовипускання. Очевидно, що важливе значення в розвитку зазначених симптомів мають гіпоксія (кров'яна і тканинна), а також зменшення вмісту міоглобіну в м'язовій тканині.
26.1.61. Що таке залізорефрактерна анемія? Які її етіологія й патогенез?
Залізорефрактерна анемія-цс анемія, що виникає в результаті порушення включення заліза в гем при зниженні активності ферментів, що каталізують синтез порфіринів і гема.
Причинами розвитку цієї анемії можуть бути:
1) генетично обумовлене зниження активності декарбоксилази копропорфірииоге-ну - ферменту, що забезпечує один з кінцевих етапів синтезу гема (успадковується рецесивно, зчеплено з Х-хромосомою);
2) зменшення вмісту піридоксальфосфату - активної форми вітаміну В6, унаслідок чого залізо не вилучається з мітохондрій еритробластів і не включається в гем;
3) блокада свинцем сульфгідрильних груп ферментів, що беруть участь у синтезі гема, при побутовому й виробничому отруєнні свинцем.
Зменшення активності ферментів, що беруть участь в утворенні порфіринів і гема, призводить до зниження утилізації заліза й порушення синтезу гема гемоглобіну, що викликає недостатність еритропоезу і розвиток гіпохромної анемії з низьким вмістом гемоглобіну.
Порушення використання заліза супроводжується підвищенням вмісту сироваткового заліза, відкладенням його у внутрішніх органах з вторинним розростанням сполучної тканини (гемосидероз).
26.2. Порушення системи лейкоцитів
26.2.1. Дайте загальну характеристику порушень системи лейкоцитів.
Патологічні зміни лейкоцитів виявляються порушеннями їх утворення в кровотворній тканині, а також кількісними та якісними змінами лейкоцитів крові. Такі порушення можуть бути наслідком первинного ураоїсення клітин гранулоцитарного, лімфоцитарного і моноцитарного рядів під впливом різноманітних патогенних факторів і виявляти себе розладами утворення, дозрівання або ж руйнування лейкоцитів
у кровотворній тканині і кровоносному руслі. Вторинні зміни лейкоцитів виникають як захисна реакція організму у відповідь на патологічні процеси, що відбуваються не [ в самій системі крові, а в органах і тканинах інших систем.
Головною ланкою в патогенезі порушень лейкоцитарного паростка крові є зміна реактивності організму, у тому числі імунологічної й алергічної. Це пояснюється розладами основних функції, що їх виконують лейкоцити, беручи участь у процесах фагоцитозу, антитілоутворення, інактивації біологічно активних речовин (гістаміну, брадикініну, серотоніну). Патологічні зміни лейкоцитів можуть супроводжуватися трофічними порушеннями тканин, місцевими мікроциркуляторними розладами. Це обумовлено тим, що одна з функцій лейкоцитів полягає в постачанні тканинам, що регенерують, поживних речовин і стимуляторів поділу клітин. Гранулоцити беруть участь у розвитку судинних порушень як переносники вазоактивних речовин (базофільні, еозинофільні лейкоцити) або ж впливають на їхній синтез і вивільнення з тканинних базофілів (нейтрофільні лейкоцити).
26.2.2. Як розподіляються лейкоцити в організмі?
Лейкоцити містяться в організмі в трьох "відсіках": червоний кістковий мозок, периферична кров, периферичні тканини. І. Червоний кістковий мозок. Тут лейкоцити утворюють 4 пули (рис. 106):
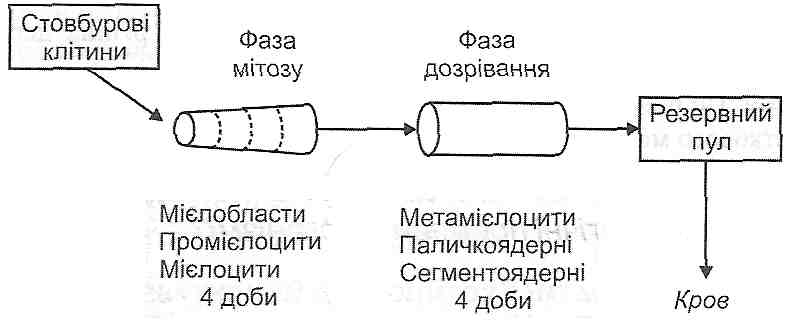
Рис. 106. Emanu лейкопоезу
1) пул стовбурових клітин, що перебувають у стані спокою. Він дуже малий і являє собою резерв кровотворення;
2) мітотичний пул. Це клітини, що перебувають у стані мітотичного поділу. Від стовбурової клітини до утворення клітин, що дозрівають, звичайно проходить від 4 до 11 поділів;
3) пул клітин, що дозрівають. Містить клітини, дозрівання яких триває в середньому 3-5 діб;
4) резервний пул. Складається зі зрілих лейкоцитів, які можуть переходити в кров.
II. Периферична кров:
1) пул циркулюючих лейкоцитів (близько 50 %);
2) пристінковий (маргінальний) пул (близько 50 %).
III. Периферичні тканини:
1) мігруючі лейкоцити,
2) лейкоцити в стані спокою.
Підраховано, що загальна маса лейкоцитів периферичної крові становить близько 10ц червоного кісткового мозку - 500 г, периферичних тканин - 600 г.
26.2.3. Які показники використовують для характеристики стану системи лейкоцитів?
1. Вміст лейкоцитів в одиниці об'єму крові. У нормі цей показник становить від 4 -109/л до 9 -109/л. Збільшення вмісту лейкоцитів у крові отримало назву лейкоцитозу, зменшення - лейкопенії.
2. Лейкоцитарна формула. Це кількісне співвідношення різних форм лейкоцитів у периферичній крові.
Для норми характерні такі значення: базофіли — 0—1 %, еозинофіли - 2-4 %, мієлоцити - 0 %, метамієлоцити (юні нейтрофіли) - 0-1 %, паличкоядерні нейтрофіли - 3-5 %, сегментоядерні нейтрофіли 50-70 %, лімфоцити- 20-35 %, моноцити - 2-8 %.
3. Абсолютний вміст кожної форми лейкоцитів в одиниці об'єму крові. Цей показник розраховують, виходячи із загального вмісту лейкоцитів і лейкоцитарної формули.
4. Якісні характеристики лейкоцитів. їх визначають на підставі вивчення мазків периферичної крові. В умовах патології можлива поява різних дегенеративних форм лейкоцитів.
5. Мієлограма. Це показник, що характеризує кількість і якісний склад клітин чер-| воного кісткового мозку.
26.2.4. Якими кількісними і якісними змінами лейкоцитів можуть виявляти себе патологічЩ'процеси в організмі?
1. Лейкоцитоз.
2. Лейкопенія.
3. Порушення співвідношення зрілих і незрілих форм лейкоцитів (зміщення лейкоцитарної формули).
4. Дегенеративні зміни лейкоцитів.
26.2.5. Що таке лейкоцитоз? Як класифікують лейкоцитози?
Лейкоцитоз - це збільшення кількості лейкоцитів в одиниці об'єму крові понад) 10 • 109/л. Лейкоцитоз не має самостійного значення, він є всього лише симптомом, що супроводжує розвиток багатьох хвороб.
Класифікація лейкоцитозів.
I. Залежно від причин розвитку виділяють фізіологічний і патологічний лейко-1 цитози.
II. Лейкоцитоз може бути абсолютним і відносним. Для абсолютного лейкоцитозу) характерне збільшення абсолютної кількості лейкоцитів в одиниці об'єму крові. Про відносний лейкоцитоз ідеться в тому випадку, коли зростає відносний вміси окремих форм лейкоцитів у периферичній крові.
III. За механізмом розвитку лейкоцитоз буває:
а) реактивним;
б) перерозподільним;
в) пухлинного походження.
IV. Залежно від видів лейкоцитів, вміст яких у крові збільшений, виділяють:
а) нейтрофільний лейкоцитоз (нейтрофільоз);
б) еозинофільний лейкоцитоз (еозинофілія);
в) базофільний лейкоцитоз;
г) лімфоцитарний лейкоцитоз (лімфоцитоз); ґ) моноцитарний лейкоцитоз (моноцитоз).
26.2.6. Наведіть приклади фізіологічного і патологічного лейкоцитозів.
Фізіологічний лейкоцитоз є фізіологічною реакцією організму на ті чи ті впливи. До його різновидів відносять:
а) емоціогенний лейкоцитоз — виникає під час сильних емоцій;
б) міогенний — розвивається під час інтенсивної фізичної роботи;
в) статичний — характерний для переходу людини з горизонтального у вертикальне положення;
г) аліментарний — розвивається після приймання їжі; ґ) лейкоцитоз у вагітних;
д) лейкоцитоз у новонароджених.
Патологічний лейкоцитоз пов'язаний з перебігом в організмі патологічних процесів. Він, як правило, розвивається при:
а) інфекційних захворюваннях;
б) запальних і алергічних процесах;
в) інтоксикації екзо- й ендогенного походження.
26.2.7. Що таке реактивний лейкоцитоз? Які механізми можуть лежати в основі його розвитку?
Реактивним називають лейкоцитоз, що виникає як реакція червоного кісткового мозку на патогенні впливи. Він закономірно розвивається при інфекційних захворюваннях, запаленні, дії низьких доз токсичних речовин.
В основі розвитку реактивного лейкоцитозу можуть лежати два механізми. І. Посилення проліферації і дозрівання лейкоцитів у червоному кістковому мозку. Це може бути пов'язано або зі збільшенням утворення в організмі лейкопоети-нів - речовин, що стимулюють утворення лейкоцитів, або зі зменшенням вмісту інгібіторів лейкопоезу. Серед лейкопоетинів найбільш вивченим нині є колонієс-тимулятивний фактор — речовина, що належить до групи цитокінів. Вона, будучи продуктом секреції активованими макрофагами, стимулює утворення гранулоцитів у червоному кістковому мозку.
На роль інгібіторів лейкопоезу претендують високомолекулярний інгібітор сироватки крові (ліпопротеїн), кейлони і лактоферин.
II. Збільшення переходу резервних лейкоцитів із червоного кісткового мозку в кров. Цьому сприяють інтерлейкін-1 і бактеріальні ендотоксини, що підвищують проникність стінки кровоносних судин червоного кісткового мозку.
26.2.8. Які особливості характерні для перерозподільного лейкоцитозу?
Перерозподільном називають лейкоцитоз, що виникає в результаті переходу лейкоцитів із пристінкового пулу в циркулюючий. Його особливостями є:
а) короткочасний характер зі швидким поверненням кількості лейкоцитів до норми після завершення дії причини;
б) збереження нормального кількісного співвідношення різних видів лейкоцитів (лейкоцитарна формула не міняється);
в) відсутність дегенеративних змін лейкоцитів. Більшість форм фізіологічного лейкоцитозу за механізмом свого розвитку є перерозподільними.
26.2.9. Наведіть приклади нейтрофільного, еозинофільного, базофільного, лімфоцитарного і моноцитарного лейкоцитозів.
Нейтрофільнш лейкоцитоз характерний для:
а) гнійно-запальних процесів, спричинених гноєтворними бактеріями (абсцеси, флегмони, сепсис);
б) важкого кисневого голодування (велика гостра крововтрата, гострий гемоліз);
в) ендогенної інтоксикації (уремія).
Еозинофільний лейкоцитоз розвивається при:
а) алергічних реакціях І типу за класифікацією Кумбса і Джелла;
б) гельмінтозах;
в) хронічному мієлолейкозі.
Базофільний лейкоцитоз буває дуже рідко. Він може супроводжувати розвиток:
а) хронічного мієлолейкозу;
б) гемофілії;
в) хвороби Вакеза (поліцитемії).
Лімфоцитарний лейкоцитоз часто буває при:
а) гострих інфекційних захворюваннях (кашлюк, вірусний гепатит);
б) деяких хронічних інфекційних хворобах (туберкульоз, сифіліс, бруцельоз);
в) хронічному лімфолейкозі.
Моноцитарний лейкоцитоз характерний для:
а) хронічних інфекцій (туберкульоз, бруцельоз);
б) інфекційного мононуклеозу;
в) інфекцій, збудниками яких є рикетсії та найпростіші (висипний тиф, малярія).
26.2.10. Що таке лейкопенія? Як класифікують лейкопенії?
Лейкопенія — це зменшення кількості лейкоцитів у периферичній крові нижче 4-109/л. Лейкопенія часто виступає симптомом якого-небудь захворювання. Однак є нозологічні одиниці, при яких саме лейкопенія є основним проявом хвороби, вона по суті визначає картину недуги та всі інші симптоми.
Класифікація лейкопепій.
І. За походженням лейкопенії бувають набутими і спадково обумовленими.
Набуті лейкопенії можуть виникати під впливом фізичних (іонізуюча радіація), хімічних (бензол, цитостатики, лікарські препарати), біологічних (віруси гепатиту, інфекційного мононуклеозу) та імунних факторів.
Прикладами спадкових лейкопеній є нейтропенія Костмана, спадкова нейтропе-нія аутосомно-домінантного типу, синдром "лінивих лейкоцитів", циклічна нейтропенія.
її. За видом лейкоцитів, кількість яких зменшена, виділяють:
а) нейтропенію;
б) лімфопенію;
в) еозинопенію.
III. За патогенезом розрізняють:
а) лейкопенії, обумовлені порушенням надходження лейкоцитів із червоного кісткового мозку в кров;
б) лейкопенії, пов'язані зі скороченням часу перебування лейкоцитів у периферичній крові;
в) перерозподільні лейкопенії.
IV. Виділяють цілий ряд клініко-гематологічних синдромів, для яких лейкопенія є провідною ознакою. Серед них агранулоцитоз, гіпопластична анемія, геморагічна алейкія.
26.2.11. Які механізми можуть лежати в основі розвитку лейкопеній, пов'язаних з порушеннями надходження лейкоцитів із червоного кісткового мозку в кров?
1. Ушкодження кровотворних клітин. При цьому розвивається так званамієлотоксична
лейкопенія. Виділяють три основних механізми ушкодження кровотворних клітин:
а) цитолітичний. Пов'язаний з дією на клітини іонізуючої радіації, цитоста-тичних препаратів, імунних факторів (антитіл, Т-лімфоцитів) та ін. Ступінь ураження червоного кісткового мозку при цьому залежить від дози й тривалості дії зазначених факторів;
б) антиметаболічний. У його основі лежить дія агентів, які втручаються в обмін пуринових і піримідинових основ, порушуючи при цьому процеси поділу стовбурових клітин. За таким принципом діють деякі протипухлинні препарати й антибіотики (левоміцетин).
в) ідіосинкразичний. Реалізується при повторному введенні лікарських препаратів, чутливість організму до яких підвищена (ідіосинкразія). Найчастіше це препарати, що містять у своїй структурі бензольні кільця. У випадку ідіосинкразії немає зв'язку між імовірністю розвитку лейкопенії і дозою, а також тривалістю дії лікарських засобів.
2. Порушення мітозу - неефективний лейкопоез. Найчастіше його причиною є:
а) дефіцит необхідних для клітинного поділу речовин, зокрема вітаміну В12 і фолієвої кислоти;
б) порушення регуляції мітозу — дефіцит лейкопоетинів.
3. Порушення дозрівання лейкоцитів. їхню основу можуть складати генетично обумовлені дефекти як самих кровотворних клітин (наприклад, нейтропенія Костмана), так і клітин "мікрооточення" (наприклад, лейкопенія у "сталевих" мишей лінії SL/SI/). При цьому дозрівання клітин крові досягає певної стадії (наприклад, промієлоцитів) і зупиняється.
4= Порушення виходу лейкоцитів із червоного кісткового мозку в кров. Подібні порушення часто пов'язані з генетичними дефектами лейкоцитів, що порушують основні їхні функції й властивості (наприклад, рухливість). Прикладами можуть бути синдром "лінивих лейкоцитів ", нейтропенія "єменських євреїв ".
5. Зменшення плацдарму лейкопоезу. Має місце при заміщенні кровотворної тканини лейкозними клітинами, метастазами пухлин та ін.
26.2.12. Які механізми можуть лежати в основі розвитку лейкопеній, пов 'язаних зі скороченням часу перебування лейкоцитів у
периферичній крові?
1. Деструкція (руйнування) лейкоцитів. Може бути обумовлена:
а) аутоімунними механізмами (ревматоїдний артрит, системний червоний вовчак);
б) гаптеновими механізмами (амідопіринова гостра нейтропенія);
в) гіперспленізмом (підвищенням фагоцитарної активності макрофагів селезінки).
2. Посилене використання лейкоцитів. Цьому передує прискорений вихід лейкоцитів із крові в тканини в умовах хронічного рецидивуючого запалення.
3. Посилене виведення лейкоцитів з організму. Виражена хронічна втрата нейтрофілів спостерігається у курців: під час ранкового кашлю з мокротинням втрачається від 0,5 до 2-Ю8 гранулоцитів і 0,8 - 1,6-108 макрофагів.
26.2.13. Що таке агранулоцитоз?
Агранулоцитоз - це клініко-гематологічний синдром, що характеризується різким зменшенням вмісту нейтрофілів нижче 0,75-109/л при зменшенні загальної кількості лейкоцитів нижче 1'109/л.
В основі розвитку агранулоцитозу можуть лежати два механізми:
а) мієлотоксичний — ураження червоного кісткового мозку;
б) імунний — руйнування клітин гранулоцитарного ряду антилейкоцитарними антитілами.
Агранулоцитоз супроводжується значним ослабленням реактивності організму у зв'язку з порушенням захисної функції лейкоцитів.
26.2.14. Що таке зміщення лейкоцитарної формули?
Зміщення лейкоцитарної формули (ядерне зміщення) - це порушення співвідношення між незрілими і зрілими формами нейтрофілів.
При дослідженні лейкограми встановлюють наявність ядерного зміщення ней-трофільних гранулоцитів уліво або вправо. Ця термінологія пов'язана з розташу-
ванням незрілих нейтрофільних гранулоцитів (мієлоцитів, метамієлоцитів, палич-коядерних нейтрофілів) у лівій частині лейкоцитарної формули (формули Арнета й Шилінга), тимчасом як зрілі, сегментоядерні нейтрофіли умовно поставлені в крайнє праве положенні. Збільшення вмісту в крові молодих форм нейтрофільних гранулоцитів свідчить про ядерне зміщення вліво, переважання зрілих нейтрофілів з великою кількістю сегментів (5-6) на тлі зникнення більш молодих клітин - про ядерне зміщення вправо.
26.2.15. Які виділяють різновиди зміщення лейкоцитарної формули вліво?
Розрізняють такі різновиди ядерного зміщення вліво.
1. Регенеративне зміщення є показником реактивної активації гранулоцитопоезу (на тлі помірного загального лейкоцитозу підвищений вміст паличкоядерних нейтрофілів та метамієлоцитів, можуть зустрічатися поодинокі мієлоцити).
2. Гіперрегенеративне зміщення відображає надмірну гіперплазію лейкопоетичної тканини з порушенням дозрівання клітин і вираженим омолодженням складу крові. При цьому різко зростає число паличкоядерних гранулоцитів та метамієлоцитів, з'являються мієлоцити, промієлоцити; загальна кількість лейкоцитів може бути збільшена, незмінена і навіть знижена внаслідок того, що розвивається виснаження мієлоїдного паростка після попередньої активізації.
3. Дегенеративне зміщення свідчить про пригнічення й глибокі порушення лейко-поезу, коли на тлі загальної лейкопенії в лейкограмі збільшується число паличкоядерних нейтрофільних гранулоцитів з дегенеративними змінами в їхній цитоплазмі та ядрі при зменшенні кількості сегментоядерних форм і відсутності метамієлоцитів.
4. Регенеративно-дегенеративне зміщення спостерігається при гіперпродукції в червоному кістковому мозку патологічно змінених лейкоцитів і порушенні їх дозрівання. У цьому випадку виявляють лейкоцитоз, а в мазку крові зростає число паличкоядерних гранулоцитів, метамієлоцитів та мієлоцитів з ознаками дегенерації.
26.2.16. Які дегенеративні зміни можуть бути характерні для лейкоцитів в умовах патології?
Дегенеративні зміни лейкоцитів можуть виявляти себе анізоцитозом, наявністю в цитоплазмі вакуоль, токсигенної зернистості, включень типу тілець Князько-ва—Деле (базофільно забарвлені грудочки цитоплазми); великою азурофільною зернистістю; зникненням звичайної зернистості, пікнозом або набряканням ядра, його гіпер- і гіпосегментацією, а також невідповідністю ступеня дозрівання ядра й цитоплазми, каріорексисом, цитолізом.
Дегенеративні зміни найчастіше спостерігають у нейтрофільних гранулоцитах і моноцитах. Причиною їх виникнення є утворення лейкоцитів з порушеним обміном речовин, що й обумовлює структурні аномалії (при лейкозі, спадковій ензимопатії), а також ушкодження лейкоцитів у кровотворних органах і крові під впливом різноманітних патогенних факторів (бактерій, вірусів, антитіл).
26.2.17. Що таке гемобластози і лейкози?
Гемобластози — це пухлинні захворювання системи крові. Інакше кажучи, це злоякісні пухлини, які ростуть із кровотворних клітин.
Залежно від того, чи уражується первинно червоний кістковий мозок, гемобластози поділяють на дві великі групи: лейкози і гемобластози, що мають первинну локалізацію поза червоним кістковим мозком. Найчастішим місцем локалізації останніх є лімфатичні вузли.
Лейкози - це злоякісні пухлини, що виникають із кровотворних клітин і первинно уражують червоний кістковий мозок.
26.2.18. Які існують докази того, що лейкози - це захворювання пухлинної природи?
1. При лейкозах, як і при злоякісних пухлинах, відбувається безмежний і нерегульо-ваний поділ клітин.
2. Лейкоз, як і будь-яка злоякісна пухлина, виникає з одної-єдиної первинно зміненої, трансформованої клітини. Усі лейкозні клітини, якими б різними вони не були, походять із однієї клітини.
3. Для лейкозів, як і для інших злоякісних пухлин, характерне явище анаплазії— поява морфологічних, біохімічних та інших властивостей, які наближають лейкозні клітини до ембріональних.
4. Для лейкозів, як і для інших злоякісних пухлин, характерна пухлинна прогресія, тобто набуття із часом лейкозними клітинами все більш і більш злоякісних властивостей.
5. Однаковість причин розвитку лейкозів і злоякісних пухлин. Одні й ті ж етіологічні фактори (фізичні, хімічні, біологічні) здатні викликати виникнення як лейкозів, так і інших злоякісних пухлин.
26.2.19. Чим лейкози відрізняються від інших злоякісних пухлин?
1. У випадку лейкозу неможливо встановити первинну локалізацію пухлини, а отже, радикально видалити її вже на ранніх етапах розвитку.
2. Лейкози - це пухлини, які з самого початку метастазують. Лейкозні клітини легко проникають у кров і розносяться нею по всьому організмі. Вони заселяють все нові й нові ділянки, на яких виникають вторинні лейкозні інфільтрати.
3. Для лейкозів характерна системність ураження. У зв'язку з раннім метастазуванням уражується вся система крові: червоний кістковий мозок, лімфатичні вузли, селезінка, печінка.
4. При лейкозах пригнічується нормальне кровотворення. Це пов'язано з витісненням нормальної кровотворної тканини лейкозними клітинами. З другого боку, має значення токсична дія продуктів лейкозних клітин на клітини гемопоезу.
5. Лейкози — це різновид пухлин, що вражають людину переважно в дитячому і молодому віці.
26.2.20. На які класи поділяють кровотворні клітини?
I клас — стовбурові поліпотентні клітини. Ці клітини дають початок усім клітинам крові.
II клас — частково детерміновані поліпотентні клітжи-попередники. Він складається з двох груп клітин: клітин-попередників лімфоцитарного ряду і клітин-по-передників мієлопоезу.
III клас — уніпотентні клітини-попередники. Клітини цього класу дають початок певному паростку крові. До них відносять клітини-попередники В-лімфоцитів, клітини-попередники Т-лімфоцитів, еритропоетинчутливі клітини, що утворюють колонії.
Клітини перших трьох класів неможливо розрізнити за допомогою відомих нині морфологічних і цитохімічних методів дослідження, тому їх об'єднують під загальною назвою - клітини, яких неможна морфологічно диференціювати.
IV клас — клітини, що проліферують. Клітини цього класу мають специфічні морфологічні й цитохімічні ознаки. До них належать бласти (плазмобласти, лімфо-бласти, монобласти, еритробласти, мегакаріобласти), проклітини (проплазмоцити, пролімфоцити, промієлоцити, пронормоцити, промегакаріоцити) і так звані цитні форми (мієлоцити, нормоцити, мегакаріоцити).
Уклас — клітини, що дозрівають. Ними є метамієлоцити, паличкоядерні гранулоцити, ретикулоцити. Загальна властивість цих клітин - втрата ними здатності до поділу.
VI клас — зрілі клітини крові.
26.2.21. Які кровотворні клітини можуть бути джерелом лейкозу?
Будь-яка пухлина може розвиватися тільки з тих клітин, що мають первісну здатність до поділу. Не є винятком і лейкози.
Джерелом лейкозів можуть бути клітини І—IV класів, тобто клітини, здатні до проліферації. Клітини V і VI класів (ті, що дозрівають, і зрілі) трансформуватися в лейкозні не можуть, оскільки втратили структури, необхідні для здійснення клітинного поділу.
26.2.22. Як класифікують лейкози?
I. Залежно від особливостей патогенезу і пов'язаної з ними гематологічної картини лейкози поділяють на гострі й хронічні.
II. Залежно від того, які кровотворні клітини втягуються в пухлинний процес, лейкози поділяють на лімфолейкози (уражується лімфоцитарний паросток), мієло-лейкози (уражується гранулоцитарний паросток), еритромієлози та ін.
III. Залежно від вмісту лейкоцитів у периферичній крові лейкози бувають лейкеміч-ними (виражений лейкоцитоз — від 20— 109/л до 100-109/л), сублейкемічними (помірний лейкоцитоз до 20-109/л), алейкемічними (вміст лейкоцитів не міняється), лейкопенічними (кількість лейкоцитів зменшується).
26.2.23. У чому полягає основна патогенетична відмінність між гострими і хронічними лейкозами?
Основною рисою патогенезу гострих лейкозів є те, що лейкозні клітини, набувши здатності до безмежного неконтрольованого росту, повністю втратили здатність дозрівати, тобто диференціюватися в наступні форми.
У той же час при хронічних лейкозах лейкозні клітини поряд зі здатністю до безмежного росту зберігають властивість дозрівати й давати наступні форми.
Таким чином, при гострих лейкозах пухлинні клітини тільки діляться і не дозрівають, при хронічних — діляться і дозрівають. З урахуванням цієї обставини гострі лейкози варто вважати більш злоякісним видом захворювання.
26.2.24. Які існують види гострих лейкозів?
Джерелом гострих лейкозів можуть бути кровотворні клітини перших чотирьох класів.
Якщо лейкоз розвивається з клітин І—III класів, що не мають специфічних морфологічних і цитохімічних ознак, то такий лейкоз називають недиферещійованим.
Якщо лейкоз розвивається з клітин IV класу, то за допомогою морфологічних і цитохімічних методів можна встановити клітину, від якої походить пухлина. Для цього використовують сім цитохімічних реакцій: 1) реакцію на пероксидазу; 2) реакцію із Суданом чорним (на ліпіди); 3) реакцію на кислу фосфатазу; 4) PAS-реакцію (на глікоген); 5) реакцію на а-нафтилацетатестеразу; 6) реакцію на хлорацетатестеразу; 7) реакцію на сульфатовані глікозаміноглікани.
Якщо джерелом лейкозних клітин є лімфобласт, то такий лейкоз називається гострим лімфобластним, якщо мієлобласт — гострим мієлобластним, якщо моно-бласт — гострим монобластним і т. д.
26.2.25. Дайте характеристику картини крові при гострих лейкозах.
Для гострих лейкозів характерні алейкемічний і лейкопенічний варіанти перебігу.
Як приклад розглянемо два види гострих лейкозів, що найчастіше зустрічаються: гострий мієлобластний і гострий лімфобластний.
Гострий мієлобластний лейкоз розвивається переважно у молодих людей і людей середнього віку. Назва лейкозу свідчить про те, що його джерелом є мієлобласт і що лейкозні клітини (мієлобласти) не здатні диференціюватися. Це визначає такі особливості картини крові (рис. 107; див. форзац).
1. Лейкозні мієлобласти постійно діляться і легко надходять у кров. Отже, у крові будуть виявлятися пухлинні клітини -мієлобласти.
2. Оскільки в червоному кістковому мозку зберігаються осередки нормального кровотворення, то вони будуть джерелом надходження у кров нормальних лейкоцитів, тобто тих, які мають бути в крові у нормі — метамієлоцитів, паличкоядерних і сегментоядерних нейтрофілів.
3. У крові відсутні перехідні форми лейкоцитів від мієлобластів до тих клітин, які виявляються в крові у нормі, тобто немає промієлоцитів~і мієлоцитів. Таке явище отримало назву лейкемічного провалу (hiatus leucemicus). Відсутність у крові пере-
хідних форм пояснюється тим, що лейкозні мієлобласти не здатні диференціюватися в зазначені клітини, а з другого боку, осередки нормального кровотворення не "випускають" у кров промієлоцитів і мієлоцитів (рис. 108).
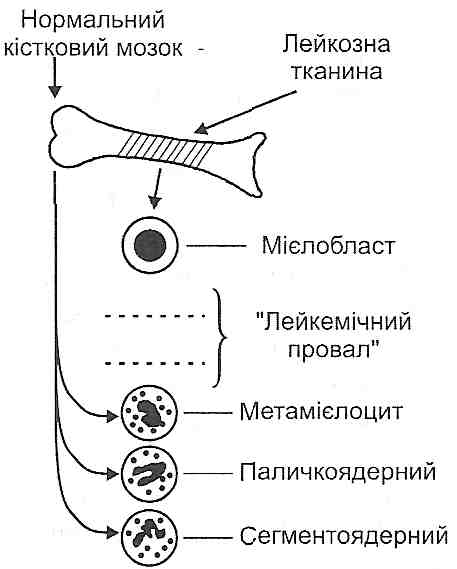
Рис. 108. Походження
"лейкемічного провалу" при
гострому мієлобластному лейкозі
Гострий лімфооластнии лейкоз. Є типовим лейкозом дитячого віку. Лейкоз розвивається з лімфобласта. Пухлинні клітини втрачають здатність до дозрівання. Тому для картини крові будуть характерні лімфобласти (пухлинного походження), а також всі ті клітини, які мають бути в нормі (з осередків нормального кровотворення).
Картина червоного кісткового мозку при всіх гострих лейкозах має ряд особливостей. По-перше, у пунктаті кісткового мозку кількість бластних клітин значно зростає і перевищує 30 %. По-друге, лейкозні бластні клітини мають якісні відмінності, що є ознакою їх анаплазії.
26.2.26. Які існують види хронічних лейкозів?
Хронічні лейкози розвиваються з кровотворних клітин IV класу.
Номенклатура хронічних лейкозів ґрунтується на тому, який тип клітин становить переважну більшість у популяції лейкозних клітин. Назва лейкозу вказує на паросток кровотворення, що виявився ураженим.
Виділяють хронічний мієлоцитарний лейкоз (хронічний мієлолейкоз), хронічний лімфоцитарний лейкоз (хронічний лімфолейкоз), хронічний моноцитарний лейкоз, хронічний еритромієлоз. При цьому зазнають уражень відповідно гранулоцитарний, лімфоцитарний, моноцитарний та еритроїдний паростки крові.
26.2.27. Дайте характеристику картини крові при хронічних лейкозах.
Для хронічних лейкозів найчастіше характерні лейкемічний і сублейкемічний варіанти перебігу.
Розглянемо два види хронічних лейкозів, що найчастіше бувають: хронічний мієлоцитарний і хронічний лімфоцитарний.
Хронічний мієлоцитарний лейкоз. Найбільш імовірним джерелом розвитку цього лейкозу є мієлобласти (іноді можуть бути промієлоцити і мієлоцити). Оскільки лейкоз хронічний, то це означає, що лейкозні мієлобласти зберігають здатність до диференціювання у наступні форми. Тому з лейкозної тканини червоного кісткового мозку в кров у великій кількості виходять всі клітини, які походять від мієлобластів, а саме промієлоцити, мієлоцити, метамієлоцити, паличкоядерні І сегментоядернг гранулоцити.
У червоному кістковому мозку переважають клітинні елементи мієлоїдного ряду. Жирова тканина повністю витісняється кровотворною (рис. 109; див. форзац).
Хронічний лімфоцитарний лейкоз. Джерелом його розвитку елімфобласти, які зберегли здатність диференціюватися в наступні форми — пролімфоцити і лімфоцити, і
Тому основна маса лейкозних клітин крові представлена лімфоцитами. їхня 1 кількість у лейкоцитарній формулі становить 80-90 %. Крім лейкозних лімфоцитів, у крові можуть виявлятися пролімфоцити і поодинокі лімфобласти. Характерною є поява так званих тіней Гумпрехта - напівзруйнованих ядер лімфоцитів, що утворюються як артефакт при готуванні мазків крові.
Лімфоцити лейкозного клону (В-лімфоцити) можуть продукувати імуноглобу-ліни однієї специфічності (моноклональні), однак при цьому пригнічується антиті-лоутворення іншими нормальними клонами В-лімфоцитів і поступово розвивається імунологічна недостатність.
У червоному кістковому мозку відбувається майже тотальне заміщення кровотворної тканини лімфоцитами (рис. ПО; див. форзац).
26.2.28. Дайте порівняльну характеристику картини крові при гострому мієлобластному і хронічному мієлоцитарному лейкозах.
1. При гострому мієлобластному лейкозі пухлинні клітини представлено лейкозни-ми мієлобластами, у той час як при хронічному мієлолейкозі основну масу становлять похідні лейкозних мієлобластів: промієлоцити, мієлоцити, метамієлоци-ти, паличкоядерні і сегментоядерні клітини.
2. При гострому мієлобластному лейкозі виявляється феномен "лейкемічного провалу", у той час як при хронічному мієлолейкозі його немає - навпаки, у крові з'являються всі перехідні форми від мієлобласта до сегментоядерних гранулоцитів.
3. Хронічний мієлолейкоз на відміну від гострого мієлобластного характеризується гіперрегенеративним зміщенням лейкоцитарної формули вліво.
4. Для хронічного мієлолейкозу характерний гіперлейкоцитоз, тобто збільшення вмісту лейкоцитів крові понад 20- 109/л. У той же час при гострому мієлобластному лейкозі кількість лейкоцитів у периферичній крові або не міняється, або зменшується.
5. При хронічному мієлолейкозі часто виникає так звана базофільно-еозинофільна асоціація - збільшується частка базофілів і еозинофілів у периферичній крові.
26.2.29. Чи може гострий лейкоз із часом перейти в хронічний, і навпаки, хронічний - у гострий?
Перехід гострих лейкозів у хронічні неможливий. Таке перетворення означало б, що лейкозні клітини, які втратили здатність до дозрівання, знову набули цю властивість. Нині немає даних про те, що такий перебіг подій можливий, хоча б принципово } з урахуванням відомих механізмів розвитку пухлин.
З другого боку, хронічний лейкоз із часом може трансформуватися в гострий. Це означає, що лейкозні клітини, які ще зберігали здатність до диференціювання, таку властивість втрачають. При цьому лейкоз із менш злоякісної перетворюється у більш злоякісну форму. Подібне явище по суті відображає пухлинну прогресію — загальну властивість усіх злоякісних пухлин.
Гематологічним проявом переходу хронічного лейкозу в гострий є так званий "бластний криз", коли в крові й червоному кістковому мозку різко зростає кількість бластних клітин, а в крові поступово зникають перехідні форми.
26.2.30. Які фактори можуть бути причиною розвитку лейкозів?
Причиною лейкозів можуть бути всі фактори, що здатні викликати розвиток злоякісних пухлин узагалі. Такими є:
1) фізичні фактори (іонізуюча радіація);
2) хімічні агенти (хімічні канцерогени);
3) фактори біологічного походження (онкогенні віруси).
26.2.31. Які існують докази того, що іонізуюча радіація може бути причиною лейкозів?
Перші докази було отримано після вивчення наслідків атомного бомбардування японських міст Хіросіми й Нагасакі в 1945 р.
Було показано, що частота лейкозів серед тих, що вижили після бомбардування, різко зросла через кілька років і досягла максимуму через 6—7 років, зберігаючись підвищеною ще 25 років.
Установлено, що частота лейкозів залежить від дози поглиненої радіації. В осіб, що перебували ближче до епіцентру вибуху, частота виникнення лейкозів була вища.
Крім того, частота лейкозів залежить від виду іонізуючої радіації. Після опромінення нейтронами ріст числа лейкозів був значно вищий, ніж після у-опромінення.
Так, на Хіросіму скинули бомбу, вибух якої був джерелом нейтронів і у-випро-мінювання (уранова бомба), а на Нагасакі скинули плутонієву бомбу, після вибуху якої виділялося тільки у-випромінювання. У Хіросімі частота лейкозів істотно збільшилася вже при поглиненій дозі 0,5 Гр, у той час як у Нагасакі навіть при дозі 1 Гр частота лейкозів не зростала.
Нині всі ми є свідками й учасниками ще одного "експерименту" - чорнобильського. Отримана в ньому інформація, безумовно, розширить наші уявлення про роль іонізуючої радіації й зокрема радіонуклідів у виникненні лейкозів.
26.2.32. Що свідчить про роль хімічних агентів у виникненні лейкозів?
1. Індукція лейкозів у тварин (миші, щури, кури) за допомогою хімічних канцерогенів (поліциклічних ароматичних вуглеводнів, нітрозосполук).
2. Виникнення лейкозів у людей після тривалої інтоксикації бензолом і його похідними, після приймання цитостатичних засобів, що використовувалися з лікувальною метою.
26.2.33. Яке значення мають віруси у виникненні лейкозів?
У 1908 р. Елерман і Бат уперше отримали лейкоз у курей після введення їм безклітинних фільтратів лейкозних клітин. У 1950 р. Гросс аналогічним чином індукував лейкоз у ссавців (дитинчат мишей). Ці експерименти довели значення вірусів у виникненні лейкозів.
Нині відомо багато вірусів, здатних викликати трансформацію кровотворних !
клітин у пухлинні. їх можна розділити на дві групи.
І. Онкогешіі віруси тварин. Викликають лейкози у різних видів тварин - птахів, мишей, щурів, кішок, мавп. Більшість з них належить до РНК-вмісних вірусів -ретровірусів. Залежно від характеру онкогенної дії їх поділяють на:
а) гостротрансформуючі віруси - викликають розвиток пухлин після короткого латентного періоду (віруси гострих лейкозів тварин). Ці віруси містять j у своїй структурі онкоген;
б) повільнотрансформуючг віруси - викликають розвиток пухлин після тривалого латентного періоду (віруси хронічних лімфолейкозів). Геном цих вірусів не містить онкогена.
її. Онкогенні віруси людиіуі. Сьогодні виявлено два таких віруси.
1. Вірус Епштейна—Барр: ДНК-вмісний вірус сімейства герпес-вірусів. Викликає розвиток лімфами Беркітта у дітей віком від 2 до 14 років у деяких країнах Центральної Африки. Лімфома Беркітта не є в буквальному значенні слова лейкозом, оскільки протікає без первинного ураження червоного кісткового мозку. Джерелом цієї пухлини є підщелепні лімфатичні вузли. Пухлина росте дуже швидко, збільшуючись у масі в 2 рази через кожні 2 дні. Через 6-12 тижнів дитина гине.
2. Вірус Т-клітинної лімфоми—лейкемії людини. Є ретровірусом, належить до так званих Т-лімфотропних вірусів (HTLV-вірусів). Викликає розвиток Т-клітинного лейкозу в Японії, країнах басейну Карибського моря, у Південній Америці, на Алясці. За своїми властивостями подібний до вірусу імунодефіциту людини (ВІЛ), що викликає СНІД.
26.2.34. Які факти свідчать про роль спадкового фактора в розвитку лейкозів?
1. У людей із хромосомними хворобами істотно збільшується частота виникнення лейкозів, Так, в осіб з хворобою Дауна (трисомія по 21-й хромосомі) частота розвитку гострих лейкозів зростає в 18-20 разів. Таку ж закономірність виявлено й серед хворих на синдроми Клайнфельтера, Шерешевського-Тернера, Фанконі.
2. Майже при всіх видах лейкозів у пухлинних клітинах виявляють ознаки хромосомних аберацій: делеція, транслокація, інверсія. Дуже характерною ознакою хронічного мієлолейкозу є поява так званої "філадельфійської" хромосоми (вона виявляється у 92 % хворих з даним видом лейкозу). Ця хромосома являє собою одну з 22-ї пари хромосом, у якої відбулася делеція приблизно половини довгого плеча. Дуже часто цей фрагмент приєднується до 9-ї хромосоми.
3. Неодноразово в літературі описано випадки так званої сімейної форми хронічних лейкозів.
4. Виведено чисті лінії тварин (мишей), у яких спостерігається висока вразливість спонтанними лейкозами.
26.2.35. Як впливають на виникнення лейкозів імунні фактори?
Показано, що виникненню лейкозів сприяють стани імунологічної недостатності.
Так, при спадково обумовлених імунодефіцитних станах, пов'язаних з ураженням В- і Т-лімфоцитів (синдроми Брутона, Луї-Барр, Віскотта-Олдрича), значно зростає частота розвитку гострого лімфобластного лейкозу й лімфосарком.
Застосування цитостатиків, що викликають розвиток імунодепресивного стану (вторинних імунодефіцитів), супроводжується збільшенням частоти виникнення ракових пухлин у 2,5 рази, а лімфоцитарних пухлин — у 35 разів.
26.2.36. Опишіть патогенез лейкозів. Які стадії розвитку проходить лейкоз?
Під впливом онкогенних вірусів, іонізуючої радіації, хімічних речовин відбувається мутація генів або епігеномне порушення регуляції процесу розмноження й дозрівання кровотворних клітин. При цьому в кістковому мозку утворюється клон пухлинних клітин, для яких характерні безмежний ріст і знижена здатність до диференціювання. Швидкий ріст лейкозних клітин призводить до поширення (метастазування) їх по всій системі крові, включаючи кровотворні органи й кров. У лейкозних клітинах, що циркулюють у крові, виявляють однакові хромосомні маркери. Для хронічного мієлолейкозу таким маркером служить "філадельфійська" хромосома.
Нестабільність генотипу лейкозних клітин призводить до виникнення мутацій, як спонтанних, так і обумовлених тривалим впливом канцерогенних факторів, у результаті чого утворюються нові пухлинні клони.
Таким чином, лейкоз проходить дві стадії свого розвитку: 1) моноклонову (відносно більш доброякісну) і 2) поліклонову (злоякіснішу, термінальну). Перехід з першої стадії в другу є показником пухлинної прогресії — лейкозні клітини набувають більшої злоякісності. Вони стають такими, що їх неможна диференціювати ані морфологічними, ані цитохімічними методами, у кровотворних органах і крові збільшується кількість бластних клітин з дегенеративними змінами ядра і цитоплазми. Лейкозні клітини поширюються за межі кровотворних органів, утворюючи лейкозні інфільтрати в різних органах. Унаслідок добору знищуються клітини тих клонів, на які діяли імунна система й гормони організму, цитостатичні засоби (хімічні, гормональні, променеві). Домінують клони пухлинних клітин, найбільш стійких до цих впливів.
26.2.37. Якими клінічними синдромами можуть виявляти себе лейкози?
Усе різноманіття клінічних ознак лейкозів можна розділити на три групи. І. Гематологічні синдроми, пов'язані із заміщенням нормальної кровотворної тканини лейкозною і пригніченням у зв'язку з цим нормального кровотворення. 1. Панцитопенія ~ зменшення вмісту всіх формених елементів крові. Особливо виражена при гострих лейкозах.
2. Анемія. Основу її патогенезу становить порушення еритропоезу Однак при деяких видах лейкозів певне значення може мати імунний гемоліз еритроцитів (наприклад, при хронічному лімфолейкозі) і кровотечі (геморагічний синдром).
3. Геморагічний синдром. Обумовлений в основному тромбоцитопенією та лейкозними інфільтратами в стінках кровоносних судин.
4. Порушення неспецифічного протимікробного захисту, у зв 'язку з чим зменшується резистентність організму до інфекцій. Основною причиною цього є зменшення вмісту функціонально повноцінних гранулоцитів.
5. Імунологічна недостатність. Розвивається як наслідок лімфопенії (при гострих лейкозах і хронічному мієлолейкозі) або неповноцінності лейкозних лімфоцитів (хронічний лімфолейкоз).
II. Синдроми, пов'язані з особливостями функціонування лейкозних клітин.
1. Гарячка. Показано, що тільки у 7-8 % хворих на лейкози підвищення температури на початку захворювання пов'язане з інфекцією. У більшості ж випадків гарячка має неінфекційне походження.
2. Інтоксикація. Велика кількість лейкозних клітин гине й вивільняє у кров свій вміст. Багато компонентів загиблих клітин мають токсичну дію на центральну нервову систему. Звідси стомлюваність, загальна слабкість, нудота та ін.
3. Аутоімунні процеси. Пов'язані зі змінами, що відбуваються в лімфоци-тарному паростку крові, а саме з появою так званих "заборонених" клонів лімфоцитів, зі зменшенням кількості й функціональної активності Т-супресорів.
III. Синдроми, пов'язані з метастазуванням лейкозних клітин і розвитком лейкозних проліфератів у різних органах і тканинах.
1. Збільшення лімфатичних вузлів, печінки й селезінки.
2. Шкірний синдром. Обумовлений появою в шкірі проліфератів лейкозних клітин - лейкемідів.
3. Виразково-некротичні ураження слизових оболонок (виразково-некротичні стоматит, ангіна, ентеропатії).
4. Кістково-суглобовий синдром, що виявляється болем у кістках і суглобах.
5. Синдром нейролейкозу. Може виявлятися менінгіальним синдромом, синдромом підвищення внутрішньочерепного тиску, різноманітними неврологічними порушеннями: парезами, паралічами, парестезіями. В основі його розвитку — поява лейкозних проліфератів в оболонках головного й спинного мозку, речовині мозку, нервових стовбурах, вегетативних гангліях.
6. Лейкозний пневмоніт. Лейкозні проліферати порушують дихальну функцію легень - розвивається недостатність зовнішнього дихання.
7. Серцева недостатність. Може бути наслідком розмноження лейкозних клітин у м'язі серця.
26=3= Порушення гемостазу
26.3.1. Що таке система гемостазу? Які існують фізіологічні механізми зупинки кровотечі?
Система гемостазу — це система, що забезпечує, з одного боку, збереження рідкого стану крові, з другого - зупинку кровотечі при ушкодженні кровоносних судин.
Існує два механізми гемостазу: судинно-тромбоцитарний і коагуляційний.
Судинно-тромбоцитарний (первинний, мікроциркуляторний) гемостаз забезпечує зупинку кровотечі з судин мікроциркуляторного русла, що мають діаметр до 100 мкм. Він обумовлений взаємодією судинної стінки з тромбоцитами. Унаслідок активації судинно-тромбоцитарного гемостазу утворюється білий тромбоцитарний тромб. Порушення цього механізму гемостазу є причиною майже 80 % кровотеч і 95 % випадків тромбоутворення.
Коагуляційний (вторинний, макроциркуляторний) гемостаз є продовженням судинно-тромбоцитарного і здійснюється на його основі. Коагуляційний гемостаз забезпечує зупинку кровотеч із судин, діаметр яких перевищує 100 мкм. У результаті його активації утворюється червоний тромб, що складається з фібрину і формених елементів крові.
26.3.2. Чим обумовлена участь судинної стінки в гемостазі?
Судинна стінка бере участь у реалізації судинно-тромбоцитарного гемостазу, початок якому завжди дає ушкодження ендотелію кровоносних судин. Воно має два важливих наслідки.
I. Активація механізмів гемостазу:
а) демаскування колагену, у результаті чого відбувається так звана контактна активація тромбоцитів і фактора Хагемана (ф.ХП);
б) вивільнення з ушкоджених клітин судинної стінки АДФ, що є сильним активатором адгезії й агрегації тромбоцитів;
в) вивільнення тканинного тромбопластину (ф. Ш), що ініціює зовнішній механізм зсідання крові й утворення невеликої кількості тромбіну безпосередньо в місці ушкодження судини;
г) вивільнення фактора Віллебранда — глікопротеїну, що утворюється в ендо-теліальних клітинах і бере участь в адгезії тромбоцитів.
II. Зменшення тромборезистентності судинної стінки:
а) зменшення утворення простацикліну — речовини, що є потужним інгібітором агрегації тромбоцитів;
б) зменшення утворення й секреції антитромбіну III - дуже сильного природного антикоагулянта;
в) зменшення здатності ендотелію фіксувати на своїй поверхні комплекс гепарин-антитромбін III;
г) порушення здатності ендотеліальних клітин утворювати й вивільняти потужні активатори фібринолізу.
26.3.3. Яка роль тромбоцитів у гемостазі?
Тромбоцити беруть участь у здійсненні гемостазу завдяки чотирьом своїм функціям.
1. Ангіотрофічна функція. Це здатність тромбоцитів підтримувати нормальну структуру й функцію стінок мікросудин. Вважають, що тромбоцити є фізіологічними "годувальниками" ендотелію. Ендотеліальні клітини поглинають тромбоцити, з яких вивільняються речовини, необхідні для підтримки структурної цілісності й функціональної активності мікросудин.
2. Вазоконстрикторна функція. Це здатність підтримувати спазм ушкоджених судин завдяки вивільненню із тромбоцитів вазоактивних речовин: адреналіну, норадреналіну, серотоніну.
3. Адгезивно-агрегаційна функція. Це здатність тромбоцитів прилипати до ушкоджених ділянок судинної стінки і склеюватися один з одним, утворюючи тромбоцитарну пробку.
4. Участь у зсіданні крові. Пов'язана з вивільненням так званих тромбоцитарних факторів зсідання крові, зокрема, фактора 3, фактора 4, фактора 6 (тромбостеніну) та ін.
26.3.4. Як здійснюється судинно-тромбоцитарний гемостаз? Судинно-тромбоцитарний гемостаз складається з таких процесів (рис. 111):
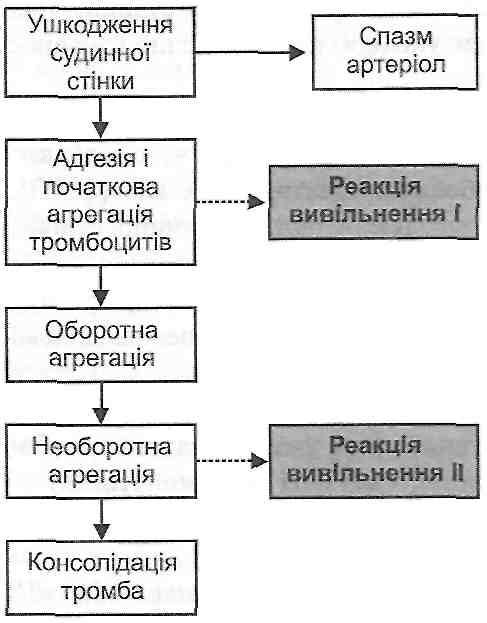
Рис. 111. Схема судинно-тромбоцитарного гемостазу
1. Спазм артеріол. Розрізняють первинний (початковий) і вторинний (відстрочений) спазм. Перший виникає відразу ж після ушкодження судинної стінки, триває кілька секунд, основний механізм його розвитку - рефлекторний. Причиною вторинного відстроченого спазму є біогенні аміни, які вивільняються тромбоцитами, — катехоламіни, серотонін.
2. Адгезія тромбоцитів — прилипання тромбоцитів до ушкоджених ділянок судинної стінки.
3. Агрегація тромбоцитів — набрякання і склеювання кров'яних пластинок.
4. Реакція вивільнення — вивільнення із тромбоцитів гранул чотирьох типів. Розрізняють реакцію вивільнення І і реакцію вивільнення II. Перша — це реакція раннього вивільнення, вона здійснюється на етапі початкової агрегації тромбоцитів. Наслідком її є вихід гранул І типу (містять біогенні аміни) і гранул II типу (містять білки, зокрема, фактор 4 пластинок, фактор Віллебранда).
Реакція вивільнення II — це реакція пізнього вивільнення, відбувається на етапі необоротної агрегації тромбоцитів. При ушкодженні тромбоцитарних мембран із пластинок виходять гранули III і IV типів, що містять лізосомні ферменти, фактор 3
5. Консолідація тромбу — його ущільнення, у результаті чого формується остаточний тромбоцитарний тромб. Відбувається в результаті в'язкого метаморфозу тромбоцитів та їх ретракції під впливом тромбостеніну.
26.3.5. Як відбувається адгезія тромбоцитів?
Основною причиною адгезії тромбоцитів є оголення (демаскування) колагену внаслідок ушкодження ендотелію судин.
Розрізняють доконтактну і контактну фази адгезії тромбоцитів.
Під час доконтактної фази в крові ще до контакту з ушкодженою стінкою судини відбувається первинна активація тромбоцитів. Спочатку змінюється форма тромбоцитів від дископодібної до сферичної (набряк). Потім викидаються довгі ниткоподібні відростки (від 3 до 10 у кожному тромбоциті).
Під час контактної фази відбувається взаємодія відростків активованих тромбоцитів з елементами базальної мембрани судинної стінки.
При цьому мають значення:
а) безпосередній контакт відростків тромбоцитів з колагеном;
б) опосередкований контакт тромбоцитів з колагеном через фактор Віллебранда;
в) реверсія електричного заряду інтими при її ушкодженні (заряд міняється з "—" на "+"), у результаті чого можлива електростатична взаємодія тромбоцитів, що мають негативний поверхневий заряд, зі стінкою судини;
г) уповільнення течії крові в ушкодженій судині.
26.3.6. Які фактори викликають агрегацію тромбоцитів? Яка динаміка цього процесу?
Причиною агрегації тромбоцитів є поява речовин-агрегантів. Вони можуть бути тромбоцитарного (виходять з активованих тромбоцитів) і нетромбоцитарного (вивільняються ушкодженими клітинами, лейкоцитами, утворюються в плазмі крові) походження.
Найбільше значення мають такі агреганти: а) АДФ. Вивільняється з ушкоджених клітин судинної стінки, гемолізованих еритроцитів, тромбоцитів у процесі їх активації;
б) тромбоксан А2 і арахідонова кислота. Тромбоксан А2 є продуктом циклоксигеназного шляху перетворення арахідонової кислоти в тромбоцитах;
в) біогенні аміни — адреналін, серотонін. їхніми джерелами є плазма крові і тромбоцити;
г) фактор агрегації тромбоцитів (ФАТ). Є речовиною ліпідної природи. Вивільняється тканинними базофілами, базофілами і нейтрофілами крові;
ґ) тромбін. Є потужним агрегантом у дозах, значно менших, якщо порівнювати з тими, що викликають зсідання крові. Такі малі кількості тромбіну завжди утворюються в місцях ушкодження судинної стінки завдяки зовнішньому механізму зсідання крові, пов'язаному з вивільненням клітинного тромбопластину;
д) тромбоспондин. Вивільняється з активованих тромбоцитів, адсорбується на їхній мембрані, взаємодіє з білками крові.
Крім речовин-агрегантів, існують фактори, що мають антиагрегантні властивості. До них, зокрема, належать простациклін, продукти фібринолізу, простацикліноза-лежний макромолекулярний білок, білковий фактор Барнес-Ліана.
У процесі розвитку агрегації тромбоцитів розрізняють такі етапи:
1) початкова агрегація. Відбувається одночасно з адгезією тромбоцитів. її викликає АДФ нетромбоцитарного походження (вивільняється з ушкоджених клітин судинної стінки);
2) оборотна агрегація. На цьому етапі агрегація може бути припинена, тромбоцити ще не ушкоджені. Оборотну агрегацію обумовлюють тромбоцитарний АДФ, а також тромбоксан А, і арахідонова кислота;
3) необоротна агрегація. У цей період тромбоцити ушкоджуються і гинуть. Вважають, що основною причиною необоротної агрегації є тромбін, що утворюється локально.
26.3.7. Які механізми лежать в основі агрегації тромбоцитів?
І. Активація тромбоцитів (рис. 112). Речовини-агреганти збільшують проникність тромбоцитарної мембрани до іонів кальцію, у результаті чого концентрація останніх у цитоплазмі кров'яних пластинок зростає. Це викликає щонайменше чотири функціонально важливих ефекти:
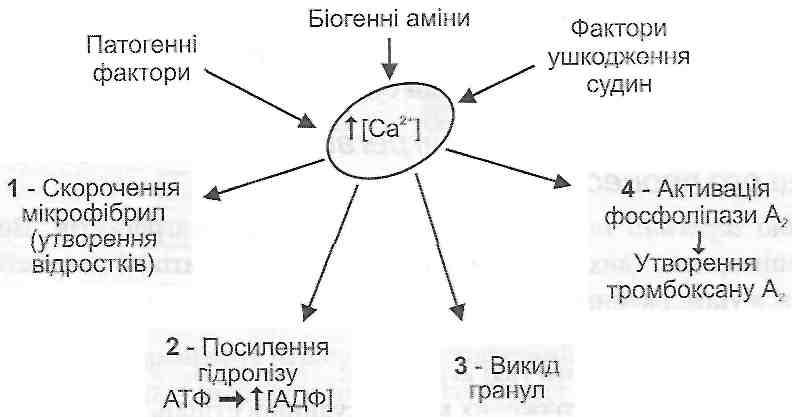
Рис. 112. Механізм активації тромбоцитів
1) скорочення мікрофібрил, у результаті чого утворюються довгі ниткоподібні відростки;
2) посилення гідролізу АТФ, наслідком чого є утворення потужного агреган-та - АДФ;
3) викид гранул тромбоцитів;
4) активація фосфоліпази А2, що викликає утворення арахідонової кислоти, а потім і тромбоксану А .
II. Власне склеювання тромбоцитів. У цьому процесі мають значення:
а) утворення між тромбоцитами "містків", що складаються з АДФ та іонів кальцію;
б) білкове склеювання — утворення "містків", що складаються з білків плазми крові. Ці білки отримали назву плазмових кофакторів агрегації. До них відносять фібриноген, альбуміни, агрексони А і В. Усі ці білки склеюють тромбоцити завдяки взаємодії з глікопротеїновими рецепторами тромбоцитів (існує 5 типів таких рецепторів) і тромбоспондином — агрегантом, адсорбованим на тромбоцитарній мембрані.
26.3.8. Що являє собою процес зсідання крові? Які фактори беруть у ньому участь?
Зсідання крові (коагуляція) є складним багатоетапним ферментативним процесом, що в кінцевому підсумку закінчується утворенням фібринового згустку.
Основу процесу зсідання крові становлять реакції протеолізу, у яких прямо або опосередковано беруть участь 12 факторів зсідання (всі вони, за винятком ф. III, мають плазмове походження) і фактор 3 тромбоцитів.
До факторів зсідання крові відносять: ф. І— фібриноген, ф. П— протромбін* ф. Ш— тканинний тромбопластин; ф. IV— іони кальцію; ф. V— проакцелерин, ф. VII — проконвертин, ф. VIII — антигемофільний глобулін, ф. IX - фактор Кріст-маса, ф. X —фактор Стюарта—Прауера, ф. XI — плазмовий попередник тромбоплас-тину, ф. ХП - фактор Хагемана, ф. ХШ - фібринстабілізуючий фактор.
За функціональними властивостями всі фактори, що беруть участь у зсіданні крові, можна поділити на такі групи:
I. Білки-ферменти. Це в основному протеолітичні ферменти: ф. II, III, VII, IX, X, XI, XII. Один фактор (ф. ХШ) є трансферазою.
Усі зазначені ферменти містяться в крові і тканинах у неактивній формі. їх активація здійснюється шляхом протеолітичного відщеплення пептидів, що закривають активні центри ферментів. Таке відщеплення відбувається за участю активованого попереднього фактора зсідання (активної протеази). Таким чином, реакції активації зсідання крові мають каскадний, ланцюговий характер.
II. Неферментні білки- акцелератори (білки-прискорювачі). Такими є ф-V і ф.УШ. Ці фактори в сотні разів прискорюють ферментативні реакції зсідання крові. На відміну від ферментів вони споживаються в процесі коагуляції крові.
III. Фосфоліпідна матриця (мікромембрана), на якій відбуваються впорядковані реакції зсідання крові. Роль такої матриці можуть виконувати:
а) фактор 3 тромбоцитів;
б) фосфоліпідні фрагменти мембран ушкоджених клітин;
в) фосфоліпідні фрагменти мембран гемолізованих еритроцитів.
IV. Іони кальцію. їхня участь у зсіданні крові полягає у фіксуванні білкових факторів до фосфоліпідних матриць.
V. Субстрат зсідання крові - фібриноген (ф.І).
Більшість факторів зсідання синтезується в печінці. їх поділяють на дві групи:
а) вітамін-К-залежні: ф. П, VII, IX, X. Вітамін К у коферментній формі входить до складу печінкових карбоксилаз, які беруть участь в утворенні зазначених факторів;
б) вітамін-К-незалежнї. ф. І, V, XI. їх утворення не вимагає вітаміну К.
26.3.9. З яких фаз складається процес зсідання крові?
Процес зсідання крові проходить три фази.
І фаза - утворення протромбінази. Існує три механізми активації цього процесу (рис. 113):
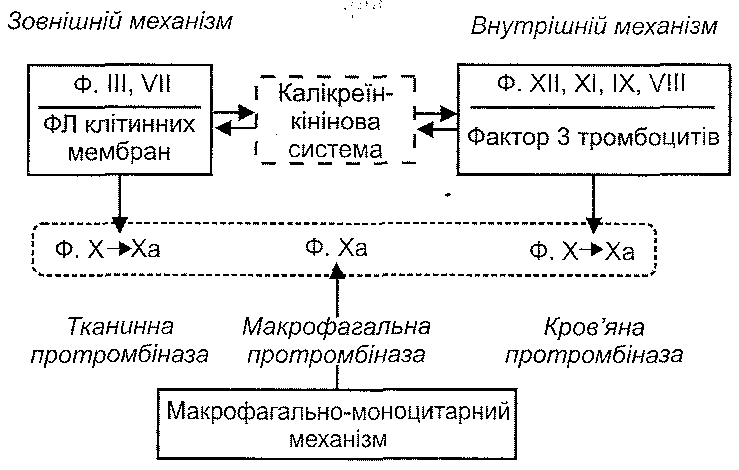
Рис. 113. Механізми активації системи зсідання крові (ФЛ- фосфоліпіди)
а) зовнішній (тканинний) механізм. Активується при ушкодженні клітин. При цьому вивільняються ферменти - тканинний тромбопластин (ф. Ш) і фосфоліпідні фрагменти мембран, що стають матрицею, на якій фіксуються фактори зсідання крові. Це дуже швидкий механізм активації зсідання. Він забезпечує зсідання крові поза кровоносними судинами (при крововиливах у тканини) і локальне утворення малих доз тромбіну, необхідного для необоротної агрегації тромбоцитів (рис. 114);
б) внутрішній (кров 'яний) механізм. Початком його є контактна або ферментативна активація ф. XII крові. Внутрішній механізм зсідання досить тривалий, тому перша його фаза по суті визначає час усього процесу коагуляції крові;
в) макрофагально-моноцитарний механізм. На відміну від двох попередніх є механізмом патологічної активації зсідання крові. Його викликає дія на макрофаги ендотоксинів бактерій, імунних комплексів, комплементу, продуктів розпаду тканин. При цьому з макрофагів вивільняється уже активна протромбіназа (ф.Ха).
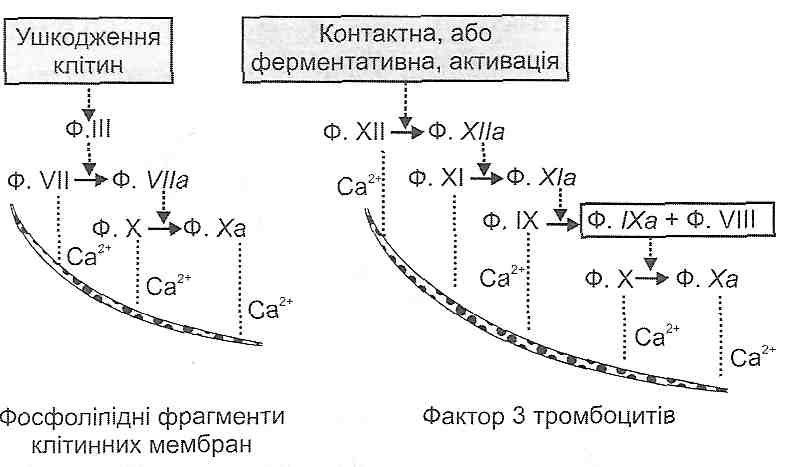
Рис. 114. Зовнішній і внутрішній механізми активації зсідання крові
Цей механізм має певне пристосувальне значення, оскільки завдяки зсіданню крові обмежується поширення патогенних факторів в організмі. її фаза — утворення тромбіну. Відбувається за участю активної протромбінази іф.У(рис.П5).
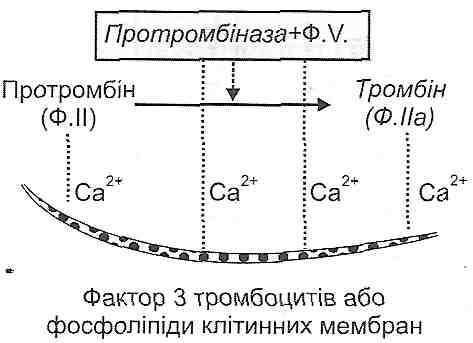
Рис. 115.7/ фаза зсідання крові
III фаза - утворення фібрину. Складається з кількох послідовних етапів (рисі 16):
а) утворення фібрину—мономеру із фібриногену під дією тромбіну;
б) утворення розчинного фібрину-полімеру (фібрину S);
в) утворення нерозчинного фібрину (фібрину І) під дією активного ф. XIII.
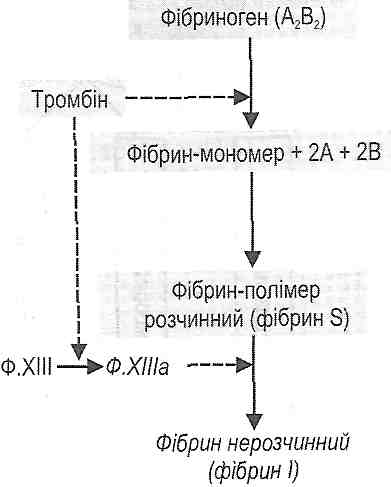
26.3.10.Як класифікують порушення гемостазу?
Порушення гемостазу (гемостазіопатії) поділяють на три групи (рис. 117):
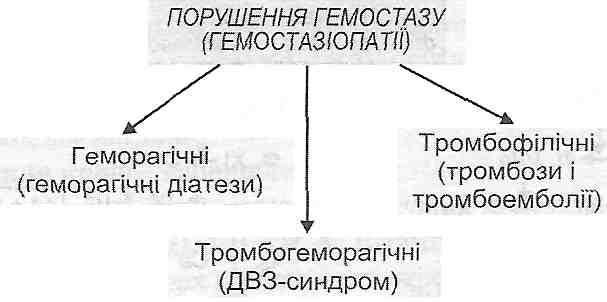
Рис. 117. Класифікація порушень гемостазу
I. Геморагічні гемостазіопатії — геморагічні діатези.
II. Тромбогеморагічні гемостазіопатії- синдром дисемінованого внутрішньосудин-ного зсідання крові (ДВЗ-синдром),
III. Тромбофілічні гемостазіопатії — тромбози і тромбоемболії.
26.3.11. Що таке геморагічні діатези? Як їх класифікують?
Геморагічний діатез — це схильність організму до повторних кровотеч і крововиливів, що виникають спонтанно або після незначних травм. Геморагічні діатези поділяють на дві великі групи (рис. 118).
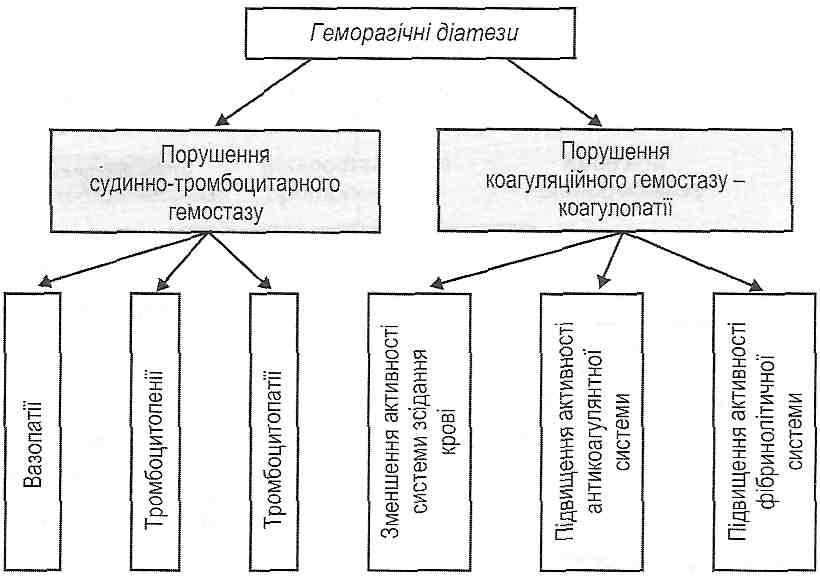
Рис. 118. Класифікація геморагічних діатезів
I. Порушення судинно-тромбоцитарного гемостазу:
а) вазопатії)
б) тромбоцитопенії;
в) тромбоцитопатії.
II. Порушення коагуляційного гемостазу - коагулопатії.
26.3.12. Що таке вазопатії? Як їх класифікують?
Вазопатії—ц& спадково обумовлені або набуті геморагічні діатези, що виникають як наслідок первинних порушень судинної стінки.
Залежно від механізму розвитку вазопатії поділяють на дві групи:
1) запальні вазопатії— васкуліти;
2) диспластичні вазопатії— ураження судин, пов'язані з порушеннями їхньої сполучної тканини (неповноцінність судинної стінки).
26.3.13. Які етіологія і патогенез запальних вазопатій?
Залежно від причин виникнення запальні вазопатії поділяють на:
1) інфекційні васкуліти. Є проявом цілого ряду інфекційних захворювань (вірусних геморагічних гарячок, висипного тифу, сепсису);
2) імунні васкуліти. Розвиваються як наслідок імунокомплексних захворювань (алергічних реакцій III типу за класифікацією Кумбса і Джелла), наприклад, при системному червоному вовчаку, вузликовому періартеріїті, геморагічному васкуліті (хвороба Шенляйн-Геноха);
3) інфекційно-імунні васкуліти. Поєднують обидва попередніх механізми.
У патогенезі запальних вазопатій провідна роль належить ушкодженню ендотелію, що може бути обумовлено:
а) цитопатичною дією ендотеліотропних вірусів;
б) токсинами мікробів, наприклад веротоксином, що його виділяють бактерії кишкової групи; *
в) комплексами антиген-антитіло і комплементом.
Наслідком ушкодження ендотелію судин є:
1) діапедез еритроцитів, що клінічно виявляється точковими крововиливами (пете-хіями);
2) інтенсивне мікротромбоутворення, що викликає порушення мікроциркуляції і живлення тканин;
3) тромбоцитопенія споживання (результат утворення мікротромбів).
26.3.14. Які етіологія і патогенез диспластичних вазопатій?
В основі диспластичних вазопатій лежать набуті або спадково обумовлені порушення сполучної тканини стінки кровоносних судин. Прикладами таких порушень є:
1. Гіповітаміноз С. Аскорбінова кислота— необхідний компонент реакції гідро-ксилювання проліну, унаслідок якої він перетворюється в оксипролін. Ця реакція вважається однією з ключових в утворенні колагену. При гіповітамінозі С з урахуванням сказаного порушується утворення повноцінного колагену (ламкість судин, випадання зубів і т. д.).
2. Телеангіектазії. Це спадково обумовлені локальні дефекти сполучної тканини судин, що обумовлюють стоншення їх стінок і розширення просвіту. Телеангіектазії є джерелом небезпечних для життя кровотеч, особливо при локалізації у внутрішніх органах.
3. Гемангіоми. Це судинні пухлини, які часто кровоточать.
4. Синдром Елерса-Данло. Його основу складають генетично обумовлені дефекти ] колагену.
У патогенезі диспластичних вазопатій мають значення:
1) стоншення стінок мікросудин і розширення їхнього просвіту;
2) неповноцінний локальний гемостаз. Через недостатню кількість або неповноцінність колагену в субендотелії судин порушується адгезія тромбоцитів;
3) легка ранимість судин.
26.3.15. Що таке тромбоцитопенія? Які механізми можуть лежати в основі розвитку тромбоцитопенії?
Тромбоцитопенія — це зменшення вмісту тромбоцитів в одиниці об'єму периферичної крові нижче 150 109/л.
На думку багатьох авторів, геморагічні прояви тромбоцитопенії з'являються при зменшенні кількості тромбоцитів нижче 50 109/л.
За походженням тромбоцитопенії можуть бути спадково обумовленими і набутими.
За механізмом розвитку виділяють такі види тромбоцитопенії. І. Тромбоцитопенії, пов'язані з порушеннями утворення тромбоцитів:
а) мієлотоксичні тромбоцитопенії— виникають унаслідок ушкодження кровотворних клітин. Дуже часто поєднуються з анемією й лейкопенією. Причинами їх розвитку є ті самі фактори, які викликають розвиток гіпопластич-ної анемії;
б) дефіцитні тромбоцитопенії— обумовлені недостатністю вітаміну В12 або фолієвої кислоти;
в) дисрегуляторні тромбоцитопенії— пов'язані з порушенням утворення тромбоцитопоетинів — речовин, що стимулюють утворення тромбоцитів;
г) тромбоцитопенії, пов'язані зі зменшенням плацдарму кровотворення, — розвиваються при лейкозах і метастазах злоякісних пухлин.
її. Тромбоцитопенії, пов'язані з посиленим руйнуванням тромбоцитів. Причиною такого руйнування можуть бути:
а) імунне ушкодження, обумовлене антитромбоцитарними антитілами на власні компоненти кров'яних пластинок або на лікарські препарати, адсорбовані на тромбоцитах. Аутоімунне ушкодження вважають найбільш імовірним механізмом розвитку так званої ідіопатичноїтромбоцитопенічноїпурпури (хвороби Верльгофа);
б) ггперспленізм — гіперфункція селезінки, що супроводжується часто спле-номегалією. У результаті підвищення фагоцитарної активності макрофагів відбувається інтенсивне руйнування всіх формених елементів крові, у тому числі й тромбоцитів;
в) механічне ушкодження тромбоцитів. Часто виникає при гемангіомах і накладенні штучних клапанів серця;
г) набутімембранопатії(гемолітична анемія Маркіафави—Мікеллі). Соматичні мутації кровотворних клітин спричиняються до утворення пулів клітин
(еритроцитів, гранулоцитів, тромбоцитів) з дефектами мембрани. У результаті збільшується чутливість таких клітин до дії комплементу й відбувається їх руйнування. III. Тромбоцитопенії споживання. Виникають у результаті посиленого використання тромбоцитів на утворення тромбів (хвороба Шенляйн—Геноха, хвороба Мошковича, ДВЗ-синдром).
26.3.16. Який патогенез порушень гемостазу в умовах тромбоцитопенії?
У патогенезі геморагічного синдрому при тромбоцитопеніях мають значення:
1) порушення ангіотрофічної функції тромбоцитів, у результаті чого виникають дистрофічні зміни в ендотелії і збільшується ламкість мікросудин. Це веде до збільшення ранимості судин, діапедезу еритроцитів, крововиливів. Останні виявляють себе петехіями на шкірі, кровотечами з ясен і носа, крововиливами в головний мозок і сітківку ока;
2) порушення адгезії й агрегації тромбоцитів. Це викликає порушення формування тромбоцитарного тромбу й призводить до збільшення часу кровотечі (проба Дюка);
3) порушення вторинного спазму ушкоджених артеріол. При тромбоцитопеніях вивільняється мало біогенних амінів (катехоламінів, серотоніну), з дією яких пов'язане скорочення гладких м'язів судин;
4) порушення зсідання крові. Обумовлено недостатнім вивільненням фактора 3 пластинок і тромбостеніну. У результаті порушується І фаза зсідання крові і ретракція згустку.
26.3.17. Що таке тромбоцитопатії? Як їх класифікують?
Тромбоцитопатії—це порушення функціональних властивостей тромбоцитів, йґ якісна неповноцінність. При цьому кількість тромбоцитів може залишатися в нормі J
За походженням тромбоцитопатії бувають спадково обумовленими і набутими.
За характером якісних дефектів кров'яних пластинок тромбоцитопатії поділяють на ендо- і екзотромбоцитарні.
Ендотромбоцитарні тромбоцитопатії обумовлені порушеннями складових частин тромбоцитів. їх, у свою чергу, поділяють на мембранопатії, гранулопатії і фер-ментопатії. Мембранопатії виникають при спадкових аномаліях мембранних пгіко-протеїнів, що виконують функції клітинних рецепторів; при блокаді цих рецепторів аномальними білками плазми крові (парапротеїнами), при ушкодженні мембрани кров'яних пластинок патогенними факторами. Гранулопатії виявляються дефіцитом гранул І і II типів. В основі ферментопатії може лежати зменшення активності ферментів циклу Кребса, гліколізу, порушення функцій АТФ-аз, циклоксигенази і тром-боксансинтетази.
При екзотромбоцитарних тромбоцитопатіях причини порушення функцій тромбоцитів лежать поза кров'яними пластинками. У зв'язку з цим екзотромбоцитарні тромбоцитопатії можуть бути:
а) пов'язаними зі змінами плазми крові (дефіцит плазмових білків, що є плазмовими кофакторами агрегації тромбоцитів);
б) пов'язаними зі змінами в судинній стінці (порушення утворення фактора Вілле-бранда ендотелієм судин, розлади зовнішнього механізму зсідання крові). Залежно від сутності порушень гемостазу виділяють:
1) тромбоцитопатії з первинним порушенням адгезії тромбоцитів^
2) тромбоцитопатії з первинними порушеннями агрегації тромбоцитів;
3) тромбоцитопатії з первинним порушенням реакцій вивільнення вмісту тромбоцитів',
4) тромбоцитопатії, пов'язані з дефіцитом або зменшенням доступності фактора З тромбоцитів.
26.3.18. Наведіть приклади тромбоцитопатій з різними механізмами порушень гемостазу.
1. Тромбоцитопатії з первинним порушенням адгезії тромбоцитів:
а) хвороба Віллебранда — ангіогемофілія. Обумовлена генетичними порушеннями синтезу фактора Віллебранда ендотеліальними клітинами (тип спадкування аутосомно-домінантний);
б) хвороба Бернара—Сульє — макротромбоцитодистрофія. Причиною є спадково обумовлений дефект глікопротеїнів тромбоцитарної мембрани (ГШЬ), що взаємодіють з фактором Віллебранда. При цьому тромбоцити набувають гігантських розмірів. Тип спадкування аутосомно-рецесивний.
2. Тромбоцитопатії з первинними порушеннями агрегації тромбоцитів - диза-грегаційні. Найчастіше буває тромбастенія Гланцмана, що виникає як наслідок дефектів мембранних глікопротеїнів І і II типів, що беруть участь в агрегації. При цьому адгезія тромбоцитів і вивільнення їхніх гранул відбуваються, а агрегація -ні, незважаючи на дію таких потужних агрегантів, як АДФ, адреналін, тромбін. Тип спадкування аутосомно-рецесивний.
3. Тромбоцитопатії з первинним порушенням реакцій вивільнення вмісту тромбоцитів:
а) порушення дегрануляції тромбоцитів — "парез реакції вивільнення ". Виникає, зокрема, при порушенні утворення тромбоксану А2 при дії ацетилсаліцилової кислоти. Показано, одо одноразове приймання аспірину необоротно збільшує час вивільнення гранул від 3,5 до 6 діб, поки не з'являться нові тромбоцити;
б) недостатність накопичення й збереження вмісту гранул тромбоцитів. Виникає, як правило, при генетично обумовлених порушеннях.
Розлади реакцій вивільнення гранул викликають порушення другої хвилі агрегації тромбоцитів. Початкова агрегація кров'яних пластинок закінчується їх дезагрегацією.
4. Тромбоцитопатії, пов'язані з дефіцитом або зменшенням доступності фактора 3 пластинок. У їхній основі можуть лежати або генетично обумовлені дефекти структури цього фактора, або порушення його вивільнення з ушкоджених тромбоцитів. При цьому порушується зсідання крові (коагуляційний гемостаз). Адгезив-но-агрегаційні властивості тромбоцитів не міняються.
26.3.19. Чим можуть бути обумовлені порушення коагуляційного гемостазу - коагулопатії?
В основі розвитку коагулопатій можуть бути:
1) зменшення активності системи зсідання крові;
2) підвищенням активності антикоагулянтної системи;
3) збільшенням активності фібринолітичної системи.
26.3.20. Що може бути причиною безпосереднього порушення І фази зсідання крові?
Залежно від характеру порушень І фази зсідання крові виділяють три групи розладів.
I. Ізольовані порушення зовнішнього механізму активації зсідання. Виникають при дефіциті ф. VII - гіпопроконвертинемії. Цей дефіцит може бути спадково обумовленим (тип спадкування аутосомно-рецесивний) або набутим (гіповітаміноз К, ураження печінки).
II. Ізольовані порушення внутрішнього механізму активації зсідання:
а) дефіцит ф.УШ- гемоф ілія А. Найчастіше виникає як генетичний дефект коагулянтної частини ф.УШ (тип спадкування зчеплений з Х-хромосомою). Можливе утворення аутоантитіл проти білкових компонентів цього фактора зсідання;
б) дефіцит ф. IX- гемофілія В. Причиною розвитку є спадкова патологія (тип спадкування зчеплений з Х-хромосомою), дефіцит вітаміну К або ураження печінки, антитіла проти ф. IX;
в) дефіцит ф. XI— гемофілія С. Виникає при генетичних порушеннях (тип спадкування аутосомно-рецесивний) або ураженнях печінки;
г) дефіцит ф. ХП. Спадкова патологія, що буває дуже рідко. Завдяки калікре-їн-кініновій системі цей дефект добре компенсується, оскільки запуск внутрішнього механізму зсідання відбувається через зовнішній;
ґ) дефіцит ф. З тромбоцитів. Є наслідком тромбоцитопенії або певних видів тромбоцитопатій.
III. Поєднані порушення зовнішнього і внутрішнього механізмів зсідання. Розвиваються при дефіциті ф. X, що може бути спадково обумовленим (тип спадкування аутосомно-рецесивний) або набутим (гіповітаміноз К, ураження печінки).
26.3.21. Що може бути причиною безпосереднього порушення II фази зсідання крові?
1. Дефіцит ф.ІІ— гіпопротромбінемія. Найчастіше має набутий характер і розвивається внаслідок гіповітамінозу К або уражень печінки.
2. Дефіцит ф. V— парагемофілія. Порушення утворення проакцелерину може бути обумовлено ураженнями печінки або аутоантитілами проти ф.У
26.3.22. Що може бути причиною безпосереднього порушення III фази зсідання крові?
1. Дефіцит фібриногену:
а) афібриногенемія - повна відсутність фібриногену (спадкове захворювання з аутосомно-рецесивним типом спадкування);
б) гіпофібрииогенемія - зменшення синтезу фібриногену в печінці при її ураженнях.
2. Дисфібршшгенемії - якісні порушення фібриногену. Розвиваються як наслідок генетичних дефектів (тип спадкування аутосомно-домінантний). Виявляються утворенням аномального фібрину.
3. Порушення полімеризації фібрину. Розвиваються б результаті утворення комплексів фібриногену з фібрином-мономером і проміжними продуктами, від яких ще повністю не відщепились пептиди А і В. При цьому утворюється так званий "заблокований фібриноген" ("тромбінрезистентний фібриноген"), що не піддається дії тромбіну.
4. Дефіцит ф. ХШ. Виникає як наслідок спадкових порушень (тип спадкування ау-тосомно-рецесивний). виявляється порушеннями перетворення розчинного фібрину (фібрину S) у нерозчинний (фібрин І).
26.3.23. Які речовини складають антикоатулянтну систему крові?
І. Первинні антикоагулянти. Постійно синтезуються в організмі й тому завжди містяться в плазмі крові. До них відносять:
1) антитромбін ПІ- основний універсальний антикоагулянт, що є інгібітором протеаз. Синтезується ендотелієм судин. Пригнічує активність усіх протеолітичних ферментів крові, у тому числі тромбіну, калікреїну, плазміну, ф. ХІІа, ф. ХІа, ф. Ха, ф. ІХа, ф.УІІа;
2) гепарин (антитромбін II) — глікозаміноглікан, що вивільняється тканинними базофілами і базофілами крові при їх дегрануляції. Антикоагулянтні властивості має не сам гепарин, а комплекс гепарину з антитромбіном III. Завдяки гепарину антитромбін III фіксується на поверхні ендотелію судин, де його антикоагулянтні властивості в багато разів зростають;
3) а -антитрипсин, а2-макроглобулін, інгібітор СІ-компонента комплементу - усі вони є неспецифічними інгібіторами протеаз, у тому числі й факторів зсідання крові.
її. Вторинні антикоагулянти. У плазмі крові в нормі не містяться. Утворюються в процесі зсідання крові й фібринолізу. До них відносять:
1) антитромбін І— фібрин, що адсорбує і в такий спосіб інактивує велику кількість тромбіну;
2) продукти фібринолізу. Перешкоджають процесам полімеризації фібрину і утворенню фібринових структур.
26.3.24. Чим може бути обумовлене підвищення активності антикоагулянтної системи крові?
Підвищення активності антикоагулянтної системи крові закономірно виникає при:
1) збільшенні вмісту гепарину в крові — гіпергепаринеміг. Це може бути обумовлено посиленою дегрануляцією тканинних базофілів і базофілів крові, зокрема, при алергічних реакціях І типу за класифікацією Кумбса і Джелла, руйнуванням базо-фільних лейкоцитів при лейкозах, введенням гепарину ззовні;
2) появою "патологічних " антикоагулянтів, до яких відносять антитромбін V, що порушує полімеризацію фібрину-мономеру; "вовчаковий" антикоагулянт, що порушує утворення протромбіназного комплексу; парапротеїни, що унеможливлюють полімеризацію фібрину.
26.3.25. Що входить у поняття "фібринолітична система"?
Фібриноліпшчна система — це система, яка забезпечує лізис (протеоліз) фібрину в кровоносному руслі. У такий спосіб вона бере участь у підтримці рідкого стану крові й у відновленні кровообігу в тромбованих судинах.
До складу системи фібринолізу входять (рис. 119):
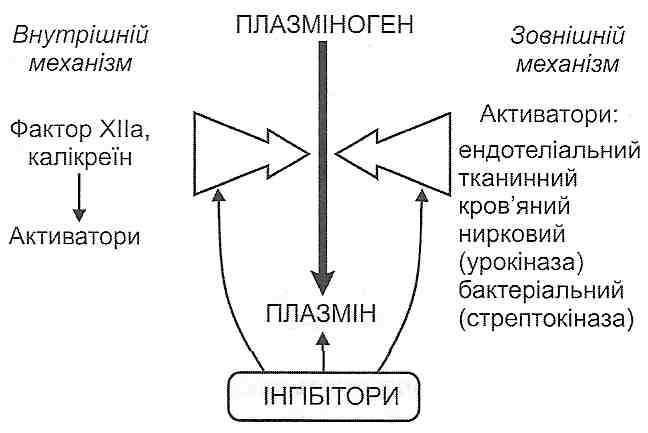
Рис. 119. Схема фібринолізу
1) плазміноген (профібринолізин) — неактивний протеолітичний фермент, що завжди міститься в плазмі крові;
2) плазмін (фібринолізин) — активна форма плазміногену. Утворюється в результаті дії активних протеаз на плазміноген і відщеплення від його молекули пептиду, який "закриває" активний центр;
3) активатори фібринолізу — велика група речовин, які або самі є протеазами і здатні перетворювати плазміноген у плазмін, або викликають появу таких протеаз;
4) інгібітори фібринолізу. До них відносять інгібітори протеаз, серед яких найбільше значення має а2-антиплазмін.
Розрізняють внутрішній і зовнішній механізми активації фібринолізу.
Внутрішній механізм обумовлений активацією фактора XII зсідання крові й утворенням калікреїну, унаслідок чого в крові з'являється велика кількість активаторів фібринолізу.
Зовнішній механізм пов'язаний з надходженням у кров готових активаторів фібринолізу: ендотеліального, клітинного, тканинного (урокіназа), бактеріального (стрептокіназа).
26.3.26. Які фактори викликають підвищення активності фібриполітичної системи крові?
1. Посилене утворення й надходження в кров активаторів фібринолізу. Відбувається при великих ушкодженнях тканин: великі травми, ушкодження клітин токсинами, операційні втручання, лейкози та ін.
2. Зменшення вмісту в крові інгібіторів протеолізу. Має місце при недостатньому їх утворенні або посиленому використанні.
26.3.27. Якими клінічними ознаками виявляють себе порушення коагуляційного гемостазу?
На відміну від порушень судинно-тромбоцитарного гемостазу для коагулопатій характерні не капілярні (точкові) кровотечі, а кровотечі з великих судин — артерій і вен. Такі кровотечі клінічно виявляють себе:
а) гематомами — великими крововиливами в м'язи, під шкіру, у порожнину суглобів (гемартрози);
б) тривалими кровотечами після операційних втручань (видалення зуба та ін.).
26.3.28. Що таке ДВЗ-синдром?
Синдром дисемінованого внутрішньосудинного зсідання крові (ДВЗ-син-дром) — це генералізоване зсідання крові всередині судин, що викликає утворення великої кількості мікрозгустків і агрегатів клітин, які порушують мікроциркуляцію в органах і тканинах.
Цей синдром часто характеризують як катастрофу для організму.
26.3.29. Що може бути причиною ДВЗ-синдрому?
Залежно від причин розвитку виділяють такі різновиди ДВЗ-синдрому:
1) інфекційно-септичний (розвивається при сепсисі);
2) посттравматичний (при краш-синдромі, опіковій хворобі, множинних переломах кісток);
3) шокогенний (при всіх видах шоку);
4) хірургічний (після операцій з великою травматизацією тканин);
5) акушерський (при передчасному відшаруванні плаценти, надходженні в кров навколоплідних вод);
6) токсигенний (після укусу змії);
7) пухлинний (при злоякісному пухлинному рості);
8) алергічний (при імунному ушкодженні тканин) та ін.
26.3.30. Що є патогенетичною основою розвитку ДВЗ-синдрому?
В основі патогенезу ДВЗ-синдрому лежить так званий "гуморальний протеазний вибух ", тобто одночасна активація всіх протеолітичних ферментів плазми крові, що входять до складу чотирьох позаклітинних біохімічних систем (рис. 120):
а) системи зсідання крові;
б) фібринолітичної системи;
в) калікреїн-кінінової системи;
г) системи комплементу.
Рис. 120. "Гуморальний протеазний вибух"
Основний принцип активації позаклітинних протеаз — відщеплення пептидів, що закривають їхні активні центри. Утворення активних протеолітичних ферментів крові має свої особливості:
а) можлива самоактивація ферментів — активний фермент, впливаючи на неактивну форму, переводить її в активну;
б) одні активні протеази здатні активувати інші (перехресна активація)',
в) ланцюговий характер активації. Теоретично поява навіть однієї молекули активної протеази може викликати активацію всіх наявних протеаз крові.
Однак у нормі реакції активації протеолітичних ферментів мають обмежений характер, що пояснюється існуванням великої групи інгібіторів протеаз.
При патології, коли у кров надходять великі кількості активних протеаз, потужність існуючих інгібіторів може виявитися недостатньою. Отоді й виявить себе ланцюговий характер активації протеолітичних систем плазми крові. Така активація набуває генералізованого характеру, втягує всі протеази крові - відбувається "гуморальний протеазний вибух".
26.3.31. Назвіть основні джерела надходження в кров активних протеаз при ДВЗ- синдромі.
Існує три основних джерела надходження протеаз у кров.
I. Ушкоджені клітини. Має значення гостре ушкодження великої кількості клітин, з яких у позаклітинний простір і кров надходять лізосомні протеази, тканинний тромбопластин.
Запалення як місцевий процес, що виникає при ушкодженні клітин, обмежує надходження продуктів розпаду в кров, локалізуючи ушкодження і в такий спосіб попереджаючи розвиток ДВЗ-синдрому.
II. Надходження в кров великої кількості позаклітинних протеаз; наприклад трипсину при гострому панкреатиті, ферментів, що містяться в навколоплідних водах.
III. Екзогенні протеази. їхніми джерелами можуть бути бактеріальні клітини при сепсисі, зміїна отрута та ін.
26.3.32. Як розгортається патогенез ДВЗ-синдрому?
У патогенезі ДВЗ-синдрому розрізняють дві фази.
I фаза - фаза гіперкоагуляції і агрегації тромбоцитів. Основу цієї фази становить генералізована активація системи зсідання крові, тобто утворення тромбіну (тромбінемія), що призводить до утворення фібрину і агрегатів тромбоцитів.
Існує три механізми запуску цієї фази:
1) ферментативний механізм - надходження в кров великої кількості активних протеаз і тканинного тромбопластину;
2) контактний механізм - активація ф. XII при контакті його з чужорідними поверхнями (екстракорпоральний кровообіг, гемодіаліз, штучні клапани серця);
3) тромбоцитарний механізм - первинна активація агрегації тромбоцитів при гене-ралізованому ушкодженні ендотелію судин, порушеннях реологічних властивостей крові, гострому внутрішньосудинному гемолізі еритроцитів.
У результаті реалізації зазначених механізмів утворюється велика кількість мі-крозгустків і агрегатів клітин, що призводить до розладів мікроциркуляції, розвитку сладж-синдрому (див. розд. 13).
Усе це веде до появи таких клінічних проявів, як гіпоксія, ацидоз, інтоксикація продуктами розпаду, гостра недостатність зовнішнього дихання (мікрозгустками закупорюються капіляри легень), гостра ниркова недостатність (забиваються капіляри клубочків), порушення мозкового кровообігу.
II фаза - фаза гіпокоагуляції (геморагічний синдром). Ця фаза розвивається як наслідок виснаження механізмів судинно-тромбоцитарного і коагуляційного гемостазу.
У її виникненні мають значення:
а) зменшення активності системи зсідання крові (споживання факторів І, V, VIII);
б) активація фібринолітичної системи (надходження в кров великої кількості активаторів фібринолізу);
в) підвищення антикоагулянтної активності крові за рахунок утворення продуктів фібринолізу;
г) розвиток тромбоцитопенії споживання;
ґ) підвищення проникності стінки судин (має значення утворення великих кількостей кінінів). Фаза гіпокоагуляції клінічно виявляє себе масивними кровотечами, що їх важко зупинити.
26.3.33. Що таке тромбофільні діатези? Що може лежати в основі їх розвитку?
Тромбофільні діатези - це захворювання і синдроми, при яких схильність до утворення тромбів обумовлена первинними порушеннями механізмів гемостазу.
Патогенетичну основу тромбофілій можуть становити: 1) ендотеліальні механізми, що обумовлюють зменшення тромборезистентності стінки судин;
2) тромбоцитарнімеханізми, пов'язані зі збільшенням агрегаційної здатності тромбоцитів;
3) зменшення антикоагулянтної активності крові (зменшення утворення антитромбіну III);
4) зниження активності фібринолітичної системи (зменшення утворення ендоте-ліального активатора плазміногену, поява потужних інгібіторів плазміну, дефіцит плазміногену);
5) порушення системи зсідання крові (дисфібриногенемії).
Клінічно тромбофільні діатези виявляють себе тромбозами і тромбоемболіями венозних і артеріальних судин у різних органах і тканинах.
27. Патологічна фізіологія серця
27.1. Що таке недостатність кровообігу? Чим вона може бути обумовлена?
Недостатність кровообігу - це стан, при якому серцево-судинна система не може забезпечити органи й тканини організму необхідною кількістю крові. Є найчастішим проявом різних порушень функцій системи кровообігу.
Недостатність кровообігу може бути обумовлена:
1) недостатністю серця;
2) недостатністю кровоносних судин;
3) серцево-судинною недостатністю, тобто одночасною недостатністю серця і судин.
27.2. Які наслідки для органів, тканин і організму в цілому має недостатність кровообігу?
1. Порушення трофічного забезпечення органів і тканин. Розвиваються як наслідок:
а) зменшення доставки кисню (циркуляторпа гіпоксія, див. розд. 19);
б) зменшення доставки поживних речовин.
2. Порушення видалення з органів і тканин кінцевих продуктів обміну речовин. Наслідком цього є розвиток:
а) негазового ацидозу;
б) інтоксикації.
27.3. Що таке недостатність серця?
Недостатність серця - це патологічний стан, обумовлений нездатністю серця забезпечити кровопостачання органів і тканин відповідно до їхніх потреб.
Це стан, при якому навантаження на серце перевищує його здатність виконувати роботу. Він виявляє себе тим, що серце не здатне переміщати в артеріальне русло всю кров, що надходить до нього по венах.
27.4. Як класифікують недостатність серця?
I. Залежно від клінічного перебігу розрізняють гостру і хронічну недостатність серця.
II. За вираженістю клінічних проявів недостатність серця може бути прихованою (компенсованою) і явною (декомпенсованою).
III. Залежно від порушення функції переважно того чи того відділу серця розрізняють лівоишуночкову, правошлуночкову і тотальну недостатність серця.
IV. За патогенезом виділяють:
а) недостатність серця від перевантаження;
б) міокардіальну недостатність серця;
в) позаміокардіальну недостатність.
27.5. Дайте коротку характеристику різних патогенетичних варіантів недостатності серця.
I. Недостатність серця від перевантаження розвивається в результаті дії на здорове серце великих навантажень опором або об'ємом, тобто коли збільшується опір серцевому виштовху або приплив крові до певного відділу серця. Це буває при вадах серця, гіпертензії великого або малого кола кровообігу, артеріовенозних фістулах або при виконанні дуже важкої фізичної роботи. При цьому до серця з нормальною скорочувальною здатністю висуваються надмірні вимоги.
II. Міокардіальна недостатність серця розвивається в результаті первинного ураження міокарда. Вона може бути пов'язана з: а) ушкодженням провідникової системи серця (аритмічна) і б) ушкодженням волокон робочого міокарда (міо-кардіопатична). Причиною її розвитку часто є інфекції, інтоксикація, гіпоксія, авітамінози, порушення вінцевого кровообігу, деякі спадкові дефекти обміну речовин. При цьому недостатність розвивається навіть при нормальному або зниженому навантаженні на серце.
III. Позаміокардіальна недостатність серця розвивається в результаті дії причин, не пов'язаних безпосередньо з міокардом. її виникнення можуть обумовлювати зменшення припливу крові до серця (гіповолемія, колапс) або перешкоди здійсненню діастоли, у результаті чого серце не може прийняти всю кров, що притікає до нього (накопичення ексудату або транссудату в порожнині перикарда, гостра тампонада серця).
27.6. Які типи перевантажень серця можуть бути причиною розвитку його недостатності?
Виділяють два типи перевантажень серця.
1. Перевантаження об'ємом виникає тоді, коли до серця або до окремих його порожнин притікає збільшений об'єм крові. У цих умовах серце або його відділ, що зазнає перевантаження, мають переміщати збільшений об'єм крові в артеріальну систему. Це досягається збільшенням хвилинного об'єму серця відповідно до збільшеного венозного повернення.
Перевантаження об'ємом виникає при:
а) збільшенні венозного повернення крові до серця, зокрема при збільшенні об'єму циркулюючої крові (гіперволемія) або збільшенні тонусу венозних судин (зменшення ємності венозної системи);
б) вадах серця — недостатності його клапанів. Так, при недостатності аортального і двостулкового клапанів розвивається перевантаження лівого шлуночка, при недостатності клапана легеневої артерії і тристулкового клапана - перевантаження правого шлуночка.
2. Перевантаження опором виникає тоді, коли серце або окремі його відділи змушені виконувати роботу проти збільшеного опору, що перешкоджає переміщенню всієї крові в артеріальну систему. При перевантаженні опором серце має зберегти свій хвилинний об'єм, незважаючи на збільшений опір вигнанню крові.
Перевантаження опором розвивається при:
а) збільшенні артеріального тиску (збільшенні периферичного судинного опору). При гіпертензії великого кола кровообігу перевантаження опором діє на лівий шлуночок, а при гіпертензії малого кола - на правий шлуночок;
б) вадах серця — стенозах клапанних отворів. 'Так, при стенозі отвору аорти розвивається перевантаження лівого шлуночка, при стенозі отвору двостулкового клапана - лівого передсердя, при стенозі отвору легеневої артерії - правого шлуночка, при стенозі отвору тристулкового клапана - правого передсердя.
27.7. Які механізми можуть забезпечувати компенсацію серця при дії на нього збільшених навантажень?
При дії на серце навантажень об'ємом і опором збільшення роботи серця забезпечується двома типами компенсаторних механізмів.
I. Негайні механізми компенсації серця. До них відносять:
а) гетерюметричтш механізм;
б) гомеометричний механізм;
в) хроноінотропний механізм;.
г) інотропна дія катехоламінів.
II. Механізми довгострокової адаптації серця - гіпертрофія міокарда.
27.8. У чому сутність тетерометричното механізму компенсації серця?
Гетерометричпиіі механізм є одним із негайних механізмів компенсації серця до дії навантажень об'ємом.
Його сутність полягає в збільшенні сили серцевих скорочень в умовах надходження до серця збільшеного об'єму крові.
Основу гетерометричного механізму становить закон Франка—Старлінга. Він має два формулювання: для окремих м'язових волокон і для серця в цілому. У першому варіанті його сутність виражено таким положенням: що більша вихідна довжина м 'язового волокна (у певних межах), то більша сила його скорочень. Для серця в цілому уживаним є формулювання: що більший кінцеводіастолічний об'єм шлуночків серця, то більший їхній ударний об'єм.
Основними проявами гетерометричного механізму компенсації є збільшення кін-цеводіастолічного тиску за рахунок збільшення надходження крові в порожнини шлуночків і збільшення ударного, а отже, і хвилинного об'ємів серця за рахунок збільшення сили серцевих скорочень. Напруга м'язових волокон міокарда при цьому не міняється. Змінюється тільки їх довжина, звідси назва механізму - гетерометричний.
27.9. У чому сутність томеометричното механізму компенсації серця?
Гомеометричний механізм є негайним механізмом компенсації серця до дії навантажень опором. Його сутність полягає в збільшенні сили серцевих скорочень в умовах збільшення опору вигнанню крові.
Нині показано, що в основі цього механізму, як і гетерометричного, закон Фран-ка-Старлінга, тобто збільшення вихідної довжини волокон міокарда і пов'язане з цим збільшення кінцеводіастолічного тиску.
Становлення гомеометричного механізму відбувається в такій послідовності. При збільшенні опору вигнанню крові різко падає ударний об'єм, унаслідок чого збільшується кінцевосистолічний об'єм шлуночків. Оскільки надходження крові в шлуночки продовжує залишатися попереднім, то в наступному циклі скорочень серця збільшується кінцеводіастолічний об'єм. А це за законом Франка-Старлінга веде до збільшення сили скорочень серця.
Для гомеометричного механізму характерні такі зміни показників кардіодинаміки:
а) збільшення кінцевосистолічного об'єму;
б) збільшення кінцеводіастолічного об'єму за рахунок первинного підвищення попереднього показника, а не за рахунок збільшення припливу крові, як при гетеро-метричному механізмі;
в) ударний, а отже, і хвилинний об'єми за рахунок збільшення сили серцевих скорочень залишаються на попередньому рівні, незважаючи на збільшення опору вигнанню крові.
При гомеометричному механізмі збільшується напруга м'язових волокон міокарда, тимчасом як їхня довжина не міняється. Звідси назва механізму компенсації — го-меометричний.
27.10. Чим характеризується хроноінотропний механізм компенсації серця?
Хроноінотропний механізм (феномен "сходинок", феномен Боудича) є одним з негайних механізмів компенсації серця до дії підвищених навантажень. Його сутність полягає в тому, що при збільшенні частоти скорочень серця збільшується сила його скорочень. При цьому одночасно зменшується час розслаблення міокарда, що сприяє швидкому наповненню шлуночків серця кров'ю.
Нині вважають, що основу хроноінотропного механізму становить збільшення надходження іонів кальцію в саркоплазму кардіоміоцитів під час потенціалів дії, сумарна тривалість яких при тахікардії зростає. Підвищення концентрації іонів кальцію в саркоплазмі призводить до збільшення кількості утворюваних кальцій-тропонінових комплексів і, як наслідок, до збільшення сили скорочень м'язових волокон.
Для хроноінотропного механізму характерні такі зміни показників кардіодинаміки:
а) ударний об'єм збільшується (при навантаженні об'ємом) або залишається постійним (при навантаженні опором). У результаті збільшення частоти серцевих скорочень зростає хвилинний об'єм серця;
б) кінцеводіастолічний об'єм зменшується, якщо приплив крові до серця постійний, або залишається без змін, якщо приплив крові зростає;
в) кінцевосистолічний об'єм зменшується.
27.11. Яка роль катехоламінів у здійсненні механізмів негайної компенсації серця?
Участь катехоламінів у здійсненні негайної адаптації серця до підвищених навантажень пов'язана зі здатністю адреналіну і норадреналіну безпосередньо збільшувати силу серцевих скорочень - з позитивним інотропним ефектом.
Установлено, що під впливом катехоламінів збільшується кількість Са-каналів сарколеми, здатних відкриватися під час потенціалу дії (катехоламіни через цАМФ-опосередкований механізм викликають фосфорування білків Са-каналів). У результаті цих процесів збільшується надходження іонів кальцію в саркоплазму, де підвищується їхня концентрація, і, як наслідок, збільшується сила скорочень кардіоміоци-тів, оскільки зростає кількість утворюваних кальцій-тропонінових комплексів.
Інотропна дія катехоламінів (а не закон Франка-Старлінга) є провідним механізмом адаптації серця до фізичних навантажень. При цьому показники кардіодинаміки змінюються таким чином:
а) збільшується ударний об'єм;
б) збільшується хвилинний об'єм серця як за рахунок збільшення ударного об'єму, так і за рахунок збільшення частоти серцевих скорочень (позитивний хронотроп-ний ефект катехоламінів);
в) зменшується кінцеводіастолічний об'єм (при рентгенівському дослідженні визначається зменшення об'єму серця);
г) зменшується кінцево систолічний об'єм.
27.12. Назвіть варіанти довгострокової адаптації серця до дії навантажень.
За Ф. Меєрсоном, виділяють три варіанти довгострокової адаптації серця.
1. Гіпертрофія серця у спортсменів ("адаптоване" серце). Розвивається при періодичних навантаженнях, інтенсивність яких поступово зростає, тобто в умовах тренувань. Є збалансованою гіпертрофією, при якій рівномірно збільшуються всі складові компоненти серця. Завдяки такій гіпертрофії істотно збільшуються функціональні резерви серця.
2. Компенсаторна гіпертрофія серця ("переадаптоване " серце). Є наслідком патологічних процесів, що мають стосунок до серця. Розрізняють два види компенсаторної гіпертрофії:
а) гіпертрофію від перевантажень (розвивається при вадах серця, артеріальній гіпертензії);
б) гіпертрофію від ушкодження (характерна для атеросклеротичних уражень, міокардіопатії).
Розвиток компенсаторної гіпертрофії серця характеризується такими особливостями:
1) патогенний фактор, що викликає гіпертрофію, діє постійно;
2) компенсаторна гіпертрофія, на відміну від гіпертрофії серця у спортсменів, є незбалансованою;
3) при компенсаторній гіпертрофії згодом розвивається недостатність серця.
3. Атрофія міокарда ("deadагітоване" серце). Характеризується зменшенням маси серця в результаті тривалої гіпокінезії і зменшення навантажень на серце.
27.13. Які механізми лежать в основі розвитку гіпертрофії серця?
При тривалому підвищенні навантаження на серце розвивається його гіперфункція, що згодом викликає структурні зміни в серці - гіпертрофію міокарда.
Найбільш доказовою теорією, що пояснює механізми переходу гіперфункції серця в його гіпертрофію, є концепція Ф. Меєрсона (рис. 121).
Рис. 121. Механізми компенсаторної гіпертрофії міокарда
Відповідно до цієї концепції, головною ланкою, що зв'язує підвищення функції клітини з роботою її генетичного апарату, є збільшення потенціалу фосфорування (ПФ):
![]()
де [АДФ], [Ф], [АТФ] - концентрації в цитоплазмі клітин відповідно АДФ, неорганічного фосфату і АТФ.
ПФ закономірно збільшується у двох випадках:
а) при посиленому використанні АТФ, що завжди спостерігається при збільшенні функціонального навантаження на клітини (при їхній гіперфункції);
б) при порушеннях утворення АТФ, що характерно для різних видів ушкодження клітин.
Збільшення показника ПФ викликає появу в клітинах речовин - регуляторів транскрипції, які, впливаючи на геном клітини, посилюють синтез інформаційної РНК на матриці генів, що кодують структуру функціонально важливих білків клі-
тини, у тому числі скорочувальних білків і ферментів. На роль речовин - регуляторів транскрипції претендує цілий ряд метаболітів, серед яких цАМФ, креатин, іони Mg2+, поліаміни (спермін, спермідин) та ін.
Таким чином, розвиток гіпертрофії серця можна описати такою послідовністю процесів: збільшення навантаження на серце (гіперфункція) → посилене використання АТФ, що перевищує інтенсивність його ресинтезу → збільшення потенціалу фосфорування → поява або збільшення концентрації в клітинах речовин - регуляторів транскрипції → зростання інтенсивності синтезу інформаційної РНК і процесів трансляції в рибосомах → посилення біосинтезу структурних, функціональних білків і білків-ферментів → збільшення маси міокарда, його гіпертрофія.
27.14. Які стадії виділяють у процесі розвитку компенсаторної гіпертрофії серця? Дайте їх характеристику.
За динамікою змін обміну, структури й функції міокарда в розвитку компенсаторної гіпертрофії серця виділяють три основні стадії (Ф. Мсєрсон).
1. Аварійна стадія. Розвивається безпосередньо після підвищення навантаження, характеризується поєднанням патологічних змін у міокарді (зникнення глікогену, зниження рівня креатинфосфату, зменшення вмісту внутрішньоклітинного калію і підвищення вмісту натрію, мобілізація гліколізу, накопичення лактату) з мобілізацією резервів міокарда і організму в цілому. У цій стадії підвищується навантаження на одиницю м'язової маси, а отже, інтенсивність функціонування структур (ІФС), відбувається швидке, протягом тижнів, збільшення маси серця за рахунок посиленого синтезу білків і потовщення м'язових волокон.
2. Стадія завершеної гіпертрофії і відносно стійкої гіперфункції. У цій стадії процес гіпертрофії завершений, маса міокарда збільшена на 100-120 % і далі не зростає, ІФС нормалізується. Патологічні зміни в обміні речовин і структурі міокарда не виявляються, споживання кисню, утворення енергії, вміст макроергічних сполук не відрізняються від норми. Нормалізуються гемодинамічні порушення. Гіпертрофоване серце пристосувалося до нових умов навантаження і протягом тривалого часу компенсує їх.
3. Стадія поступового виснаження і прогресуючого кардіосклерозу. Характеризується глибокими метаболічними і структурними змінами, які поволі накопичуються в енергоутворюючих і скорочувальних елементах клітин міокарда. Частина м'язових волокон гине й заміщається сполучною тканиною, ІФС знову зростає. Порушується регуляторний апарат серця. Прогресуюче виснаження компенсаторних резервів призводить до виникнення хронічної недостатності серця, а надалі - до недостатності кровообігу.
27.15. Які особливості гіпертрофованого серця є передумовою розвитку його декомпенсації?
Гіпертрофоване серце відрізняється від нормального низкою обмінних, функціональних і структурних ознак, які, з одного боку, дають йому можливість тривалий час долати підвищене навантаження, а з другого - створюють передумови для виникнення патологічних змін.
1. Збільшення маси серця відбувається за рахунок потовщення кожного м'язового волокна, що супроводжується зміною співвідношення внутрішньоклітинних структур. Об'єм клітини при цьому збільшується пропорційно кубу лінійних розмірів, а поверхня — пропорційно їх квадрату, що призводить до зменшення клітинної поверхні на одиницю маси клітини. Відомо, що через поверхню клітини відбувається її обмін з позаклітинною рідиною — поглинання кисню, поживних речовин, виведення продуктів метаболізму, обмін води і електролітів. Зазначені вище зміни створюють умови для погіршення постачання м 'язового волокна, особливо його центральних відділів.
2. Клітинна мембрана відіграє важливу роль у проведенні збудження і спряженні процесів збудження і скорочення, що здійснюється через тубулярну систему і сар-коплазматичний ретикулум. Оскільки ріст цих утворень при гіпертрофії м'язового волокна також відстає, то створюються передумови для порушення процесів скорочення і розслаблення кардіоміоцитів: унаслідок уповільнення виходу іонів кальцію в саркоплазму погіршується скорочення, а в результаті утруднення зворотного транспорту іонів кальцію в саркоплазматичний ретикулум - розслаблення, іноді можуть виникати локальні контрактури окремих кардіоміоцитів.
3. При гіпертрофії об'єм клітини зростає у більшій мірі, ніж об'єм ядра. Оскільки роль ядра полягає в забезпеченні білкового синтезу, а отже, і процесів відновлення внутрішньоклітинних структур, то відносне зменшення об'єму ядра (якщо порівнювати з цитоплазмою) може призводити до порушення синтезу білків і погіршення пластичного забезпечення клітини.
4. У процесі розвитку гіпертрофії маса мітохондрій спочатку збільшується швидше, ніж маса скорочувальних білків, створюючи умови для достатнього енергетичного забезпечення і належної компенсації функції серця. Однак надалі, у міру посилення процесу, збільшення маси мітохондрій починає відставати від росту маси цитоплазми. Мітохондрії починають працювати з граничним навантаженням, у них розвиваються деструктивні зміни, знижується ефективність їхньої роботи, порушується окисне фосфорування. Це веде до погіршення енергетичного забезпечення гіпертрофованої клітини.
5. Збільшення маси м'язових волокон найчастіше не супроводжується адекватним збільшенням капілярної мережі, особливо у випадках швидкого розвитку недостатності серця. Великі вінцеві артерії також не можуть забезпечити необхідне пристосування. Тому під час навантаження погіршується судинне забезпечення гіпертрофованого міокарда.
6. При розвитку гіпертрофії міокарда в процес обов'язково втягується нервовий апарат серця. Спостерігається посилене функціонування внутрішньосерцевих і екстракардіальних нервових елементів. Однак ріст нервових закінчень відстає від збільшення маси скорочувального міокарда. Відбувається виснаження нервових клітин, порушуються трофічні впливи, зменшується вміст норадреналіну в міокарді, що веде до погіршення його скорочувальних властивостей, утруднення мобілізації його резервів. Отже, порушується (регуляторне забезпечення серця.
7. Гіпертрофоване серце за рахунок збільшення маси його скорочувального апарату і систем енергозабезпечення здатне тривалий час виконувати значно більшу роботу, ніж серце нормальне, зберігаючи при цьому нормальний метаболізм. Однак здатність пристосовуватися до навантажень, що змінюються, діапазон адаптаційних можливостей у гіпертрофованого серця обмежені, зменшений функціональний резерв. Це робить гіпертрофоване серце через зазначену вище незбалансованість внутрішньоклітинних і тканинних структур більш уразливим у різних несприятливих обставинах.
27.16. Чим може бути обумовлений розвиток міокардіальної недостатності серця?
Міокардіальна недостатність серця розвивається внаслідок первинних уражень міокарда. Оскільки міокард складається з двох типів м'язових волокон - атипових, що становлять провідникову систему серця, і скорочувальних клітин робочого міокарда— то розвиток недостатності серця може бути пов'язаний з ушкодженням як перших, так і других. Звідси можна виділити два варіанти міокардіальної недостатності серця:
а) обумовлену ураженням провідникової системи серця — аритмічну;
б) обумовлену ушкодженням робочого міокарда. Іноді її називають міокардіопатичною.
27.17. Що таке аритмії серця? Як їх класифікують?
Аритміями серця називають порушення частоти, ритму, узгодженості й послідовності його скорочень.
Аритмія – порушення ЧСС, ритмічності скорочень, локалізації водія ритму чи провідності (Мурашко В.В., Електрокардіографія).
Розвиток аритмій може бути пов'язаний з порушеннями основних функцій провідникової системи серця: автоматизму, збудливості і провідності. На цьому ґрунтується класифікація аритмій, відповідно до якої виділяють:
I. Аритмії, обумовлені порушеннями автоматизму.
II. Аритмії, пов'язані з порушеннями збудливості.
III. Аритмії, обумовлені порушеннями провідності.
IV. Аритмії, пов'язані з поєднаними порушеннями збудливості і провідності.
27.18. Які аритмії серця можуть виникати в результаті порушення функції автоматизму?
Розрізняють дві групи аритмій, пов'язаних з порушенням автоматизму серця.
I. Номотопні аритмії. Генерація імпульсів до скорочення, як і в нормі, відбувається в синусно-передсердному вузлі. До цієї групи відносять:
а) синусну тахікардію - збільшення частоти серцевих скорочень;
б) синусну брадикардію - зменшення частоти серцевих скорочень;
в) синусну (дихальну) аритмію - зміну частоти серцевих скорочень у різні фази дихального циклу (прискорення при вдиху і уповільнення при видиху).
II. Гетеротопні аритмії - синдром слабкості синусно-передсердного вузла. Генерація імпульсів до скорочення відбувається не в синусно-передсердному вузлі, а в інших структурах провідникової системи, що є водіями ритму II і III порядку.
Синдром розвивається в результаті зменшення активності або припинення діяльності синусно-передсердного вузла при ушкодженні його клітин або первинних функціональних порушеннях. При цьому можуть розвиватися такі види патологічних ритмів серця:
а) передсердний повільний ритм — водій ритму міститься в структурах лівого передсердя, частота серцевих скорочень менше 70/хв.;
б) атріовентрикулярний ритм — джерелом імпульсів є водії ритму II порядку (верхня, середня або нижня частина атріовентрикулярного вузла), частота серцевих скорочень залежно від місця генерації імпульсів зменшується від 70 до 40/хв;
в) ідіовентрикулярний шлуночковий ритм — генерація імпульсів відбувається у водіях ритму III порядку (пучок Гісса або його ніжки), частота скорочень серця менше 40/хв.
27.19. Які причини і механізми розвитку синусної тахі- і брадикардії?
Синусні тахікардія і брадикардія належать до групи номотопних аритмій, пов'язаних з порушеннями функції автоматизму.
Здатність до автоматичного утворення імпульсів залежить від клітин, розташованих у провідниковій системі серця (β-клітини), у яких відбувається спонтанна повільна деполяризація клітинної мембрани в період діастоли (рис. 122). У результаті, при досягненні певного критичного рівня, виникає потенціал дії. Частота генерації імпульсів залежить від трьох чинників: а) максимального діастолічного потенціалу цих клітин; б) рівня критичного потенціалу, після досягнення якого виникає потенціал дії; і в) швидкості діастолічної деполяризації.
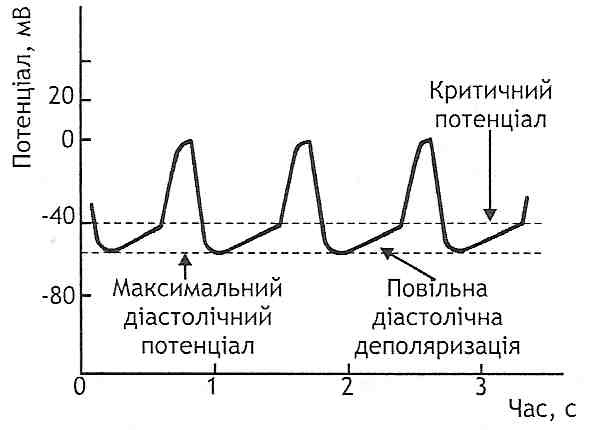
Рис. 122. Спонтанна генерація потенціалів дії в клітинах синусно-передсердного вузла
Зміна рівня максимального діастолічного потенціалу, критичного потенціалу або швидкості діастолічної деполяризації в той чи той бік веде до зміни частоти генерації імпульсів або до появи інших джерел імпульсації, якщо ці зміни виникають в інших, здатних до збудження ділянках серця і призводять до появи в них потенціалів дії. При зменшенні рівня максимального діастолічного потенціалу клітин синусно-передсердного вузла, при наближенні до нього критичного потенціалу або збільшенні швидкості повільної діастолічної деполяризації імпульси генеруються частіше, розвивається тахікардія. Це відбувається під впливом підвищеної температури тіла, при розтягненні ділянки синусно-передсердного вузла, при збудженні симпатичних нервів і дії катехоламінів.
Навпаки, зменшення швидкості повільної діастолічної деполяризації, гіперпо-ляризація в діастолі й віддалення критичного потенціалу від рівня максимального діастолічного, як це спостерігається при подразненні блукаючого нерва, супроводжуються уповільненням генерації імпульсів, а отже, і скорочень серця — брадикардією. Періодичні зміни тонусу блукаючого нерва під час акту дихання можуть викликати дихальну аритмію (прискорення серцебиття при вдиху, уповільнення — при видиху). Дихальна аритмія в нормі буває у дітей, але зрідка може спостерігатися і у дорослих.
27.20. Які аритмії виникають у результаті порушення збудливості міокарда? Який механізм їх розвитку?
В основі аритмій, пов'язаних з порушеннями функції збудливості, лежить поява розташованих поза синусно-передсердним вузлом так званих ектопічних осередків збудження, що генерують позачергові імпульси до скорочення.
У патологічних умовах може виявитися власний автоматизм нижчерозташова-них відділів провідникової системи серця (потенційних водіїв ритму). Такі умови можуть виникнути при зниженні автоматизму синусно-передсердного вузла або при підвищенні здатності до генерації імпульсів в інших ділянках міокарда. У цих випадках частота імпульсів, що генеруються нормальним водієм ритму, виявляється недостатньою для пригнічення автоматизму інших відділів, що призводить до появи додаткових імпульсів з ектопічно розташованих осередків збудження.
Іншим механізмом, що призводить до появи ектопічних осередків збудження, може бути виникнення різниці потенціалів між розташованими поруч міоцитами внаслідок, наприклад, неодночасного закінчення реполяризації в них, що може викликати збудження у волокнах, які вже вийшли з фази рефрактерності. Це явище спостерігається при локальній ішемії міокарда і при отруєнні серцевими глікозидами.
Серед аритмій цієї групи найчастіше бувають екстрасистолія і пароксизмальна тахікардія.
27.21. Що таке екстрасистолія? Назвіть види екстрасистол і їх основні електрокардіографічні характеристики.
Екстрасистолія - це вид аритмій, обумовлених порушеннями функції збудливості, що виявляє себе виникненням позачергових скорочень серця або тільки шлуночків. Такі позачергові скорочення отримали назву екстрасистол.
Залежно від локалізації осередку, з якого виходить позачерговий імпульс, розрізняють кілька видів екстрасистол: синусну (або номотопну), передсердну, передсерд-но-шлуночкову і шлуночкову (рис. 123). Оскільки хвиля збудження, що виникла в незвичному місці, поширюється в зміненому напрямку, це відображається на структурі електричного поля серця й знаходить відображення на електрокардіограмі. Кожний
вид екстрасистоли має свою електрокардіографічну картину, що дає можливість визначити місце ектопічного осередку збудження.
Синусна екстрасистола виникає внаслідок передчасного збудження частини клітин синусно-передсердного вузла. Електрокардіографічно вона не відрізняється від нормального скорочення, за винятком укорочення діасто-лічного інтервалу Т-Р. Унаслідок укорочення діастоли й зменшення наповнення шлуночків пульсова хвиля при екстрасистолі зменшена.
Передсердна екстрасистола спостерігається при наявності осередку ектопічного збудження в різних ділянках передсердь. Характеризується зміною форми зубця Р (знижений, двофазний, негативний) при збереженому комплексі QRS і деякому подовженні діастолічного інтервалу після екстрасистоли. Це обумовлено тим, що, ідучи ретроградним шляхом, збудження передчасно розряджає нормальний синусний імпульс, який збігається у часі зі збудженням шлуночків. Наступний передсердний імпульс, що виникає через нормальний інтервал, виявляється трохи віддаленим у часі від моменту закінчення збудження шлуночків - неповна компенсаторна пауза.
Передсердно-шлуночкова екстрасистола спостерігається при виникненні додаткового імпульсу в передсердно-шлуночковому вузлі. Хвиля збудження, що виходить з верхньої і середньої частин вузла, поширюється у двох напрямках: у шлуночках — у нормальному, у передсердях — у ретроградному. При цьому негативний зубець Р може накладатися на комплекс QRS. Діастолічний інтервал після екстрасистоли трохи подовжений. Екстрасистола може супроводжуватися також одночасним скороченням передсердь і шлуночків. При передсердно-шлуночковій екстрасистолі, що виходить із нижньої частини вузла, виникає компенсаторна пауза, така ж, як і при шлуночковій екстрасистолі.
Для шлуночкової екстрасистоли характерна наявність повної компенсаторної паузи після позачергового скорочення. Вона виникає внаслідок того, що збудження, яке охопило шлуночки, не передається через передсердно-шлуночковий вузол назад на передсердя, водночас черговий нормальний імпульс збудження, що йде від синусно-передсердного вузла, не поширюється на шлуночки, бо вони перебувають у фазі рефрактерності. Наступне скорочення шлуночків виникає тільки після приходу до них чергового нормального імпульсу. Тому тривалість компенсаторної паузи, разом з інтервалом, що передує екстрасистолі, дорівнює тривалості двох нормальних діасто-лічних пауз. Однак якщо скорочення серця настільки рідкі, що до моменту приходу чергового нормального імпульсу шлуночки встигають вийти зі стану рефрактерності, то компенсаторної паузи не буває. Позачергове скорочення потрапляє в інтервал між двома нормальними і в цьому випадку воно має назву вставної екстрасистолії. Оскільки хвиля збудження при шлуночковій екстрасистолії поширюється по шлуночках як у прямому, так і в ретроградному напрямках, це супроводжується значними змінами форми комплексу QRS, його деформацією.
Рис. 123. Екстрасистоли: а — передсердна, б — шлуночкова
27.22. Що таке пароксизмальна тахікардія? Чим вона характеризується?
Пароксизмальна тахікардія - це аритмія, обумовлена порушеннями функції збудливості, яка виявляється виникненням групи екстрасистол, що швидко повторюються і повністю пригнічують фізіологічний ритм.
При пароксизмальній тахікардії нормальний ритм серця раптово переривається нападом скорочень частотою від 140 до 250 ударів за хвилину. Тривалість нападу може бути різною - від кількох секунд до кількох хвилин, після чого він так само раптово припиняється і установлюється нормальний ритм.
Найчастіше спостерігається передсердна форма пароксизмальної тахікардії. А оскільки тривалість потенціалів дії і тривалість рефрактерних періодів збільшується по ходу провідникової системи, то дистально розташовані ділянки її не завжди здатні відтворити частоту імпульсації, що надходить з проксимальних відділів. Тому більша частина імпульсів при передсердній тахікардії не може проводитися передсердно-шлуночковим вузлом. Оскільки тривалість рефрактерних періодів і потенціалів дії у волокнах правої ніжки пучка Гісса більші, ніж у лівій, при високій частоті імпульсів частіше порушується проведення збудження саме до правого шлуночка.
27.23. Які аритмії виникають у результаті порушень функції провідності міокарда ?
Виділяють дві групи аритмій, пов'язаних з порушеннями провідності.
1. Блокади серця. Це аритмії, обумовлені уповільненням або повним припиненням проведення імпульсів по провідниковій системі.
Причиною блокади може бути ушкодження провідникових шляхів, що веде до подовження рефрактерного періоду і уповільнення або повного припиненням проведення імпульсу.
Порушення провідності можуть виникати між синусно-передсердним вузлом і передсердями, усередині передсердь, між передсердями і шлуночками, а також в одній із ніжок пучка Гісса. Тому виділяють такі види блокад:
а) внутрішньопередсердну;
б) передсердно-шлуночкову;
в) внутрішньошлуночкову.
2. Прискорене проведення імпульсів- синдром Вольфа-Паркінсона-Уайта (див. запит. 27.1.25).
27.24. Дайте характеристику передсердно-шлуночкової блокади серця.
Передсердно-шлуночкова, або поперечна, блокада серця може бути повною і неповною. У неповній блокаді серця розрізняють три ступені (рис. 124).
Передсердно-шлуночкова блокада І ступеня характеризується збільшенням часу проведення імпульсу від передсердь до шлуночків, що супроводжується подовженням інтервалу P-Q (0,2-0,5 с).
Блокада II ступеня (періоди Венкебаха) характеризується прогресуючим збільшенням інтервалу P-Q доти, доки одне із збуджень, найчастіше восьме-десяте, не проводиться. Після випадіння скорочення шлуночка інтервал P-Q відновлюється, поступово подовжуючись із кожним наступним скороченням серця. Вважають, що цей феномен пов'язаний з прогресуючим утрудненням проведення імпульсів через вузол.
При блокаді III ступеня спостерігають випадання кожного другого-третього скорочення або, навпаки, проводиться тільки кожне друге, третє або четверте збудження передсердь.
При повній передсердно-шлуночковій блокаді передсердя і шлуночки скорочуються незалежно одне від одного, кожне у своєму ритмі: передсердя з частотою близько 70, шлуночки — близько 35 скорочень в 1 хв. (ідіовентрикулярний ритм).
Особливе значення має момент переходу неповної блокади в повну, коли до шлуночків не надходять імпульси від передсердь. Повільна діастолічна деполяризація в потенційних водіях ритму виникає тільки через якийсь час після припинення надходження імпульсів від синусно-передсердного вузла. Цей період носить назву пре-автоматичної паузи, під час якої спостерігається асистолія шлуночків. При цьому внаслідок припинення надходження крові до головного мозку виникає втрата свідомості, судоми (синдром Морганьї—Едемса—Стокса). Можлива смерть, але найчастіше при поновленні скорочень шлуночків зазначені явища проходять. Синдром може повторюватися багаторазово.
Рис. 124. Передсердно-шлуночкова
блокада: а — І ступеня; б—II ступеня;
в — III ступеня; г - повна поперечна
блокада
27.25. Чим може бути обумовлений розвиток синдрому Вольфа-Паркінсона-Уайта ?
Синдром Вольфа—Паркінсона—Уайта характеризується прискореним проведенням імпульсів від передсердь до шлуночків, у результаті чого відбувається передчасне збудження останніх, розвивається тахікардія, зменшується інтервал P-Q на електрокардіограмі.
Причиною розвитку цього синдрому вважають існування додаткових шляхів проведення імпульсів. До таких шляхів відносять (рис. 125):
а) пучок Паладіно—Кента: видозмінена міокардіальна тканина, локалізована в зоні атріовентрикулярного кільця. Може існувати два пучки (лівий і правий), що містяться відповідно у кільцях двостулкового і тристулкового клапанів. Зазначені пучки проводять імпульси від передсердь до шлуночків, минаючи перед-сердно-шлуночковий вузол;
Рис. 125. Додаткові провідникові шляхи серця:
1 — пучок Паладіно— Кента;
2 — пучок Махайма;
3 — пучок Джеймса
б) пучок Махайма. З'єднує верхню частину пучка Гісса зі шлуночками;
в) пучок Джеймса. Зв'язує передсердя з нижньою частиною передсердно-шлуночкового вузла або з пучком Гісса.
По додаткових провідникових шляхах імпульси проводяться швидше і досягають шлуночків раніше імпульсів, які йдуть звичайним чином через передсердно-шлуноч-ковий вузол. Це призводить до передчасної активації частини шлуночків, інша їх частина збуджується пізніше імпульсами, що проходять через передсердно-шлуноч-ковий вузол. ,
27.26. Які аритмії виникають у результаті одночасного порушення функцій збудливості й провідності?
1. Тріпотіння передсердь (частота скорочень передсердь — 250-400/хв.).
2. Миготіння передсердь (частота імпульсів, що виникають у передсердях, становить 400-600/хв.).
Тріпотіння й миготіння передсердь мають однакові причини розвитку і можуть переходити одне в одне. Тому ці два види порушень ритму серця поєднують одним поняттям — миготлива аритмія.
3. Тріпотіння шлуночків (частота скорочень шлуночків — 150—300/хв.).
4. Миготіння (фібриляція) шлуночків (частота імпульсів у шлуночках- 300-500/хв, серце не скорочується).
27.27. Якими ознаками виявляє себе миготлива аритмія? Які механізми її розвитку?
При наявності численних ектопічних осередків збудження і такої зміни проведення імпульсу, при якій порушується швидкість його проведення по різних ділянках міокарда або відбувається поширення імпульсу тільки в одному напрямку, створюються умови для
тривалої циркуляції хвилі збудження в певному відділі серця, виникають розлади ритму —миготлива аритмія, що може виявлятися тріпотінням [миготінням передсердь (рис. 126).
![]()
Рис. 126. Миготлива аритмія
При тріпотінні передсердь частота їхніх скорочень досягає 250—400/хв. При цьому внаслідок нездатності шлуночків відтворювати високий ритм передсердь розвивається відносна серцева блокада; шлуночки відповідають скороченням на кожне друге, третє або четверте скорочення передсердь, тому що інші хвилі збудження потрапляють у фазу рефрактерності. Скорочення шлуночків можуть виникати раніше, ніж настане достатнє наповнення їх кров'ю - це викликає важкі порушення кровообігу.
Якщо кількість імпульсів у передсердях досягає 400-600/хв, говорять про миготіння, або фібриляцію передсердь. При цьому скорочуються лише окремі м'язові волокна, а все передсердя перебуває в стані неповного скорочення, його участь у перекачуванні крові припиняється. Імпульси, що безладно приходять до передсерд-но-шлуночкового вузла по окремих м'язових волокнах передсердя, здебільшого нездатні викликати його збудження, тому що застають вузол у стані рефрактерності або не досягають критичного рівня. Тому передсердно-шлуночковий вузол збуджується нерегулярно, і скорочення шлуночків носять випадковий характер. Як правило, число скорочень шлуночків за хвилину перевищує нормальне. Нерідко скорочення шлуночків відбуваються до їх наповнення кров'ю й не супроводжуються пульсовою хвилею. Тому частота пульсу стає меншою за частоту скорочень серця (дефіцит пульсу). Найчастіше причиною розвитку миготливої аритмії є (правило 03):
а) стеноз лівого атріовентрикулярного отвору,
б) тиреотоксикоз;
в) атеросклеротичний кардіосклероз.
Нині найбільш визнаною теорією, що пояснює механізми розвитку миготливої аритмії (і екстрасистолії!), є теорія повторного входження імпульсів (re-entry) (рис. 127). Відповідно до неї, тріпотіння і миготіння виникають як наслідок порушень провідності, при яких поширення імпульсів припиняється тільки в одному (антеградному) напрямку і зберігається у зворотному (ретроградному). При цьому створюються умови для постійного руху імпульсів по колу в міокарді. У нормальних умовах хвиля збудження, виникнувши в одному місці, поширюється в обидва боки серцевої камери. Досягнувши протилежної стінки, вона згасає, зустрівшись з іншою хвилею, що залишила за собою зону рефрактерності. Якщо ж унаслідок виникнення тимчасового блоку або запізнення приходу збудження по деяких волокнах міокарда збудження приходить до місця, що вже вийшло зі стану рефрактерності, то створюються умови для тривалеї циркуляції імпульсу, що виник.
Рис. 127. Механізм повторного входження імпульсів
27.28. Що таке фібриляція шлуночків? Коли вона виникає й чим виявляє себе?
При деяких патогенних впливах (проходження електричного струму через серце, наркоз хлороформом або циклопропаном, закупорка вінцевих артерій або інші випадки різкої гіпоксії, травма серця, дія токсичних доз наперстянки і кальцію) виникає фібриляція шлуночків. При цьому внаслідок хаотичного скорочення окремих м'язових волокон пропульсивна сила скорочень практично зникає, кровообіг припиняється, швидко настає втрата свідомості і смерть. Фібриляції сприяють зменшення концентрації внутрішньоклітинного калію, що веде до зниження мембранного потенціалу кардіоміоцитів і легшого виникнення в них деполяризації та збудження, а також зміна вмісту нервових медіаторів, особливо катехоламінів.
При лікуванні фібриляції шлуночків найефективнішим є пропускання через серце короткого сильного одиничного електричного розряду (дефібриляція). При цьому відбувається одночасна деполяризація всіх волокон міокарда і припиняються асинхронні збудження м'язових волокон. Як захід, що попереджає розвиток фібриляції, застосовують корекцію електролітного складу крові.
27.29. Чим може бути обумовлений розвиток порушень скорочувальної функції міокарда ?
У клітинах робочого міокарда відбувається п'ять процесів, які визначають скорочувальну функцію серця. Усі вони можуть порушуватися і становити основу розвитку недостатності серця.
Розлади скорочувальної функції міокарда можуть бути обумовлені порушеннями:
1) збудження кардіоміоцитів;
2) електромеханічного спряження;
3) процесів власне скорочення;
4) процесів розслаблення;
5) енергозабезпечення міокарда.
27.30. Які фактори впливають на характер потенціалу дії і збудливість кардіоміоцитів? Якими електрофізіологічними ознаками виявляють себе зміни їхньої збудливості?
Основні характеристики потенціалу дії (тривалість, амплітуда, крутість наростання деполяризації) і збудливість кардіоміоцитів залежать від таких чинників (рис. 128):
Рис. 128. Потенціал дії скорочувальних клітин міокарда: І—фаза швидкої деполяризації; II- фаза повільноїреполяризації (фаза "плато "); III — фаза швидкої реполяризації
а) характеру електричної імпульсації, що надходить до кардіоміоцитів по провідниковій системі серця. При аритміях можливе порушення основних параметрів потенціалу дії волокон робочого міокарда і його збудливості;
б) стану (властивостей) сарколеми кардіоміоцитів, і насамперед іонних каналів. Нині відомі хімічні агенти, здатні вибірково порушувати провідність цих каналів. Це блокатори Na-каналів, Са-каналів, К-каналів;
в) концентрації позаклітинних іонів, що беруть участь у формуванні електричних потенціалів на мембрані кардіоміоцитів. В умовах in vivo найбільше значення має позаклітинна концентрація іонів калію.
Зміни збудливості волокон міокарда можуть виявляти себе:
1) змінами тривалості потенціалів дії, а отже, і сили серцевих скорочень. Зменшення тривалості потенціалів дії виникає при збільшенні частоти стимуляції, збільшенні позаклітинної концентрації іонів калію, при дії ацетилхоліну. Збільшення тривалості потенціалу дії характерно для охолодження;
2) змінами тривалості періодів абсолютної і відносної рефрактерності, а отже, здатності сприймати імпульсацію (засвоювати ритм). Тривалість цих періодів залежить від тривалості потенціалів дії.
27.31. Як впливають зміни позаклітинної концентрації іонів калію на збудливість міокарда?
Позаклітинна концентрація іонів калію впливає на збудливість міокарда через рівень потенціалу спокою кардіоміоцитів.
Збільшення концентрації іонів калію — гіперкаліємія - викликає деполяризацію мембрани м'язових волокон. При цьому характер змін збудливості міокарда залежить від рівня гіперкаліємії. Виділяють такі діапазони концентрацій іонів калію, для яких характерні певні картини порушень:
1) від 4 до 8 ммоль/л. Відбувається незначна деполяризація, мембранний потенціал падає від -90 мВ до -80 мВ. При цьому стан Na-каналів сарколеми істотно не змінюється. Унаслідок того, що величина мембранного потенціалу наближається до критичного рівня деполяризації, збудливість м'язових волокон і швидкість проведення імпульсів зростають;
2) від 8 до 35 ммоль/л. Мембранний потенціал падає від -80 мВ до -40 мВ. При такому рівні деполяризації значно зменшується провідність швидких потенціал-залежних Na-каналів (період відносної рефрактерності). Наслідком є зменшення збудливості і провідності, а також зміни характеру потенціалів дії (зменшення тривалості, амплітуди, крутості);
3) понад 35 ммоль/л. Мембранний потенціал стає менше -40 мВ. При цьому всі швидкі потенціалзалежні Na-канали перебувають у стані інактивації (абсолютна рефрактерність) — відбувається зупинка серця.
Зменшення позаклітинної концентрації іонів калію — гіпокаліємія — викликає гі-перполяризацію мембрани. Якщо гіпокаліємія досягає рівня 2—3 ммоль/л, дещо збільшується поріг деполяризації, зменшується збудливість, збільшуються тривалість потенціалів дії і сила скорочень серця.
27.32. Що таке кардіоплегія? Які використовують підходи до її здійснення?
Кардіоплегія — це штучна зупинка серця, яку застосовують під час операцій на "сухому" серці.
Розроблено такі підходи до здійснення кардіоплегії: а) ішемічна кардіоплегія. Перетискають аорту до відходження вінцевих артерій. Оскільки через 30 хв. починаються необоротні зміни в міокарді, цей підхід можна використати при короткочасних операціях (до 10—15 хв.);
б) хімічна кардіоплегія. У вінцеві артерії вводять кардіоплегічні розчини, до складу яких входять високі концентрації іонів калію, ацетилхолін і деякі інші препарати. Цей метод дозволяє проводити операції на "сухому" серці протягом 40-60 хв.;
в) холодова кардіоплегія. її досягають зрошенням серця охолодженим до +4 °С... — 5 °С фізіологічним розчином. Тривалість операцій на серці при цьому може становити 60 хв.
27.33. Які фактори впливають на характер електромеханічного спряження в міокарді?
1. Тривалість потенціалу дії.
2. Частота потенціалів дії.
3. Концентрація іонів кальцію в позаклітинній рідині.
4. Стан Са-каналів сарколеми кардіоміоцитів.
5. Стан систем видалення іонів кальцію із цитоплазми м'язових волокон.
27.34. Як зміни тривалості і частоти потенціалів дії впливають на силу серцевих скорочень?
Зміни тривалості потенціалів дії в кардіоміоцитах відбуваються за рахунок укорочення або подовження фази "плато ", під час якої іони Са2+ через потенціалзалеж-ні Са-канали сарколеми надходять у саркоплазму.
З урахуванням того, що концентрація іонів Са2+ у цитоплазмі м'язових волокон визначає кількість актоміозинових комплексів, що утворюються, а отже, і силу скорочень, можна зробити такий висновок. При зменшенні тривалості потенціалу дії зменшується надходження іонів Са2+ у саркоплазму і, як наслідок, зменшується сила скорочення м'язового волокна. При збільшенні тривалості потенціалу дії — все навпаки.
Тривалість потенціалу дії кардіоміоцитів зменшується при гіперкаліємії, при дії на м'язові волокна ацетилхоліну. Збільшення тривалості потенціалу дії характерне для гіпокаліємії, охолодження серця.
При збільшенні частоти потенціалів дії, незважаючи на те, що тривалість кожного окремого потенціалу, а отже, і фази "плато" зменшується, сумарна тривалість зазначеної фази за одиницю часу збільшується. Це означає, що в кардіоміоцитах створюються більш високі концентрації Са2+ і, як наслідок, збільшується сила скорочення м'язових волокон. Цим, зокрема, пояснюється хроноінотропний механізм негайної компенсації серця (див. запит. 27.10).
При зменшенні частоти потенціалів дії (наприклад, при брадикардії) сумарна тривалість фази "плато" за одиницю часу зменшується, що веде до зменшення концентрації Са2+ у саркоплазмі і зменшення сили скорочень серця.
27.35. Як впливає позаклітинна концентрація іонів кальцію на силу скорочень серця? Що таке "кальцієвий парадокс"?
На початку XX ст. Рінгером було показано, що в розчині, позбавленому іонів кальцію, ізольоване серце швидко зупиняється. Нині відомо, що причиною цього є повне роз'єднання збудження і скорочення. В умовах норми іони кальцію, що надходять під час фази "плато" потенціалу дії з позаклітинного середовища в саркоплазму
кардіоміоцитів, "запускають" вивільнення Са2+ із саркоплазматичного ретикулуму й поповнюють його запаси в цих структурах м'язових волокон. При зменшенні позаклітинної концентрації Са2+ його запаси в саркоплазматичному ретикулумі швидко виснажуються, концентрація Са2+ у саркоплазмі падає й, отже, зменшується сила серцевих скорочень.
"Кальцієвий парадокс" — це експериментальний феномен, що розвивається після того, як у безкальцієвий розчин, яким здійснювали перфузію серця протягом декількох хвилин, вносять іони кальцію. При цьому розвивається необоротне ушкодження міокарда: зменшується вміст АТФ і креатинфосфату, з кардіоміоцитів виходять білки, у тому числі ферменти (міоглобін, креатинкіназа), руйнується саркоплаз-матичний ретикулум.
"Кальцієвий парадокс" пояснюють тим, що в безкальцієвому розчині відбувається розшарування глікокалікса кардіоміоцитів (зовнішній шар глікокалікса відокремлюється від внутрішнього), у результаті чого значно зростає проникність сарколеми до іонів кальцію. При наступному внесенні в середовище іонів Са2+ відбувається їх масивне надходження у клітини, різко зростає його концентрація в саркоплазмі, що "запускає" кальцієві механізми ушкодження (див. розд. 11).
27.36. Які фактори впливають на стан кальцієвих каналів сарколеми кардіоміоцитів? Чим можуть бути обумовлені їх активація і блокада?
У сарколемі кардіоміоцитів є потенціолзалежні повільні Са-канали. Це білкові молекули, вмонтовані в плазматичну мембрану і здатні пропускати через себе іони кальцію в тому випадку, коли вони відкриті. Відкриття Са-каналів відбувається при деполяризації мембрани. Відкриватися здатні не всі наявні канали, а тільки фосфо-ровані (активовані).
Під активацією Са-каиалів розуміють їх фосфорування, у результаті чого збільшується кількість Са-каналів, здатних відкриватися при виникненні потенціалу дії. В основі фосфорування Са-каналів лежить збільшення концентрації цАМФ у саркоплазмі м'язових волокон. Усі фактори, що викликають утворення цАМФ, ведуть до активації Са-каналів. До таких, зокрема, належать:
а) катехоламіни і фармакологічні агенти, що активують fi-адренорецептори;
б) інгібітори фосфодіестерази (метилкеантини: теофілін, кофеїн та ін.). Зазначені агенти збільшують вміст цАМФ або через активацію аденілатциклази (через 0-адренорецептори), або через пригнічення руйнування цАМФ (інгібітори фосфодіестерази).
З активацією Са-каналів пов'язаний позитивний інотропний ефект катехола-мінів/Q основі його така послідовність подій: катехоламіни → активація р-адрено-рецепторів → активація аденілатциклази → утворення цАМФ → активація проте-їнкіназ → фосфорування Са-каналів сарколеми → збільшення надходження Са2+ у саркоплазму під час потенціалів дії → збільшення концентрації Са2+ у саркоплазмі → збільшення сили скорочень серця.
В умовах блокади Са-капалів зменшується надходження іонів Са в саркоплазму кардіоміоцитів і, отже, зменшується сила серцевих скорочень.
Блокаду Са-каналів викликають:
а) ендогенні фактори — дефіцит АТФ, дефіцит цАМФ, іони водню (ацидоз);
б) екзогенні фактори — двовалентні іони (Ni2+, Со2+, Мп2+), деякі тривалентні іони (La3+), органічні сполуки, що їх застосовують у практичній медицині (верапаміл, ніфедипін та ін.).
27.37. Які фактори впливають на стан систем видалення іонів кальцію з кардіоміоцитів і, як наслідок, на силу серцевих скорочень?
До таких чинників відносять:
а) серцеві глікозиди — фармакологічні препарати рослинного походження, що підвищують силу скорочень серця (препарати дигіталісу, строфантин та ін.);
б) ендогенні дигіталісоподібні і строфантиноподібні фактори. Вважають, що серцеві глікозиди, яких здавна використовують в медицині, імітують дію цих, відносно недавно відкритих, ендогенних природних факторів. Фізіологічне значення останніх сьогодні ще не встановлено.
Механізм дії серцевих глікозидів і подібних до них ендогенних факторів пов'язаний з пригніченням активності Na-K-АТФ-ази кардіоміоцитів. Наслідком цього є порушення роботи Na-K-насосів сарколеми, зменшення градієнта концентрацій іонів Na+ по обидва боки плазматичної мембрани м'язових волокон. Це призводить до порушення Na-Ca-обмінного механізму в кардіоміоцитах, у результаті чого зменшується видалення Са2+ із саркоплазми в позаклітинне середовище і збільшується внутрішньоклітинна концентрація Са2+. Останнє і обумовлює збільшення сили серцевих скорочень.
27.38. Від чого залежить сила скорочень окремих кардіоміоцитів?
На силу скорочень м'язових волокон серця впливають:
1) концентрація іонів Са2+у саркоплазмі. Залежність тут така: що вищий вміст Са2+ у саркоплазмі, то більше утворюється комплексів Са2+ з тропоніном С, то більше вивільняється центрів зв'язування (активних центрів) на актинових міофіламентах, то більше утворюється "містків" між актином і головками міозину, то більшою буде сила скорочення м'язового волокна. При зменшенні концентрації Са2+ у саркоплазмі — навпаки;
2) ступінь спорідненості тропоніну С до іонів кальцію. Іони водню й неорганічного фосфату, зв'язуючись з тропоніном С, унеможливлюють взаємодію цього білка з Са2+, у результаті чого сила скорочень кардіоміоцитів зменшується;
3) стан скорочувальних білків — актину і міозину. Велике значення має взаємне розташування актинових і міозинових міофіламентів. Воно лежить в основі залежності, що її описує закон Франка-Старлінга. При дуже сильному розтягненні м'язових волокон кількість утворюваних актоміозинових "містків" зменшується - зазначений закон не "спрацьовує", сила скорочень серця падає;
4) концентрація АТФ, енергія гідролізу якого забезпечує ковзання м'язових філамен-тів один відносно одного.
27.39. Що лежить в основі розслаблення кардіоміоцитів? Які механізми забезпечують видалення іонів кальцію із саркоплазми волокон міокарда? Що може бути причиною порушень розслаблення серцевого м'яза?
Основний процес, що визначає розслаблення кардіоміоцитів, — це видалення іонів кальцію із саркоплазми, у результаті чого концентрація Са2+ у ній зменшується і стає нижче 10"7 моль/л. При цьому комплекси Са2+ з тропоніном С розпадаються, тро-поміозин зміщується по відношенню до актинових філаментів і закриває їхні активні центри — скорочення припиняється.
Існує три механізми видалення іонів Са2 + із саркоплазми кардіоміоцитів:
1) Са-насоси плазматичної мембрани і саркоплазматичного ретикулуму. Видаляють Са2+ у позаклітинне середовище і цистерни саркоплазматичного ретикулуму. Складовою їх частиною є Са-АТФ-аза, яка для здійснення активного транспорту іонів Са2+ використовує енергію АТФ;
2) Na-Ca-обмінний механізм. Видаляє іони Са2+ у позаклітинне середовище. Є різновидом вторинного активного транспорту (антипорту). Використовує енергію градієнта концентрацій іонів натрію по обидва боки плазматичної мембрани, тому залежить від роботи Na-K-насоса, що створює цей градієнт;
3) Са-акумулятивна функція мітохондрій. Активується тільки при значному підвищенні вмісту іонів Са2+ у саркоплазмі, що найчастіше буває в умовах патології. Видалення Са2+ із саркоплазми в матрикс мітохондрій відбувається за рахунок енергії, що вивільняється в процесі транспорту електронів по дихальному ланцюгу. Використання цієї енергії на активний транспорт іонів Са2+ у мітохондрії є альтернативою окисному фосфоруванню.
Основними причинами порушень розслаблення кардіоміоцитів є:
а) дефіцит АТФ. При цьому порушується енергозабезпечення Са- і Na-K-насосів, а також не відбувається розщеплення актоміозинових "містків", що утворилися в процесі скорочення;
б) порушення роботи Са-транспортних систем. Відомі спадково обумовлені дефекти білків Са-насосів, що призводять до розвитку кардіопатії.
Порушення розслаблення кардіоміоцитів виявляють себе розвитком м'язових контрактур.
27.40. Яке значення має АТФ у забезпеченні функцій клітин міокарда? Чим можуть бути обумовлені порушення енергетичного обміну в серцевому м 'язі?
Енергія гідролізу АТФ у функціонально активних кардіоміоцитах забезпечує:
1) механічну роботу скорочень міофібрил (ковзання міофіламентів один відносно одного);
2) осмотичну роботу — активний транспорт іонів Са2+, Na +, К+ проти градієнтів концентрацій (робота іонних насосів);
3) фосфорування білків Са-каналів, фосфоламбану (білка Са-насосів саркоплазматичного ретикулуму).
Розлади енергозабезпечення кардіоміоцитів можуть бути пов'язані з порушеннями:
а) ресинтезу АТФ (гіпоксія, голодування, дефіцит вітамінів, зменшення активності ферментів енергетичного обміну);
б) транспорту АТФ із мітохондрій до місць його використання (порушення креатин-кіназної транспортної системи);
в) утилізації АТФ (зменшення АТФ-азної активності структур кардіоміоцитів).
27.41. Дайте порівняльну характеристику гіпо- і гіперкальцієвого варіантів недостатності серця.
Сучасний рівень знань про молекулярні механізми скорочувальної функції серця дозволяє виділити два принципово різних патогенетичних варіанти недостатності серця: гіпокальцієвий і гіперкальцієвий, для яких характерне відповідно зменшення і збільшення концентрації іонів Са2+ у саркоплазмі кардіоміоцитів.
Гіпокальцієвий варіант розвивається в результаті порушень збудження і електромеханічного спряження у волокнах міокарда. Це буває при аритміях (брадикардії різного походження, блокади), короткочасній ішемії міокарда (порушується фосфорування Са-каналів у результаті дефіциту АТФ), ацидозі (блокада Са-каналів іонами водню), гіпокальціємії. Виявляє себе зменшенням сили серцевих скорочень. Основний принцип лікування — підвищення вмісту іонів Са2+ у саркоплазмі кардіоміоцитів під час систоли серця. Для цього використовують: а) серцеві глікозиди; б) катехола-міни і р-адреноміметики; в) парні електричні стимули.
Гіперкальцієвий варіант розвивається в результаті посиленого надходження іонів Са2+ у саркоплазму кардіоміоцитів з позаклітинного середовища (усі види ушкодження сарколеми, при яких підвищується її проникність) або внаслідок порушень видалення Са2+ із саркоплазми (дефіцит АТФ, порушення функції Са-транспортних систем). Виявляється розвитком контрактури (перескорочення) міофібрил, у результаті чого стає неможливим розслаблення м'язових волокон, а отже, і наступне їх скорочення. Основний принцип лікування — зменшення вмісту іонів Са2+ у саркоплазмі кардіоміоцитів. Для цього використовують: а) β-адреноблокатори; б) блокатори Са-каналів.
27.42. Коли розвивається позаміокардіальна недостатність серця? Які компенсаторні механізми характерні для неї?
Позаміокардіальна недостатність розвивається в тих випадках, коли до серця притікає мало крові по венах або коли воно не в змозі прийняти всю кров, що надходить. Перше буває при гіповолемії (крововтрата) або різкому розширенні судин (колапс), друге - при накопиченні рідини в порожнині перикарда, що утруднює розширення порожнин під час діастоли.
Накопичення рідини в порожнині перикарда може відбуватися швидко і повільно. Швидке накопичення відбувається внаслідок крововиливу при пораненні або розриві серця, при перикардиті, що швидко розвивається. Через погану розтяжність перикарда в порожнині підвищується тиск, що перешкоджає діастолічному розширенню сер-
ця, виникає гостра тампонада серця. В експерименті цей процес моделюють введенням рідини в порожнину перикарда (О. Фохт). При цьому спостерігають, як зменшується кро-вонаповнення порожнин серця, знижується ударний об'єм і падає артеріальний тиск. Між внутрішньоперикардіальним і артеріальним тиском існує чітка обернена залежність: що більший внутрішньоперикардіальний тиск, то нижчий артеріальний. Венозний тиск при цьому підвищується.
Вмикання компенсаторних механізмів при перикардиті відбувається рефлекторно за участю сигналів, що надходять з трьох рецепторних полів:
1) отворів порожнистих і легеневих вен — підвищення тиску на шляхах припливу;
2) аорти і сонних синусів (синокаротидні зони) — зниження тиску на шляхах відтоку;
3) перикарда — підвищення внутрішньоперикардіального тиску. При перетинанні блукаючих і депресорних нервів, а також при вимиканні рецепторних полів за допомогою новокаїну пристосувальні реакції не здійснюються - порушення кровообігу мають набагато тяжчий перебіг. При тампонаді серця мобілізація потужних механізмів компенсації, які ведуть до посилення скорочень серця (гомео- і гетерометричні механізми, інотропний ефект катехоламінів), малоефективна або неможлива. Тому діють тільки відносно малопотужні і енергетично невигідні механізми компенсації і підтримки артеріального тиску - збільшення частоти серцевих скорочень і звуження периферичних судин. Цим і пояснюється важкий клінічний перебіг гострої тампонади серця.
При повільнішому накопиченні рідини в перикарді робота компенсаторних механізмів виявляється ефективнішою, підвищення внутрішньоперикардіального тиску протягом деякого часу може компенсуватися. Повільне накопичення рідини, що спостерігається при хронічному ексудативному перикардиті і гідроперикарді, супроводжується поступовим розтягуванням перикарда і збільшенням об'єму навколосерцевої сумки. Унаслідок цього внутрішньоперикардіальний тиск змінюється порівняно мало, а порушення кровообігу не виникають тривалий час.
27.43. Які зміни показників кардіо- і гемодинаміки характерні для недостатності серця?
При декомпенсації серця показники кардіодинаміки змінюються в такий спосіб:
1) зменшується серцевий виштовх (ударний об'єм);
2) збільшується кінцевосистолічний об 'єм;
3) збільшується кінцеводіастолічний об 'єм;
4) збільшується кінцеводіастолічний тиск, у результаті чого розвивається міогенна дилатація серця (розширення його порожнин);
5) зменшуються серцеві індекси, тобто показники, що характеризують скорочувальну активність серця. Серед них велике значення має зниження dP/dt — максимальної швидкості приросту тиску під час періоду ізоволемічного скорочення;
6) збільшується частота серцевих скорочень (тахікардія). Розвивається рефлекторно в результаті збудження рецепторів устя переповнених кров'ю порожнистих вен (рефлекс Бейнбріджа). Має також значення безпосереднє збудження клітин синусно-передсердного вузла в результаті підвищення тиску крові в порожнині правого передсердя. Розлади гемодинаміки характеризуються такими змінами:
1) зменшується хвилинний об'єм крові;
2) якщо недостатність серця розвивається за лівошлуночковим типом, то:
а) зменшується артеріальний тиску великому колі кровообігу;
б) збільшується загальний периферичний опір (реакція, спрямована на зменшення падіння артеріального тиску);
в) збільшується тиск крові в малому колі кровообігу — гіпертензія малого кола, що призводить до застою крові в легенях;
3) якщо недостатність серця розвивається за правошлуночковим типом, то:
а) зменшується артеріальний тиску малому колі кровообігу;
б) збільшується опір судин малого кола;
в) збільшується центральний венозний тиск— застій крові у великому колі кровообігу;
4) збільшується об'єм циркулюючої крові - розвивається гіперволемія. Вона є результатом затримки води в організмі і поліцитемії.
27.44. Якими клінічними синдромами й ознаками виявляє себе недостатність серця?
1. Циркуляторна гіпоксія (див. розд. 19).
2. Задишка. У її розвитку мають значення:
а) вплив надлишку іонів водню на хеморецептори судин і безпосередньо на дихальний центр;
б) набряк інтерстиціальної тканини легень і пов'язане з цим збудження J-pe-цепторів.
3. Ціаноз. Обумовлений збільшенням концентрації відновленого гемоглобіну в результаті більш повного вилучення кисню тканинами.
4. Набряки. При правошлуночковій недостатності розвиваються набряки нижньої половини тіла; при лівошлуночковій - інтерстиціальний набряк легень (синдром серцевої астми) або альвеолярний набряк (синдром набряку легень).
5. Кардіальний цироз печінки. Характерний для правошлуночкової недостатності серця. Виявляється порушеннями функції печінки й синдромом портальної гіпертензії (див. розд. 31).
6. Порушення кислотно-основного стану. Можливі такі їх варіанти:
а) негазовий ацидоз — у крові накопичуються кислі продукти обміну речовин, зокрема, молочна кислота. Є відображенням гіпоксії;
б) газовий ацидоз - може бути проявом альвеолярного набряку легень. У результаті розвитку недостатності зовнішнього дихання виникає гіперкапнія;
в) негазовий алкалоз — може бути результатом вторинного гіперальдостероніз-му і обумовленої ним гіпокаліємії;
г) газовий алкалоз — може бути наслідком рефлекторної задишки (гіпервенти-ляції), що призводить до гіпокапнії.
7. Вторинний гіперальдостеронізм (див. розд. 33). Обумовлює електролітні порушення в організмі — гіпернатріємію, гіпергідрію, гіпокаліємію.
8. Поліцитемічна гіперволемія (див. розд. 26.1).
27.45. Якими особливостями характеризується вінцевий кровообігу серці?
1. Високий рівень екстракції кисню в капілярах серця. У серці екстрагується 70—75 % 02, що надходить з артеріальною кров'ю, тимчасом як у тканинах головного мозку — 25 %, у скелетних м'язах і печінці — близько 20 %, у нирках — 10 %. Високий рівень вилучення 02 у серці пояснюється значною довжиною його капілярів і у зв'язку з цим — більшим часом контакту крові зі стінкою капілярів.
При збільшенні потреби серця в кисні вона не може бути задоволена збільшенням екстракції 02 (як у скелетних м'язах), оскільки остання і так є максимально можливою в стані спокою. Тому для забезпечення збільшених енергетичних потреб серця залишається тільки один шлях — збільшення вінцевого кровообігу.
2. Високий базальний тонус вінцевих судин. Він дає можливість у стані спокою забезпечувати вінцевий кровообіг на рівні 250-300 мл/хв, що становить близько 5 % хвилинного об'єму крові.
Високий базальний тонус вінцевих судин обумовлює високий резерв вінцевого кровообігу. Так, при зменшенні базального тонусу судин серця Інтенсивність кровообігу у них може зростати в 7—10 разів.
3. Фазний характер вінцевого кровообігу, пов'язаний з періодами серцевого циклу. Під час систоли відбувається здавлювання інтрамуральних судин - кровообіг мінімальний і становить близько 15 % загального вінцевого кровообігу. Під час діастоли здавлювання судин припиняється і кровообіг стає максимальним (близько 85 % загальної величини).
Фазність вінцевого кровообігу найбільш виражена в субендокардіальній зоні міокарда (найбільше здавлювання судин) і найменш виражена - у субепікардіальній зоні.
4. Підпорядкованість вінцевого кровообігу метаболічним потребам серця і відносна незалежність його від нервових регуляторних впливів. В умовах патології ця підпорядкованість часто порушується і збільшується чутливість вінцевих судин до нервових імпульсів.
5. Винятково висока чутливість вінцевих судин до зменшення напруги кисню в крові. Зменшення р02 артеріальної крові всього лише на 5 % істотно збільшує інтенсивність вінцевого кровообігу.
6. Недостатній розвиток колатеральних судин. При несприятливих умовах колате-ралі в серці не можуть компенсувати порушення течії крові у вінцевих судинах, тому колатеральний кровообіг тут функціонально неповноцінний.
27.46. Як здійснюється регуляція вінцевого кровообігу?
У регуляції вінцевого кровообігу розрізняють міогенну ауторегуляцію, метаболічну і нервову регуляцію.
1. Міогенна ауторегуляція. її основу становить закон Бейліса, відповідно до якого при розтягненні гладких м 'язів кровоносних судин збільшується сила їх скорочення. Міогенна ауторегуляція забезпечує сталість вінцевого кровообігу і відносну незалежність його від змін артеріального тиску. Так, при збільшенні тиску крові в аорті збільшується розтягування гладком'язових клітин вінцевих артерій, що веде до їх скорочення, підвищення тонусу артерій і збереження сталості кровообігу. При зменшенні артеріального тиску вінцевий кровообіг підтримується на постійному рівні завдяки розслабленню гладких м'язів і розширенню артерій.
Нині показано, що при розтягуванні гладком'язових клітин судин збільшується проникність їхньої плазматичної мембрани до іонів кальцію. Останні проникають у клітини і викликають їх скорочення.
2. Метаболічна регуляція. Підпорядковує вінцевий кровообіг метаболічним потребам серця. Здійснюється за допомогою цілого ряду іонів і метаболітів, серед яких іони водню, калію, молочна кислота, простагландини. Однак найбільше значення мають два фактори: зменшення напруги 02 в артеріальній крові і аденозин. Останній утворюється в результаті гідролізу аденінових нуклеотидів при гіпоксії і при посиленій роботі серця. Будучи природним блокатором Са-каналів, аденозин зменшує надходження іонів Са2+ у цитоплазму гладком'язових клітин вінцевих судин, унаслідок чого зменшується ступінь їхнього скорочення і падає базальний тонус - вінцеві судини розширюються, коронарний кровообіг зростає (рис. 129).
Рис. 129. Один з механізмів метаболічної регуляції вінцевого кровообігу
3. Нервова регуляція. Нейрогенний тонус вінцевих судин незначний, про що свідчить майже повна відсутність змін вінцевого кровообігу після повної денерва-ції судин серця. Набагато більше значення має опосередкований вплив нервової системи на коронарний кровообіг. Він здійснюється через зміни роботи серця та інтенсивності обміну речовин у ньому.
В експерименті показано можливість і безпосереднього впливу нервів на тонус вінцевих судин. Так, при подразненні парасимпатичних нервів і введенні ацетилхоліну відбувається незначне розширення вінцевих артерій. Медіатори симпатичної нервової системи (катехоламіни) при дії на а-адренорецептори викликають звуження судин, а впливаючи на р-адренорецептори — розширення. Оскільки в нормі у вінцевих судинах переважають β-адренорецептори, то загальний ефект симпатичних впливів — незначне розширення судин серця.
27.47. Що таке недостатність вінцевого кровообігу? Чим відрізняються відносна і абсолютна коронарна недостатність?
Недостатність вінцевого кровообігу - це патологічний стан, що характеризується нездатністю вінцевих судин забезпечувати кровопостачання серця відповідно до його енергетичних потреб. Інакше кажучи, виникає невідповідність між енергетичними потребами серця і доставкою кисню і поживних речовин вінцевими судинами.
Недостатність вінцевого кровообігу може бути відносною і абсолютною.
Відносна коронарна недостатність виникає у випадку первинного збільшення енергетичних потреб серця (збільшення навантаження на серце при фізичній роботі, артеріальній гіпертензії). За таких обставин інтенсивність вінцевого кровообігу може зростати, але цього буває замало, щоб задовольнити потреби серця.
Абсолютна коронарна недостатність виникає у випадку первинного порушення вінцевого кровообігу, у результаті чого зменшується доставка кисню і поживних речовин міокарду як у стані спокою, так і при збільшенні енергетичних потреб серця.
27.48. Які патогенетичні фактори можуть обумовлювати розвиток абсолютної недостатності вінцевого кровообігу?
Інтенсивність вінцевого кровообігу визначається формулою
![]()
де Q — об'ємна швидкість течії крові; Ра і Рвсн — тиск крові відповідно на початку і в кінці системи вінцевих судин; (Ра - Р ) - перфузійний тиск; R - опір вінцевих судин.
Оскільки абсолютна коронарна недостатність характеризується зменшенням інтенсивності вінцевого кровообігу (зменшенням Q), можливими є два патогенетичних варіанти її розвитку.
І. Зменшення перфузійного тиску. При цьому розвивається коронарна недостатність центрального походження. її причинами можуть бути:
а) артеріальна гіпотензія (зменшення Ра п), наприклад, при всіх видах шоку, колапсі, недостатності аортальних клапанів. При зменшенні артеріального тиску спрацьовують механізми міогенної ауторегуляції вінцевого кровообігу, що підтримують його на постійному рівні. Однак, коли артеріальний тиск падає нижче 70 мм рт. ст., ці механізми виявляються неспроможними;
б) порушення венозного відтоку (збільшення Рвен). В експерименті моделюють перев'язуванням вен серця. Може мати значення при декомпенсованій недостатності правого шлуночка, коли збільшується центральний венозний і кінцеводіастолічний тиск.
II. Збільшення опору вінцевих судин. При цьому розвивається коронарна недостатність місцевого походження. Оскільки
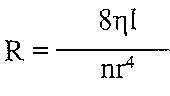
де tj — в'язкість крові; 1 — довжина судин; г — радіус судин, то збільшення опору може бути обумовлене:
а) збільшенням в 'язкості крові при порушенні її реологічних властивостей (зневоднення, поліцитемія, ДВЗ-синдром). При цьому розвиваються порушення мікроциркуляції, аж до сладж-синдрому та істинного капілярного стазу; : б) зменшенням радіуса судин. Це основний фактор розвитку абсолютної коронарної недостатності. Він викликає ішемію серця.
27.49. Які механізми можуть лежати в основі розвитку ішемії міокарда?
I. Обтураційний механізм — зменшення просвіту вінцевих артерій. Його причинами можуть бути:
а) стенозуючш атеросклероз (є причиною ішемії міокарда у 90 % випадків);
б) тромбоз вінцевих артерій (найчастіше є наслідком атеросклерозу);
в) емболія вінцевих артерій;
г) запальні процеси в стінці судин серця — коронаріїти. Бувають при ревматизмі, сифілісі.
Для розвитку клінічних ознак ішемії міокарда має значення величина "критичного стенозу ". Це мінімальне зменшення просвіту судин, при якому виникає ішемія. У людини цей показник становить 75 % (у свиней - менше, у собак - більше).
II. Ангіоспастичний механізм- спазм вінцевих судин. Його причинами можуть бути:
а) збудження а-адренорецепторів на тлі блокади р-адренорецепторів;
б) вазопресин;
в) ангіотензин II;
г) тромбоксан А^;
ґ) гіпокапнія;
д) ендотелій - біологічно активна речовина ендотеліального походження. Існує клінічна форма коронарного ангіоспазму — стенокардія Принцметала.
III. Компресійний механізм — здавлювання вінцевих судин. Може мати місце при тахікардії (збільшується загальна тривалість періоду здавлювання вінцевих судин під час систоли серця). Іноді його причиною є рубці і пухлини. В експерименті використовують для моделювання ішемії й інфаркту міокарда шляхом накладення лігатури на вінцеві артерії.
27.50. Які патогенетичні фактори впливають на міокард в умовах коронарної недостатності?
1. Гіпоксія.
2. Ацидоз. Розвивається в результаті накопичення кислих продуктів обміну речовин унаслідок порушення відтоку крові і активації гліколізу.
3. Збільшення позаклітинної концентрації іонів калію в зоні ішемії. Виявляють від самого початку порушень вінцевого кровообігу (через кілька хвилин), коли ще немає ушкодження кардіоміоцитів. У цей період з невідомих ще причин відбувається пасивний вихід іонів калію з клітин (можливо, збільшується проникність К-каналів), і його позаклітинна концентрація зростає до 8 ммоль/л і більше. Через декілька годин "витікання" іонів К+ із клітин може бути обумовлене порушенням роботи Na-K-насосів (дефіцит АТФ) та збільшенням проникності ушкоджених мембран.
27.51. Які наслідки для міокарда може мати недостатність вінцевого кровообігу?
1. Порушення скорочувальної здатності міокарда з розвитком недостатності серця.
2. Поява аномальної електричної активності — електрична нестабільність серця, розвиток аритмій.
3. Ушкодження кардіоміоцитів, обумовлене ішемією (див. розд. 11).
4. Реперфузійний синдром.
27.52. Які порушення скорочувальної функції міокарда можуть виникати при його ішемії?
При ішемії міокарда розрізняють ранні і пізні порушення його скорочувальної функції.
Ранні порушення виникають дуже рано, вже при незначному дефіциті АТФ. Вони є відображенням гіпокальцієвого варіанта порушень скорочувальної функції серцевого м'яза і розвиваються в результаті блокади Са-каналів сарколеми кардіоміоцитів. Порушення провідності Са-каналів в умовах ішемії має щонайменше два механізми:
а) дефіцит АТФ → порушення фосфорування білків Са-каналів;
б) активація гліколізу → накопичення в клітинах іонів Н+ → безпосередня блокада Са-каналів.
Наслідком зазначених порушень є зменшення надходження іонів Са2+ у саркоплазму м'язових волокон, розлади електромеханічного спряження, зменшення сили скорочень кардіоміоцитів.
Пізні порушення скорочувальної функції виникають при тривалих розладах вінцевого кровообігу - понад ЗО хв. їх відносять до гіперкальцієвого (контрактурного) типу порушень. Концентрація іонів кальцію в саркоплазмі кардіоміоцитів збільшується через дві основні причини:
а) порушення видалення іонів Са2+ із саркоплазми внаслідок дефіциту АТФ;
б) збільшення надходження іонів Са2+ у м'язові волокна через ушкоджену плазматичну мембрану клітин.
У результаті збільшення вмісту Са2+ порушуються процеси розслаблення кардіоміоцитів, настає контрактура міофібрил, порушується скорочувальна здатність серця.
27.53. Які механізми можуть обумовлювати розвиток аритмій при ішемії міокарда ?
Розвиток аритмії пов'язаний з електричною нестабільністю серця, що виникає вже через кілька хвилин від початку розвитку ішемії міокарда. Основна її причина — вихід іонів калію з кардіоміоцитів, у результаті чого збільшується їх позаклітинна концентрація. Це викликає часткову деполяризацію сарколеми клітин, що перебувають поруч. Залежно від рівня підвищення вмісту позаклітинного калію можливі такі наслідки:
а) у ділянках міокарда, де концентрація позаклітинного калію збільшується до 8 ммоль/л, мембранний потенціал зменшується від —90 мв до -80 мв, наближаючись до рівня критичного потенціалу деполяризації. Тому в цих ділянках збільшується збудливість клітин і швидкість проведення імпульсів;
б) у ділянках міокарда, де позаклітинна концентрація калію перевищує 8 ммоль/л, мембранний потенціал м'язових волокон стає менше -80 мв, зменшується провідність Na-каналів (відбувається їх інактивація), у результаті зменшується збудливість кардіоміоцитів і швидкість проведення імпульсів.
Одночасне існування різних ділянок міокарда з різними типами електрофізіологічних порушень (з одного боку, збільшення збудливості й провідності, з другого боку —зменшення цих характеристик) створює умови для реалізації механізмів повторного входження імпульсів (re-entry), у результаті чого можуть розвиватися тріпотіння й фібриляція шлуночків, що призводять до смерті.
Фібриляція шлуночків, що виникає при ішемії міокарда, є основною причиною так званої раптовоїсмерті. При цьому на розтині, як правило, жодних ознак ішемічного ушкодження міокарда не виявляють.
27.54. Що таке реперфузійний синдром? У чому його сутність? Які механізми його розвитку?
Реперфузійний синдром — це синдром, що виникає внаслідок поновлення кровообігу в ішемізованій ділянці міокарда, тобто в результаті реперфузії.
Поновлення вінцевого кровообігу може бути обумовлене припиненням коронарного ангіоспазму, лізисом тромбу, руйнуванням агрегатів клітин крові, хірургічним видаленням .тромбу, зняттям лігатури.
Клінічно реперфузійний синдром виявляє себе значним збільшенням інтенсивності ушкодження міокарда відразу ж після поновлення вінцевого кровообігу. Внаслідок цього стан хворого різко погіршується. Мінімальна тривалість ішемії, після якої виникає виражений реперфузійний синдром, становить 40 хв. Якщо тривалість ішемії менше 20 хв., зазначений синдром не розвивається (така тривалість характерна для нападів стенокардії). При тривалості ішемії 20-40 хв. іноді можуть розвиватися реперфузійні ушкодження серця.
Патогенетичною основою реперфузійного синдрому є так званий "кисневий парадокс". Якщо здійснювати перфузію серця розчином, що не містить кисню (або містить його мало), а через 40 хв. і більше перейти на перфузію розчином з нормальною напругою Ог, то в результаті такої перфузії порушення, обумовлені попередньою
гіпоксією, не тільки не зменшуються, як цього варто було б очікувати, а стають більш вираженими (парадокс!). В основі зазначеного парадокса різка активація процесів пероксидного окиснення ліпідів, обумовлена надходженням кисню в клітини, у яких міститься велика кількість відновлених компонентів дихального ланцюга. Відбувається скидання електронів в обхід дихального ланцюга безпосередньо на молекули кисню, внаслідок чого утворюється велика кількість вільних радикалів. Останні ініціюють реакції пероксидного окиснення ліпідів, що є важливим молекулярним механізмом ушкодження клітинних мембран (див. розд. 11).
27.55. Що таке ішемічна хвороба серця? Які існують її клінічні форми?
Ішемічна хвороба серця — це хвороба, що розвивається в результаті абсолютної недостатності вінцевого кровообігу й виявляється ушкодженнями міокарда різного ступеня тяжкості.
її основними клінічними формами є:
1) стенокардія — напади короткочасної (до 20 хв.) гострої коронарної недостатності, які супроводжуються больовим синдромом, відчуттям страху і пов'язаними з цим вегетативними реакціями.
Розрізняють стенокардію напруги, стенокардію спокою і стенокардію Принцме-тала (спазм вінцевих артерій);
2) передінфарктний стан (проміжний коронарний синдром, або гостра вогнищева дистрофія міокарда) — розвивається при тривалості'ішемії міокарда від 20 до 40 хв.;
3) інфаркт міокарда— некроз серцевого м'яза, обумовлений порушеннями вінцевого кровообігу. Виникає при оборотній (транзиторній) ішемії, що триває понад 40-60 хв.? або при необоротних порушеннях коронарного кровообігу;
4) кардіосклероз — склеротичні зміни серцевого м'яза. Можуть бути дифузними (атеросклеротичний кардіосклероз) і вогнищевими (постінфарктний кардіосклероз).
27.56. Назвіть основні причини розвитку інфаркту міокарда.
1. Атеросклероз вінцевих артерій (див. розд. 28). Його розвиток супроводжується порушенням постачання міокарда киснем.
2. Збільшення навантаоїсення на серце (фізична напруга, артеріальна гіпертензія). При цьому збільшується потреба серця в кисні.
3. Стрес (див. розд. 33).
27.57. Яка роль катехоламінів у розвитку інфаркту міокарда?
У розвитку інфаркту міокарда можуть мати значення такі ефекти катехоламінів.
1. Порушення вінцевого кровообігу. Це пов'язане з тим, що катехоламіни:
а) сприяють розвитку атеросклерозу (стрес і катехоламіни, що беруть участь у його реалізації, є фактором ризику цієї хвороби);
б) викликають контрактурний спазм гладких м'язів вінцевих артерій (катехо-ламінове ушкодження клітин судинної стінки);
в) активують тромбоутворення і зсідання крові.
2. Збільшення потреби серця в кисні. Енергетичні потреби серця зростають у результаті реалізації позитивного іно- і хронотропного ефектів катехоламінів та
збільшення загального периферичного опору, внаслідок чого збільшується навантаження на серце. 3. Некоронарогенні катехоламіновіушкодження кардіоміоцитів. Вони не пов'язані з порушенням вінцевого кровообігу. Виникають у результаті того, що великі дози катехоламінів активують ліпідні і кальцієві механізми ушкодження клітин (див. розд. 11).
27.58. Які клінічні синдроми характерні для інфаркту міокарда ?
1. Больовий синдром.
2. Гостра серцева недостатність. Розвивається при уражені великих ділянок міокарда. Може виявляти себе синдромом серцевої астми і набряку легень або карді-огенним шоком.
3. Аритмічний синдром. Можливий розвиток усіх видів аритмій. Найнебезпечнішою є поява фібриляції шлуночків.
4. Резорбційно-некротичний синдром.
27.59. Які механізми розвитку і значення больового синдрому при інфаркті міокарда?
У розвитку больового синдрому при некрозі серцевого м'яза мають значення:
а) хімічні фактори, що з'являються в тканинах при ушкодженні клітин. Серед них іони Н+, К+, простагландини, лізосомні ферменти;
б) зміни скорочувальних властивостей ішемізованої ділянки міокарда, у результаті чого відбувається патологічне розтягування (пролабування) стінки серця при його скороченні. Це веде до подразнення механорецепторів серця і розвитку болю.
Патогенетичне значення больового синдрому в розвитку інфаркту міокарда полягає в тому, що:
1) біль є потужним чинником ініціації стресу і активації симпатоадреналової системи. Великі дози катехоламінів, що вивільняються при цьому, сприяють ушкодженню міокарда (див. запит. 27.57);
2) сильний біль викликає спочатку збудження, а потім і перезбудження життєво важливих центрів головного мозку (дихального, серцево-судинного). Ця обставина є важливим чинником розвитку кардіогенного шоку.
27.60. Що таке кардіотенний шок? У яких формах він може виявляти себе?
Кардіогенний шок — це шок, що виникає в результаті різкого падіння нагнітальної (насосної) функції серця. Це - найнебезпечніше ускладнення інфаркту міокарда, що часто призводить до смерті.
Розрізняють 4 форми кардіогенного шоку. 1. Рефлекторна форма (больовий шок). Основним механізмом її розвитку є тривалий біль, що викликає активацію симпатоадреналової системи, яка переходить у гальмування. Це призводить до депресії скорочувальної функції серця, брадикардії, зменшення тонусу периферичних судин і падіння артеріального тиску.
2. Гіпокінетична форма (істинний кардіогенний шок). Основним фактором її розвитку є різке зменшення скорочувальної функції серця в результаті ішемічного ушкодження кардіоміоцитів. Істинний кардіогенний шок розвивається, коли площа ураженого міокарда перевищує 40 %.
3. Дискінетична форма. Виникає в результаті асинергії (неузгодженості) скорочень міокарда. Причиною такої асинергії є грубі ушкодження серця - аневризми, розрив міжшлуночкової перегородки, відрив хорд клапанів.
4. Аритмічна форма. Є наслідком важких аритмій.
27.61. Який патогенез кардіогенного шоку?
У патогенезі кардіогенного шоку розрізняють кілька етапів.
I етап - первинне падіння артеріального тиску. Всі патогенетичні фактори кардіогенного шоку (рефлекторна депресія, збільшення площі ушкодженого міокарда, асинергія серцевих скорочень, аритмії) викликають зменшення серцевого ви-штовху. Це, за законами гемодинаміки, призводить до зменшення хвилинного об'єму серця і падіння артеріального тиску.
II етап - компенсаторний спазм артеріол. Характеризується активацією сим-патоадреналової системи, надходженням у кров катехоламінів, вазопресину, глкжо-кортикоїдів, утворенням ангіотензину II. Вивільнення потужних судинозвужувальних факторів викликає генералізований спазм артеріол, у результаті чого збільшується загальний периферичний опір. Зазначена реакція є компенсаторною і спрямована на попередження подальшого падіння артеріального тиску.
III етап - вторинне падіння артеріального тиску. Тривалий спазм артеріол у периферичних тканинах викликає порушення мікроциркуляції і гіпоксію. Наслідком кисневого голодування є:
а) ацидоз, що викликає депресію скорочувальної функції міокарда;
б) розширення артеріол, що виникає в результаті накопичення в тканинах метаболі-тів-вазодилататорів ( "метаболічний симпатоліз ");
в) надходження у кров із тканин так званих ішемічних токсинів. Серед них велике патогенетичне значення має фактор депресії міокарда, що вивільняється з підшлункової залози.
Усі зазначені зміни, погіршуючи скорочувальну функцію серця й "знімаючи" компенсаторний спазм артеріол, викликають подальше падіння артеріального тиску.
IV етап - термінальні зміни. У результаті істотного падіння артеріального тиску (нижче 40 мм рт. ст.):
а) ще більше порушується коронарний кровообіг і збільшується ішемія міокарда -зменшення скорочувальної функції міокарда прогресує;
б) розвивається гостра ниркова недостатність (повністю припиняється клубочко-ва фільтрація, виникають анурія, інтоксикація);
в) порушується мозковий кровообіг, розвивається гіпоксія головного мозку, виникають розлади функції життєво важливих центрів.
Сукупність зазначених змін призводить до смерті.
27.62. У чому сутність резорбційно-некротичного синдрому, що розвивається при інфаркті міокарда?
Резорбційію-некротіїчний синдром при інфаркті міокарда є наслідком надходження в кров продуктів розпаду змертвілої тканини серця. Він виявляє себе такими ознаками:
а) гарячкою (див. розд. 15);
б) нейтрофільним лейкоцитозом;
в) збільшенням швидкості осідання еритроцитів (ШОЕ);
г) ферментемією — появою в крові ферментів, що надходять із ушкоджених кардіоміо-цитів (креатинкіназа, аспартатамінотрансфераза, лактатдегідрогеназа І типу та ін.);
j) аутоімунним синдромом (синдромом Дреслера). Розвивається в результаті кон-формаційних змін білків міокарда. Виявляється запаленням серозних оболонок організму - полісерозитом (перикардитом, плевритом, перитонітом).
27.63. Що таке некоронарогенні некрози серця? Як їх моделюють в експерименті?
Некоронарогеншіми називають некрози серця, що виникають не в результаті недостатності вінцевого кровообігу, а через інші причини.
Існує кілька експериментальних моделей некрозу серцевого м'яза, причина виникнення якого не пов'язана з патологією вінцевих судин. Ці моделі певною мірою відбивають ситуацію, що спостерігається в природних умовах.
1. "Гіпоксичний некроз міокарда. Може бути відтворений за допомогою різних видів гіпоксії: гіпоксичної, гемічної. При цьому на тлі загальної недостатності кисню в організмі, що сама по собі веде до підвищення навантаження на систему кровообігу, розвивається некротичне ушкодження м'язових волокон серця. Розвитку некрозу сприяє фіксація тварини в незручній позі, наприклад, розтягування у верстаті, або додаткове навантаження — біг у тредбані.
2. Електролітно-стероїдна кардіопатія з некрозом. За спостереженнями Сельє, при введенні щурам значної кількості солей натрію разом з деякими аніонами (сульфатними, фосфатними) у серці з'являються осередки ушкодження дегенеративно-некротичного типу, що часто супроводжуються гіалінозом судин інших органів. Ці ушкодження стають більшими або виникають при введенні меншої кількості солей, якщо одночасно вводити деякі стероїдні гормони надниркових залоз. На цьому тлі легше розвиваються і мають тяжчий перебіг ушкодження серця, викликані іншими причинами. Так, введення навіть невеликих доз норадреналіну, похідних кальциферолу, гіпоксія, м'язова напруга або, навпаки, значне обмеження рухливості ведуть до розвитку великого некрозу міокарда. Солі калію і магнію при цьому мають захисну дію.
3. Імунні ушкодження серця. Можливі при введенні в організм експериментальної тварини гетерогенної сироватки, що містить антитіла проти білків серця тварини даного виду (кардіоцитотоксини). Доведено також, що в організмі за певних умов можуть виникати антитіла і сенсибілізовані лімфоцити, які діють на тканини власного серця і спричиняють його ушкодження. Цьому сприяє проникнення в
кров денатурованих компонентів некротизованих кардіоміоцитів. В експерименті аналогічний процес можна викликати введенням тварині суспензії міокарда зі стимулятором імунологічної реакції (ад'ювантом Фрейнда). Серце може бути ушкоджене і циркулюючими імунними комплексами антиген-антитіло, а також при фіксації на його структурах цитофільних антитіл типу IgE з наступною їх реакцією з антигеном. 4. Нейрогенні ураження серця. Дистрофічні зміни і некроз міокарда можна відтворити гострим або хронічним подразненням шийно-грудного вузла симпатичного стовбура, блукаючого нерва, гіпоталамуса, мозкового стовбура або інших відділів головного мозку. Введення в кров великих доз адреналіну або норадреналіну також веде до ураження серця. В основі механізму нейрогенних ушкоджень лежить невідповідність між рівнями функції, метаболізму і кровопостачання. Подразнення серцевих симпатичних нервів супроводжується значним збільшенням споживання кисню міокардом. При цьому збільшення вінцевого кровообігу є недостатнім (відносна коронарна недостатність), а тому розвивається гіпоксія міокарда. При склерозуванні вінцевих артерій невідповідність інтенсивності кровообігу рівневі обміну речовин виявляється ще в більшій мірі, що може виявитися катастрофічним як для серця, так і для організму в цілому.
28. Патологічна фізіологія кровоносних судин
28.1. Як класифікують кровоносні судини залежно від функцій, що вони їх виконують? Які патологічні процеси характерні для різних типів судин?
Відповідно до функціональної класифікації, кровоносні судини поділяють на такі групи:
1. Компенсаційні судини - аорта і артерії еластичного типу. їхня функція полягає насамперед у тому, щоб перетворювати поштовхоподібні викиди крові із серця у рівномірну течію крові. Еластичні і колагенові структури цих судин визначають напругу їхніх стінок, необхідну для протидії значній розтягувальній дії крові. При цьому важливо те, що підтримання постійної функціональної напруги за рахунок зазначених структур не вимагає витрат енергії.
2. Резистивні судини, або судини опору, - артеріоли і венули, розташовані в пре- і посткапілярних ділянках судинного русла. Опір течії крові у зазначених судинах здійснюється завдяки їхнім структурним особливостям (відносно товста стінка, якщо порівнювати з величиною просвіту), а також здатності м'язових структур стінки перебувати в стані постійного тонусу і активно змінювати величину просвіту під дією додаткових нейрогуморальних впливів. Цим забезпечується відповідність просвіту резистивних судин об'єму крові, що перебуває в них, а також сталість і адекватність кровопостачання органів і тканин.
3. Судини обміну - капіляри і венули. На рівні цих судин здійснюється двосторонній обмін між кров'ю і тканинами водою, газами, електролітами, необхідними поживними речовинами і метаболітами.
4. Ємнісні судини - переважно дрібні вени. Вони депонують кров з метою її розподілу і повернення до серця. Основна маса крові (75—80 %) зосереджена саме в цих судинах. Викид крові з ємнісних судин здійснюється як активним скороченням м'язових волокон, так і пасивно-еластичною віддачею.
5. Судини перерозподілу- судини-сфінктери і артеріо-венозні шунти. Регулюють кровонаповнення органів і тканин.
З патологічними змінами в різних типах судин пов'язаний розвиток тих чи тих захворювань. Так, атеросклероз характеризується інфільтративно-проліферативни-ми змінами в судинах еластичного і еластично-м'язового типу. Тому він є хворобою переважно компенсаційних судин. Відповідно артеріальну гіпер- і гіпотензію відносять до патології тонусу резистивних судин, а порушення проникності стінки судин - до характерних проявів патології судин обміну. Порушення ємнісних судин виявляються розладами центральної гемодинаміки широкого діапазону - від розвитку артеріальної гіпертензії до виникнення колапсу.
28.2. Як класифікують склеротичні ураження артеріальних судин?
Відповідно до рекомендацій ВООЗ (1955), усі склеротичні ураження артерій поділяють на дві групи.
I. Власне артеріосклероз. Охоплює такі форми, як атеросклероз, артеріосклероз Менкеберга, артеріолосклероз, вікові склеротичні зміни артерій.
II. Захворювання артерій запальної і запально-алергічної природи. До них відносять сифілітичний аортит, облітеративний ендартеріїт, алергічні васкуліти, ревматоїдний артеріїт та ін.
28.3. Які процеси становлять патогенетичну сутність артеріосклерозу?
Артеріосклероз являє собою комбінацію чотирьох процесів: інфільтрації, проліферації, дегенерації і склерозування. Різні поєднання цих процесів у різних судинах визначають "мозаїчний" характер артеріосклеротичних уражень.
1. Інфільтрація - проникнення із плазми крові в судинну стінку і відкладення в ній ліпідів, складних вуглеводів і білків.
2. Проліферація — розмноження гладком'язових клітин артеріальної стінки, у результаті чого формуються так звані фіброзні "бляшки", що виступають у просвіт артерій і порушують течію крові в них.
3. Дегенерація— цим терміном позначають ушкодження і загибель клітин судинної стінки, а також розвиток дистрофічних змін, у тому числі кальцинозу.
4. Склерозування — посилене утворення сполучної тканини, що виявляється синтезом її основної інтерстиціальної речовини й волокнистих структур.
28.4. Що таке атеросклероз?
Спочатку поняття "атеросклероз", запропоноване Маршаном у 1904 p., використовували для позначення лише двох типів змін: накопичення ліпідів у вигляді кашкоподібних мас у внутрішній оболонці артерій (від грецьк. athere — каша) і власне склерозу - сполучнотканинного ущільнення стінки артерій (від грецьк. scleras -твердий).
Сучасне тлумачення атеросклерозу набагато ширше. За визначенням ВООЗ, атеросклероз — це різні поєднання змін інтими артерій, що виявляються у вигляді осередкового відкладення ліпідів, складних сполук вуглеводів, елементів крові і циркулюючих у ній продуктів, утворення сполучної тканини і відкладення кальцію.
28.5. Що таке артеріосклероз Менкеберга?
Описана в 1903 р. Менкебергом на прикладі артерій нижніх кінцівок людини форма артеріосклерозу характеризується ураженням середньої оболонки (медії) артерій еластичного і еластично-м'язового типу і виявляється тріадою ознак: медіанекро-зом, медіакальцинозом і медіасклерозом.
28.6. Дайте порівняльну характеристику атеросклерозу й артеріосклерозу Менкеберга.
Показник |
Атеросклероз |
Артеріосклероз Менкеберга |
1.Уражується |
Інтима |
Медіа |
2. Переважають процеси |
Інфільтрації і проліферації |
Дегенерації і склерозування |
3. Переважно відкладаються |
Ліпіди (холестерол) |
Солі кальцію (розвивається кальциноз) |
4. Прояви: |
Стенозування артерій (утворення «бляшок») |
Зменшення еластичності артеріальної стінки |
5. Наслідки: |
Ішемія органів і тканин |
Аневризми судин (розшарування стінки і розрив) |
28.7. Чим виявляють себе склеротичні зміни кровоносних судин?
Склеротично змінені судини вирізняються підвищеною щільністю й крихкістю. Унаслідок зниження еластичних властивостей вони не в змозі адекватно змінювати свій просвіт залежно від потреби органа або тканини у кровопостачанні.
Спочатку функціональна неповноцінність склеротично змінених судин, а отже, органів і тканин виявляється тільки при підвищенні до них вимог, тобто при збільшенні навантаження. Подальше прогресування атеросклеротичного процесу може призвести до зниження працездатності і у стані спокою.
Сильний ступінь атеросклеротичного процесу, як правило, супроводжується звуженням і навіть повним закриттям просвіту артерій. При повільному склерозу ванні артерій в органах з порушеним кровопостачанням відбуваються атрофічні зміни з поступовим заміщенням функціонально активної паренхіми сполучною тканиною.
Швидке звуження або повне перекриття просвіту артерій часто веде до змертвіння ділянки органа з порушеним кровообігом, тобто до інфаркту. Інфаркт міокарда — найчастіше і найнебезпечніше ускладнення атеросклерозу вінцевих артерій.
28.8. Як в експерименті моделюють атеросклероз?
У 1912 р. М. Анічков і С. Халатов запропонували спосіб моделювання атеросклерозу у кролів шляхом уведення всередину холестеролу (через зонд або домішуючи його до звичайного корму). Виражені атеросклеротичні зміни розвиваються через декілька місяців при щоденному використанні 0,5 г холестеролу на 1 кг маси тіла. Як правило, їх супроводжує підвищення рівня холестеролу в сироватці крові (у 3-5 разів, якщо порівнювати з вихідними величинами), що стало підставою для припущення про провідну патогенетичну роль у розвитку атеросклерозу гіперхолес-теролемії. Цю модель легко відтворюють не тільки у кролів, але і в курей, голубів, мавп, свиней.
У собак і щурів, резистентних до дії холестеролу, атеросклероз відтворюють шляхом комбінованого впливу холестеролу і метилтіоурацилу, що пригнічує функцію щитоподібної залози. Таке поєднання двох факторів (екзогенного і ендогенного)
веде до тривалої і вираженої гіперхолестеролемії. Додавання до їжі вершкового масла і солей жовчних кислот також сприяє розвитку атеросклерозу.
У курей (півнів) експериментальний атеросклероз аорти розвивається після тривалої дії діетилстильбестролу. У цьому випадку атеросклеротичні зміни виявляються на тлі ендогенної гіперхолестеролемії, що виникає внаслідок порушення гормональної регуляції обміну речовин.
28.9. Що таке фактори ризику атеросклерозу? Що до них відносять?
Факторами ризику атеросклерозу називають сукупність внутрішніх і зовнішніх умов, які в багато разів підвищують імовірність розвитку цього захворювання у людини.
Проведені в багатьох країнах світу епідеміологічні дослідження дають можливість виділити цілий ряд факторів ризику атеросклерозу.
1. Вік. Різке збільшення частоти і тяжкості атеросклеротичних уражень судин у зв'язку з віком, особливо помітне після ЗО років, стало підставою для того, щоб деякі дослідники вважали атеросклероз функцією віку і винятково біологічною проблемою (І. Давидовський). Більшість учених, однак, дотримуються думки, що вікові і атеросклеротичні зміни судин — це різні форми артеріосклерозу, особливо на пізніх стадіях їхнього розвитку. При цьому вікові зміни судин сприяють розвитку атеросклербтичних уражень. Зазначений аспект проблеми атеросклерозу знайшов своє відображення в роботах М. М. Горева і очолюваної ним лабораторії Інституту геронтології НАН України.
2. Стать. У віці 40—70 років на атеросклероз та інфаркт міокарда атеросклеротичної природи чоловіки хворіють частіше, ніж жінки (у середньому в 3-4 рази). Після 70 років захворюваність на цю недугу серед чоловіків і жінок приблизно однакова. Зазначені відмінності, мабуть, пов'язані, з одного боку, з нижчим вихідним рівнем холестеролу і тим, що у жінок він міститься в сироватці крові в основному у фракції неатерогенних ліпопротеїдів високої густини, а з другого — з антисклеротичною дією жіночих статевих гормонів.
3. Спадковість. Роль спадкового фактора у виникненні атеросклерозу підтверджують статистичні дані про високу частоту ішемічної хвороби серця в окремих родинах, а також у однояйцевих близнюків. Мова йде про спадкові форми гіперліпо-протеїнемії і спадково обумовлені дефекти метаболізму артеріальної стінки.
4. Надлишкове харчування. Досвід країн з високим життєвим рівнем (США, Швеція, Чехія та ін.) переконливо доводить таку закономірність: що більше потреба в енергії задовольняється за рахунок тваринних жирів і продуктів, які містять хо-лестерол, то вищий вміст холестеролу в крові й відсоток захворюваності на атеросклероз. Навпаки, у країнах, де на частку жирів тваринного походження припадає незначна частина енергетичної цінності добового раціону (близько 10 %), захворюваність на атеросклероз низька (Японія, Китай).
Існує також залежність між захворюваністю на атеросклероз і кількістю споживаного цукру. Якщо додати до цього, що 75-85 % хворих на цукровий діабет хворіють на атеросклероз і вмирають від нього, у 4/5 хворих на атеросклероз установлене зниження толерантності до глюкози, а 1/3 з них перебуває в предіабетичному стані, то певну роль у виникненні атеросклерозу варто відвести надмірному споживанню вуглеводів і порушенню їх утилізації.
5. Стрес. Є спостереження, які свідчать про те, що захворюваність на атеросклероз вища серед людей "стресових професій", тобто професій, що вимагають тривалої і сильної нервової напруги (лікарі, учителі, викладачі, працівники управлінського апарату, льотчики та ін.).
У цілому захворюваність на атеросклероз вища серед міського населення, якщо порівнювати з сільським. Це може пояснюватися тим, що в умовах великого міста людина частіше зазнає нейрогенних стресових впливів.
6. Гіподинамія. Малорухливий спосіб життя, різке зменшення фізичного навантаження (гіподинамія) - ще один важливий фактор атерогенезу. Про це, зокрема, свідчать менша захворюваність на атеросклероз серед працівників фізичної праці і більша- в осіб, робота яких пов'язана з розумовою працею; швидша нормалізація рівня холестеролу в сироватці крові, після надмірного його надходження ззовні, під дією фізичних навантажень.
В експерименті виявлено виражені атеросклеротичні зміни в артеріях кролів після того як вони тривалий час перебували у спеціальних клітках, що значно обмежували рухову активність тварин. Особливу атерогенну небезпеку являє собою поєднання малорухливого способу життя і надлишкового харчування. 1. Інтоксикація. Вплив алкоголю, нікотину, інтоксикація бактеріального походження та інтоксикація, викликана різними хімічними речовинами (фториди, CO, H2S, свинець, бензол, сполуки ртуті), також є факторами, що сприяють розвитку атеросклерозу. Більшість наведених тут інтоксикацій супроводжувалася не тільки загальними порушеннями жирового обміну, властивими атеросклерозу, але й типовими дистрофічними та інфільтративно-проліферативними змінами в артеріальній стінці.
8. Артеріальна гіпертензія. Підвищений артеріальний тиск набуває значення фактора, що сприяє розвитку атеросклерозу в комбінації з іншими, особливо якщо він перевищує 160/90 мм рт. ст. Так, при однаковому рівні холестеролу захворюваність на інфаркт міокарда при гіпертензії в п'ять разів вища, ніж при нормальному артеріальному тиску. В експерименті на кролях, у їжу яких додавали холестерол, атеросклеротичні зміни розвиваються швидше і досягають більшого ступеня на тлі артеріальної гіпертензії.
9. Гормональні порушення, хвороби обміну речовин. У деяких випадках атеросклероз виникає на тлі попередніх гормональних порушень (цукровий діабет, мікседема, зниження функції статевих залоз) або хвороб обміну речовин (подагра, ожиріння, спадкові форми гіперліпопротеїнемії і гіперхолестеролемії). Про етіологічну роль гормональних розладів у розвитку атеросклерозу свідчать і досліди з експериментального відтворення цих порушень у тварин шляхом впливу на ендокринні залози.
28.10. Які існують концепції патогенезу атеросклерозу?
Відомі нині теорії патогенезу атеросклерозу можна звести до двох, принципово різних концепцій, що відрізняються між собою відповіддю на питання: що первинне, а що вторинне при атеросклерозі, інакше кажучи, що є причиною, а що наслідком - ліпоїдоз внутрішньої оболонки артерій чи дегенеративно-проліферативні зміни останньої.
Відповідно до уявлень R Вірхова і його послідовників, при атеросклерозі спочатку розвиваються дистрофічні зміни внутрішньої оболонки стінки артерій, а відкладення ліпідів і солей кальцію — явище вторинного порядку. Перевагою даної концепції є те, що вона спроможна пояснити розвиток спонтанного і експериментального атеросклерозу як у тих випадках, коли є порушення ліпідного обміну, так і в тих (що особливо важливо), коли їх немає. Першорядну роль автори цієї концепції відводять артеріальній стінці, тобто субстрату, який безпосередньо втягується в патологічний процес.
На противагу цим поглядам ще відтоді, коли М. Анічков і С. Халатов провели перші свої експерименти, успішно розвивається концепція про роль у розвитку атеросклерозу загальних метаболічних порушень в організмі, що супроводжуються гі-перхолестеролемією і гіперліпопротеїнемією. З цих позицій атеросклероз — наслідок первинної дифузної інфільтрації ліпідів, зокрема холестеролу, у незмінену внутрішню оболонку артерій. Подальші зміни в судинній стінці (явища мукоїдного набухання, дистрофічні зміни волокнистих структур і клітинних елементів субендотеліаль-ного шару, продуктивні зміни) розвиваються у зв'язку з відкладенням у ній ліпідів, тобто є вторинними.
28.11. У чому сутність плазмової теорії патогенезу атеросклерозу?
Відповідно до плазмової теорії, основу патогенезу атеросклерозу та інфільтрації судинної стінки зокрема, становлять зміни хімічного складу плазми крові, що виникають унаслідок загальних порушень ліпідного обміну в організмі.
Пріоритет у становленні цієї теорії належить М. М. Ані-чкову та його послідовникам.
Плазмова теорія у своєму розвитку пройшла два етапи. ^
Перший етап — холестероловий. Сутність теорії на цьому етапі її розвитку зводилася до положення: "без холестеро-лу немає атеросклерозу". Вважалося, що причиною виникнення інфільтративних змін артеріальної стінки (ліпоїдозу) є збільшення вмісту холестеролу в плазмі крові — гіперхолес-теролемія.
Після того, як стало відомо, що транспорт ліпідів, у тому числі і холестеролу, здійснюється у складі лшопротеїдів, виявилося, що для розвитку атеросклерозу має значення не стільки гіперхолестеролемія, скільки кількісні і якісні зміни ліпо-~~ протеїдів плазми крові. Настав другий етап розвитку плазмової теорії —ліпопротеїновий. Основою його стало положення: "без атерогенних ліпопротеїдів немає атеросклерозу".
28.12. Які функції виконує в організмі холєстєрол? Що доводить його роль у розвитку атеросклерозу?
Про важливе фізіологічне значення холестеролу свідчать такі факти:
а) щодня з їжею в організм людини надходить 300-500 мг холестеролу;
б) в організмі синтезується (в основному в печінці) ще 700-1000 мг холестеролу щодоби;
в) кожна клітина організму, за невеликим винятком, має власні системи синтезу холестеролу, здатні забезпечувати потреби клітин у цій речовині.
Така висока надійність забезпечення організму холестеролом пояснюється його важливими функціями. Серед них:
1. Мембранна функція. Холєстєрол є важливим компонентом плазматичних мембран усіх клітин. Його наявність у мембранах забезпечує такі їхні властивості:
а) механічну міцність;
б) рідинний стан ліпідів мембрани;
в) проникність мембран для іонів і метаболітів;
г) електроізоляційні властивості;
ґ) активність деяких мембранних ферментів;
д) здатність мембран до злиття.
2. Метаболічна функція. Холєстєрол є попередником цілого ряду сполук, що виконують важливі функції в організмі. Серед цих речовин:
а) жовчні кислоти. На їх утворення використовується 600 мг холестеролу щодоби;
б) стероїдні гормони (глюко- і мінералокортикоїди, чоловічі й жіночі статеві гормони). їхній синтез вимагає близько 40 мг холестеролу щодоби;
в) вітамін Dy Синтезується в шкірі під дією ультрафіолетового випромінювання з 7-дегідрохолестеролу.
Роль холестеролу у виникненні атеросклерозу доводить насамперед "холесте-ролова" модель цього захворювання, запропонована М. Анічковим і С. Халатовим (див. запит. 28.8).
Принципово можливими є такі механізми розвитку гіперхолестеролемії:
1) надлишкове надходження холестеролу в організм у складі їжі;
2) надлишковий синтез холестеролу в самому організмі;
3) порушення виведення холестеролу з організму в складі жовчі;
4) порушення використання холестеролу периферичними клітинами.
28.13. Які зміни ліпопротеїдів плазми крові сприяють розвитку атеросклерозу?
Нині показано, що для виникнення атеросклерозу велике значення мають кількісні і якісні зміни ліпопротеїдів плазми крові. Для характеристики таких змін послуговуються узагальненим терміном "дисліпопротеїнемія атерогенного характеру". Цей стан виявляє себе такими ознаками:
а) збільшенням вмісту в плазмі крові багатих холестеролом і тригліцеридами ліпопротеїдів низької (ЛПНГ) і дуже низької густини (ЛПДНГ);
б) появою в крові не властивих для норми ліпопротеїдів, що одержали назву "модифікованих". До них відносять глікозильовані, ацетоацетильовані ліпопротеїди, ліпопротеїди, зв'язані з продуктами ПОЛ; комплекси ліпопротеїд-антитіло та ін.;
в) зменшенням вмісту в плазмі крові ліпопротеїдів високої густини (ЛПВГ).
ЛПДНГ і "модифіковані" ліпопротеїди одержали назву атерогениих, а ЛПВГ -антиатерогенних.
28.14. Яке значення має рецепторний апарат клітин судинної стінки в розвитку її атеросклеротичних змін?
У 60-ті роки XX ст. лауреати Нобелівської премії Гольдштейн і Браун відкрили рецепторний механізм надходження ліпопротеїдів плазми крові в периферичні клітини. Було показано, що в організмі існує два механізми транспорту ЛПНГ в клітини: рецепторопосередкований (специфічний) і неспецифічний.
Основні відмінності цих механізмів представлено в таблиці.
Рецепторопосередкований механізм |
Неспецифічний механізм |
Пов'язаний зі специфічними рецепторами до ЛПНГ |
Не пов'язаний зі специфічними рецепторами до ЛПНГ, є різновидом піноцитозу |
Є у всіх клітинах |
Є тільки в клітинах макрофагального ряду |
Інтенсивність його регулюється кількістю рецепторів |
Інтенсивність його не регулюється |
Мета: задоволення потреби клітин у холестеролі |
Мета: очищення крові від надлишку ліпопротеїдів (гомеостатична функція) |
Не є причиною накопичення холестеролу в клітинах |
Є причиною накопичення холестеролу в клітинах |
Гольдштейн і Браун показали, що порушення рецепторного апарату клітин можуть бути причиною розвитку спадкового атеросклерозу (рецепторна теорія). Було встановлено, що у хворих зі спадковою гіперліпопротеїнемією Па типу (збільшений вміст ЛПНГ) має місце генетично обумовлений дефект специфічних рецепторів до ЛПНГ—у гетерозигот їх менше, ніж у нормі, а у гомозигот вони взагалі відсутні. Внаслідок цього холестерол плазми не може надходити в периферичні клітини (останні забезпечують ним самі себе завдяки власним системам синтезу), вміст його у крові збільшується. Це призводить до активації неспецифічного механізму транспорту в макрофагах і, можливо, у гладком'язових клітинах судинної стінки. У результаті в зазначених клітинах накопичується велика кількість холестеролу і тригліцеридів, і вони перетворюються у так звані "пінисті" клітини. Переповнені ліпідами клітини гинуть, і холестерол вивільняється в тканину. Там він у вигляді кристалів знову стимулює активність макрофагів, і процес повторюється. Згодом відбувається накопичення холестеролу в інтимі артерій (ліпоїдоз) і розростання сполучної тканини, у результаті чого формуються атеросклеротичні бляшки.
28.15. Яке значення має ушкодження ендотелію судинної стінки в розвитку її атеросклеротичних змін?
Росе і Гломзет запропонували теорію, відповідно до якої інфільтративні зміни при атеросклерозі розвиваються не внаслідок порушень ліпопротеідного складу плазми крові, а в результаті первинного ушкодження ендотелію судинної стінки (теорія "відповіді на ушкодження"). При цьому істотно зростає проникність артеріальної стінки, і нормальні складові частини плазми крові - ліпопротеїди, альбуміни, фібриноген - проникають в інтиму артерій.
Ушкодження ендотелію судин може бути обумовлене:
а) механічними і фізичними факторами (тиск крові, її турбулентний рух, іонізуюча радіація);
б) хімічними сполуками (нікотин, гомоцистеїн, великі дози вітаміну D);
в) ендотеліотропними вірусами, токсинами бактерій (наприклад, веротоксин);
г) імунними факторами.
Як доказ цієї теорії наводять такі факти:
1) в експерименті в місцях руйнування ендотелію спеціальним катетером розвивається ліпоїдоз артеріальної стінки;
2) у людини ліпоїдоз найчастіше виникає в тих ділянках артерій, які зазнають дії гемо-динамічних факторів, таких як кров'яний тиск, турбулентна течія крові, удар пульсової хвилі. Це дуга і біфуркація аорти, місця відходження і розгалуження артерій;
3) у хворих на артеріальну гіпертензію значно зростає ймовірність розвитку атеросклерозу.
28.16. Якими механізмами може бути обумовлений розвиток проліферативних змін у судинній стінці при атеросклерозі?
Для пояснення механізмів розмноження клітин у судинній стінці в процесі формування атеросклеротичних бляшок було запропоновано ряд теорій.
1. Теорія факторів росту (Росе). У результаті ушкодження ендотелію судинної стінки відбувається адгезія й агрегація тромбоцитів, унаслідок чого останні вивільняють фактор росту тромбоцитарного походження. Він викликає активацію в гладком'язових клітинах специфічних рецепторів, пов'язаних з тирозиновою про-теїнкіназою. Наслідком цього є розмноження зазначених клітин.
Як доказ цієї теорії наводять той факт, що у свиней зі спадковою хворобою Вілле-бранда (ендотеліальні клітини не утворюють фактора Віллебранда) не відбувається адгезія і агрегація тромбоцитів, не вивільняється фактор росту тромбоцитарного походження, ніколи не розвиваються спонтанні й індуковані атеросклеротичні бляшки.
2. Мембранна теорія (Джексон, Готто). Коли в клітинах міститься велика кількість вільного холестеролу, він перетворюється в естерифіковану форму. Якщо для цього виявляється недостатньо жирових кислот, то надлишковий холестерол спрямовується в плазматичні мембрани, до складу яких він входить. У результаті змінюється рідинний стан мембрани, її основні властивості. Інформація про це — у поки що нез'ясований спосіб - надходить у ядро, і клітина починає ділитися, щоб утилізувати надлишковий холестерол, "пустивши" його на утворення мембран нових клітин.
Доказом теорії є те, що in vitro при інкубації гладком'язових клітин у середовищі з підвищеним вмістом ЛІШГ активуються процеси клітинного поділу.
3. Моноклональна теорія (Бендітт). Відповідно до цієї теорії, атеросклеротичні бляшки є по суті доброякісними пухлинами. Під дією мутагенних факторів (тютюновий дим, віруси та ін.) відбуваються зміни геному якоїсь одної гладком'язової клітини, а згодом під впливом промоторних факторів (артеріальна гіпертензія, гі-перхолестеролемія) ця клітина починає проліферувати. Доказом теорії служить той факт, що всі клітини атеросклеротичної бляшки походять з однієї клітини. Це було встановлено при вивченні ізоферментного складу ряду ферментів, зокрема глюкозо-6-фосфатдегідрогенази, у нормальних і атеросклеротично змінених су^ динах деяких популяцій людей.
28.17. Які механізми можуть лежати в основі розвитку дегенеративних змін судинної стінки при атеросклерозі?
Основу дегенеративних змін у судинній стінці становить ушкодження її клітин. Історично сформувалося кілька теорій, що пояснюють механізми такого ушкодження.
1. Теорія локального запалення (Вірхов). Численні патогенні агенти (фізичного, хімічного, біологічного походження) ушкоджують клітини та інтерстиціальну речовину сполучної тканини артерій. Унаслідок альтерації розвивається локальне запалення судинної стінки і дистрофічні зміни в ній.
Існування великої кількості агентів, здатних викликати ушкодження стінки артерій і розвиток артеріосклеротичних змін в експерименті, є одним з вагомих доказів цієї теорії.
2. Теорія енергодефіциту (Ю. Биць, О. Атаман). Багато патогенних факторів, що діють на судинну стінку (гіпоксія, голодування, дефіцит вітамінів, отрути, токси-
ни та ін.), первинно порушують процеси енергетичного обміну в ній. Це викликає обумовлене дефіцитом АТФ ушкодження клітин і зменшує активну резистентність судин до дії інших ушкоджувальних чинників. Кінцевим результатом є розвиток дегенеративних змін в артеріальній стінці (зокрема, кальцинозу) з наступним її склерозуванням. Докази цієї концепції див. запит. 28.18. 3. Пероксидна теорія (О. Воскресенський). Ушкодження клітин судинної стінки та її позаклітинних компонентів, зокрема еластичних і колагенових волокон, відбувається внаслідок активації реакцій вільнорадикального окиснення і утворення великої кількості пероксидних сполук. Причиною появи останніх можуть бути:
а) самоокиснення ліпідів;
б) дія факторів, що активують вільнорадикальне окиснення (радіація, речо-вини-окислювачі);
в) недостатність антиоксидантів. Доказами теорії служать такі факти:
а) тривале перебування кролів (протягом 4-6 місяців) на штучній дієті, повністю позбавленій антиоксидантів, супроводжується появою в артеріальній стінці тварин виражених дегенеративних змін;
б) в осередках дегенеративних змін у судинній стінці завжди виявляється велика кількість проміжних та кінцевих продуктів пероксидного окиснення ліпідів;
в) у людини реєструється сезонний характер загострень атеросклерозу. Навесні, коли у багатьох розвивається стан антиоксидантної недостатності, такі загострення виникають частіше.
28.18. Які є докази ролі первинних порушень енергозабезпечення судинної стінки в розвитку її дегенеративних і склеротичних змін?
Є дві групи доказів. І. Аналіз численних наукових даних дозволив виявити певну закономірність. Сутність її можна визначити таким положенням: що нижча інтенсивність енергетичного обміну стінки кровоносних судин, то вища їх чутливість до розвитку дегенеративних і склеротичних уражень. Про це свідчить цілий ряд фактів:
а) види тварин з високою інтенсивністю енергетичного обміну в стінці артерій (щури, собаки) характеризуються високою резистентністю до артеріосклерозу, і навпаки, кролі й людина, у яких відзначають низький рівень енергозабезпечення судин, є високосприйнятливими до розвитку склеротичних уражень;
б) молоді тварини, у яких інтенсивність енергетичного обміну судинної стінки значно вища, ніж у старих, стійкіші до розвитку артеріосклерозу;
в) артерії з нижчим рівнем енергетичного обміну (грудна й черевна аорти) уражуються частіше, якщо порівнювати з артеріями, у яких енергетичний обмін інтенсивніший (легенева артерія);
г) вени, інтенсивність енергозабезпечення яких у 4-5 разів вища, ніж великих артерій, ніколи не уражуються атеросклерозом.
II. Експериментальні докази:
а) первинне порушення енергетичного обміну артеріальної стінки отрутами, що є інгібіторами метаболізму (монойодацетат, фторид натрію, етилмер-курхлорид), викликає розвиток виражених дистрофічно-склеротичних уражень артеріальних судин (Ю. Биць);
б) пригнічення енергетичного обміну венозної стінки тими ж отрутами істотно зменшує, а інколи і повністю знімає, високу резистентність вен до дії ушкоджувальних факторів (великих доз вітаміну D, адреналіну) (О. Атаман).
28.19. Які механізми можуть лежати в основі власне склерозування судинної стінки?
Складні механізми розвитку сполучної тканини в стінці судин намагається пояснити ряд теорій.
1. Тромбогенна теорія (Рокитанський, Дьюгід). Прихиль-
ники цієї теорії першопричиною розвитку фіброзних бляшок в артеріальній стінці вважають утворення тромбів на внутрішній поверхні артерій. Згодом відбувається організація цих тромбів, і вони перетворюються у фіброзні бляшки, що зменшують просвіт судин. Постійне виявлення в атеросклеротичних бляшках організму людини продуктів крові, зокрема фібрину, робить цю концепцію життєздатною і нині.
2. Аноксемічна теорія (Купер). Стінка артерій характеризується поганими умовами кисневого забезпечення, оскільки у внутрішній і середній її оболонках відсутні vasa vasorum. Ця обставина робить її досить уразливою до дії факторів, що погіршують постачання киснем. Так, утворення ліпідної плівки на поверхні інтими в умовах гі-перліпідемії може привести до зменшення напруги кисню в судинній стінці нижче критичного рівня, після чого стимулюється розростання сполучної тканини (склерозування). Той факт, що всі види гіпоксії сприяють розвитку артеріосклерозу, варто розглядати як один із доказів цієї теорії.
3. Теорія старіння (І. Давидовський). Артеріосклероз є одним з місцевих проявів загального процесу старіння, для якого характерні зміни сполучної тканини у всіх органах і тканинах. З цього погляду артеріосклероз є обов'язковим атрибутом старіння.
28.20. Чим можуть виявляти себе порушення функціїрезистивних судин (судин опору)? Які зміни регуляторних механізмів можуть їх обумовлювати?
Порушення функції резистивних судин ведуть до змін периферичного судинного опору і виявляються розладами артеріального тиску: його збільшенням (артеріальною гіпертензією) і зменшенням (артеріальною гіпотензією).
Зазначені порушення можуть бути обумовлені змінами функції регуляторних систем організму, що забезпечують сталість артеріального тиску. Збільшення артеріального тиску може бути пов'язане з:
а) збільшенням активності пресорних систем організму.
б) зменшенням активності депресорних його систем.
Відповідно розвиток артеріальної гіпотензії може бути обумовлений:
а) зменшенням активності пресорних систем;
б) збільшенням активності депресорних систем організму.
28.21. Які нейрогуморальні системи беруть участь у регуляції артеріального тиску?
Залежно від спрямованості дії всі системи, що беруть участь у регуляції артеріального тиску, поділяють на дві групи: ті, що підвищують артеріальний тиск (пресо-рні), і ті, що понижують його (депресорні).
До пресорних відносять:
а) симпатоадреналову систему;
б) ренін-ангіотензинну систему;
в) альдостерон-вазопресинову систему;
г) глюкокортикоїди.
Депресорну дію виявляють:
а) рефлексїа з барорецепторів дуги аорти і синокаротидної зони (барорецепторні рефлекси);
б) простагландини А, Е, І;
в) калікреїн-кінінова система;
г) передсердний натрійуретичний гормон (атріопептин).
28.22. Що таке первинна і вторинна артеріальна гіпертензія?
Розрізняють первинну і вторинну артеріальну гіпертензію.
При первинній артеріальній гіпертензії підвищення артеріального тиску не може бути пов'язане з конкретним захворюванням або патологічним процесом у тих чи тих органах і системах: причина підвищення артеріального тиску залишається неясною. Для позначення цієї форми гіпертензії в різних країнах використовують два рівнозначних терміни: "есенціальна гіпертонія " і "гіпертонічна хвороба ".
Вторинна артеріальна гіпертензія виникає як наслідок патологічних процесів у різних органах і системах. Вона характерна для:
а) захворювань нирок (гломерулонефрит, пієлонефрит, полікістоз нирок та ін.);
б) пухлин надниркових залоз (феохромоцитома, альдостерома);
в) уражень серця й судин (деякі вади серця, коарктація аорти);
г) захворювань нервової системи (бульбарний поліомієліт, енцефаліти; травми, струси мозку та ін.).
У всіх цих випадках причина гіпертензії ясна. її усунення, як правило, веде до нормалізації артеріального тиску.
28.23. Які виділяють гемодинамічні варіанти артеріальної гіпертензії?
Оскільки величина артеріального тиску визначається загальним законом гемоди-наміки, відповідно до якого
P = QR,
де Р — артеріальний тиск; Q — хвилинний об'єм серця; R — загальний периферичний опір,
то артеріальна гіпертензія може бути обумовлена збільшенням хвилинного об'єму серця (Q), збільшенням загального периферичного опору (R) або тим та іншим одночасно.
Відповідно до цього виділяють три гемодинамічних варіанти артеріальної гіпертензії.
1. Гіперкінетичнш тип. Обумовлений істотним збільшенням роботи серця, у результаті чого зростає його хвилинний об'єм (Q).
2. Еукінетичний тип. Виникає при помірному збільшенні хвилинного об'єму серця (Q) і загального периферичного опору (R).
3. Гіпокінетичний тип. Його розвиток пов'язаний з істотним збільшенням загального периферичного опору (R).
28.24. Які існують експериментальні моделі артеріальної гіпертензії?
Жодна хвороба людини не має такої великої кількості різних експериментальних моделей, як артеріальна гіпертензія. Нині артеріальну гіпертензію вивчають на мишах, щурах, кролях, кішках, собаках, свинях, мавпах.
За методами відтворення всі моделі артеріальної гіпертензії можна поділити на кілька великих груп.
I. Порушення функції центральної нервової системи:
а) зіткнення процесів умовного збудження і гальмування, що призводить до розвитку у тварин (собак, мавп) неврозу;
б) моделювання психоемоційної напруги шляхом створення зоосоціального конфлікту (у мавп), змін біоритмів, іммобілізації тварин;
в) електрична і хімічна стимуляція лімбічних структур головного мозку.
II. Порушення мозкового крово- і лімфообігу:
а) одно- і двостороннє перев'язування сонних і вертебральних артерій, що живлять мозок (центрально-ішемічна артеріальна гіпертензія)',
б) блокада лімфовідведення по периневральних і периваскулярних лімфатичних шляхах за допомогою каоліну, що його вводять у велику цистерну мозку.
III. Порушення функції депресорних регуляторних систем:
а) двостороннє перетинання у кролів і собак депресорних і синусних нервів, у результаті чого знімаються гальмівні впливи з барорецепторів рефлексогенних зон дуги аорти і каротидного синуса (рефлексогенна гіпертензія, або гіпертензія розгальмування);
б) центральна деаферентація барорецепторів, що її викликають ушкодженням ядра солітарного тракту;
в) пригнічення синтезу простагландинів за допомогою індометацину.
IV. Порушення функції нирок:
а) звужування обох ниркових артерій або звужування однієї ниркової артерії з видаленням другої контрлатеральної нирки (реноваскулярна гіпертензія). Виникнення артеріальної гіпертензії в цьому випадку пов'язане з активацією ренін-ангіотензинної системи;
б) видалення обох нирок і переведення тварин на гемодіаліз для запобігання уремії (ренопривна гіпертензія). її розвиток пояснюють припиненням депресорних функцій нирок;
в) обгортання нирок целофаном, шовком. При цьому виникає перинефрит:' здавлюється ниркова паренхіма, розвивається венозний застій і гіпоксія нирок, активується ренін-ангіотензинна система.
V. Порушення гормонального стану:
а) введення тваринам адреналіну;
в) введення вазопресину;
в) субтотальне видалення кори надниркових залоз. При цьому відбувається посилення регенерації залозистої тканини з посиленою продукцією кортикостероїдів, особливо альдостерону (надшрнико-регенераційна гіпертензія).
VI. Порушення водно-сольового обміну:
а) введення тваринам великої кількості кухонної солі (сольова гіпертензія)',
б) введення мінералокортикоїдів (дезоксикортикостерону, альдостерону) - мі-нералокортикоїдна гіпертензія;
в) поєднане введення кухонної солі й мінералокортикоїдів.
VII. Моделі генетично обумовленої артеріальної гіпертензії. У багатьох лабораторіях світу виведено чисті лінії щурів, характерною рисою яких є гіпертензія -ознака, що спадкується. Це, зокрема, щури зі спонтанною гіпертензією (лінія Окамото - Аокі); щури, схильні до інсультів; новозеландські щури, міланські щури; щури, чутливі до сольової дієти, та ін.
28.25. Яка етіологія первинної артеріальної гіпертензії?
Нині виділяють ряд факторів, що мають безпосередній стосунок до виникнення
гіпертонічної хвороби (есенціальної гіпертензії).
1. Психоемоційна перенапруга. Установлено більшу поширеність артеріальної гіпертензії серед людей, характер роботи яких пов'язаний з постійною психоемоційною напругою, наприклад серед телефоністок і телеграфістів, студентів у період екзаменаційної сесії, у дітей і підлітків, що навчаються у спеціалізованих математичних і деяких інших школах з напруженим режимом занять. Відомо, що під час другої світової війни в Ленінграді під час облоги виникла була ціла "гіпертонічна" епідемія.
Російський учений Ланг уперше висловив думку, що першопричиною гіпертонічної хвороби є психоемоційні перенапруги, які ведуть до розвитку невротичних
порушень вищої нервової діяльності і підвищення артеріального тиску як вегетативного компонента цих порушень. Основну роль Ланг відводив так званим "не-відреагованим" негативним емоціям, тобто таким, при яких сильні вегетативні реакції, зокрема з боку серцево-судинної системи, не супроводжуються адекватними руховими реакціями.
2. Спадковий фактор. Як доказ значення спадковості в етіології гіпертонічної хвороби наводять той факт, що артеріальну гіпертензію виявляють у парах однояйце-вих близнюків набагато частіше, ніж у парах близнюків двох'яйцевих. Крім того, поширеність артеріальної гіпертензії серед родичів хворих на гіпертонічну хворобу є значно більшою, ніж в популяції в цілому. Нарешті, експериментальні моделі генетично обумовленої гіпертензії у тварин за основними своїми характеристиками є найближчими до первинної артеріальної гіпертензії людини.
3. Надмірне споживання кухонної солі. В епідеміологічних дослідженнях відзначено низьку захворюваність на гіпертонічну хворобу в представників деяких етнічних груп, що живуть у різних регіонах нашої планети: гренландських ескімосів, аборигенів гір Китаю та Австралії, деяких племен індіанців, що живуть у Панамі. Загальним для всіх цих народностей є споживання малих кількостей кухонної солі (1—2 г на добу), якщо порівнювати з іншими людьми, що споживають її 10-1'5 г на добу.
Поряд з цим, там, де солі споживається більше, первинна артеріальна гіпертензія виявляється частіше. Високу захворюваність на гіпертонічну хворобу виявлено серед негритянського населення Багамських островів, у деяких регіонах Японії, де споживання солі досягає 20-50 г на добу.
В експерименті артеріальну гіпертензію можна одержати шляхом згодовування тваринам кухонної солі.
28.26. Які існують концепції патогенезу первинної артеріальної гіпертензії?
Нині уявлення про патогенез гіпертонічної хвороби розвиваються в основному в рамках двох концепцій: дисрегуляторної і мембранної.
Дисрегуляшорна концепція пояснює виникнення первинної артеріальної гіпертензії порушеннями механізмів регуляції артеріального тиску.
В основі мембранної концепції лежить положення про те, що первинна артеріальна гіпертензія виникає як наслідок первинних порушень у гладком'язових клітинах артеріол.
28.27. Який патогенез первинної артеріальної гіпертензії з погляду дисрегуляторної концепції її розвитку?
У патогенезі гіпертонічної хвороби розрізняють дві фази: гіперкінетичну і гіпо-кінетичену.
Гіперкінетична фаза характеризується переважно збільшенням хвилинного об'єму серця, наслідком чого і є підвищення артеріального тиску (рис. 132).
У її розвитку виділяють ряд послідовних етапів.
I етап — активація симпатоадреналової системи. Відбувається в результаті частих стресів, психоемоційних перенапруг, що зумовлюють появу осередків постійного тривалого збудження в центральній нервовій системі (патологічна домінанта). Катехоламіни, які виділяються при цьому, викликають щонайменше три важливих для подальшого розвитку гіпертензії ефекти:
а) збільшують хвилинний об'єм серця;
б) збільшують загальний периферичний опір;
в) викликаючи спазм приносних артеріол нирок і безпосередньо діючи на клітини юкстагломерулярного апарату, сприяють виділенню в кров реніну.
II етап — активація ренін-ангіотензинної системи. Надходження реніну в кров зумовлює ряд послідовних біохімічних реакцій, у результаті яких утворюється ангіо-тензин Пі ангіотензин III. З цими пептидами пов'язані такі зміни:
а) скорочення гладких м'язів артеріол (ангіоспазм);
б) збудження структур центральної нервової системи, що беруть участь у регуляції артеріального тиску;
в) вивільнення в кров альдостерону клітинами клубочкової зони кори надниркових залоз.
III етап — активація альдостерон-вазопресинової системи. Надходження в кров альдостерону, а також надмірне надходження в організм хлориду натрію викликають розвиток гіпернатрієміїІ підвищення у зв'язку з цим осмотичного тиску плазми крові. Збудження центральних і периферичних осморецепторів, що настає в цих умовах, активує секрецію вазопресину (антидіуретичного гормону) у ядрах гіпоталамуса. Вазопресин, впливаючи на нирки, викликає збільшення факультативної реабсорбції води.Це, у свою чергу, веде до збільшення об'єму циркулюючої крові (гіперволемія), хвилинного об'єму серця, а отже, і артеріального тиску. Зазначений механізм доповнюється безпосередньою судинозвужувальною дією вазопресину.
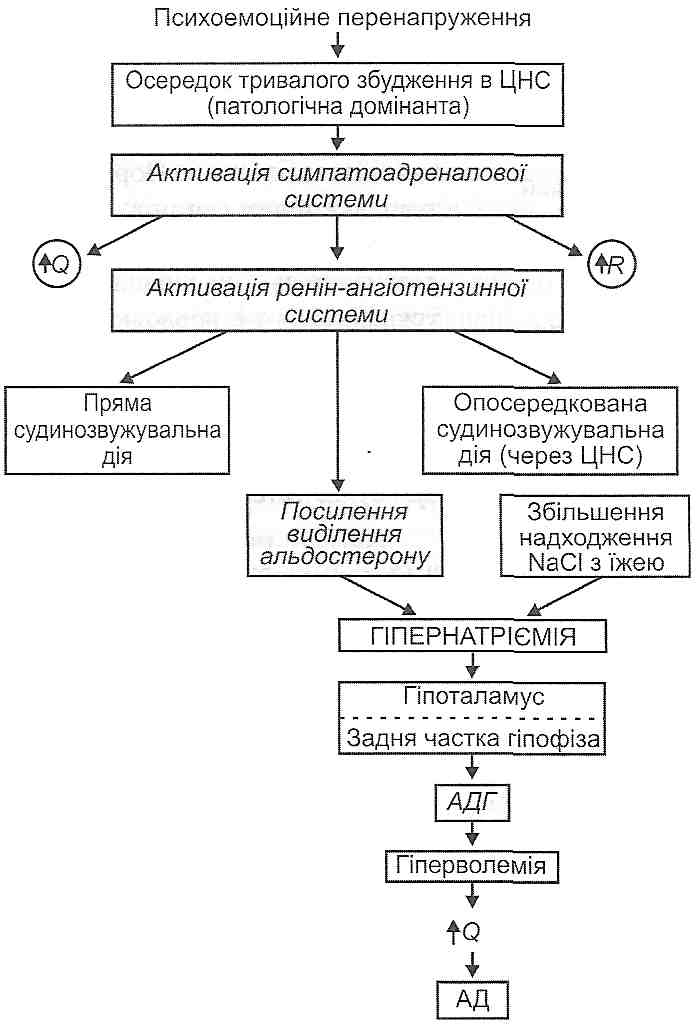
Рис. 132. Механізми гіпєркінетичноїфази розвитку первинної артеріальної гіпертензії: Q - хвилинний об'єм серця; R - загальний периферичний опір; AT— артеріальний тиск; АДГ
- антидіуретичшй гормон
Гіпокінетична фаза характеризується необоротними структурними змінами ре-зистивних судин, у результаті чого загальний периферичний опір і артеріальний тиск постійно збільшені (рис. 133).
У розвитку цієї фази можна виділити ряд послідовних стадій:
1) ауторегуляторний спазм артеріол. Виникає як наслідок збільшення хвилинного об'єму серця. Є реакцією, спрямованою на підтримку сталості кровообігу в тканинах (попереджає надходження надлишкової кількості крові);
2) гіпертрофія гладких м 'язів артеріол. Є структурним проявом гіперфункції гладких м'язів, що виникає при часто повторюваних спазмах;
3) артеріолосклероз. Гіпертрофовані гладком'язові клітини поступово зазнають дистрофічних змін і гинуть, відбувається їх заміщення сполучною тканиною - розви-
вається артеріолосклероз. Артеріоли перетворюються у ригідні сполучнотканинні трубки, не здатні ні до скорочення, ні до розслаблення. Загальний периферичний опір, а отже, і артеріальний тиск постійно збільшені. Порушується живлення життєво важливих органів: головного мозку, серця, нирок. Можливий розрив змінених артеріол — тоді розвивається крововилив. Найнебезпечнішим ускладненням є крововилив у мозок — геморагічний інсульт.
Рис. 133. Механізми
гіпокінетичної фази розвитку
первинної артеріальної гіпертензії
28.28. У чому сутність мембранної концепції патогенезу первинної артеріальної гіпертензії?
Автори мембранної концепції (Ю. Постнов, Р. Орлов) головну роль у розвитку гіпертонічної хвороби відводять спадково обумовленим порушенням іонних насосів мембран гладком'язових клітин.
У рамках цієї концепції нині розвиваються два напрями, що вивчають роль відповідно Са- і Na-K-насосів у порушенні функції гладком'язових клітин артеріол.
1. Дефекти Са-насосів клітинних мембран спричиняються до порушень видалення іонів кальцію із цитоплазми клітин і збільшення їхньої внутрішньоклітинної концентрації. Це викликає постійну контрактуру (перескорочення) гладких м'язів артеріол, що виявляється збільшенням загального периферичного опору і артеріального тиску. Крім того, надлишок іонів Са2+ у цитоплазмі клітин викликає їх ушкодження (див. розд. 11) і є, таким чином, передумовою розвитку артеріоло-склерозу.
2. Пригнічення роботи Na-K-насосів плазматичної мембрани гладком'язових клітин є однією з ознак первинної артеріальної гіпертензії. Про це, зокрема, свідчить той факт, що у багатьох хворих на гіпертонічну хворобу виявляють так званий ендогенний строфантиноподібний фактор, що, як і відомі серцеві глікозиди (строфантин, оуабаїн), пригнічує роботу Na-K-насосів.
У результаті порушень діяльності Na-K-насосів у цитоплазмі поступово збільшується концентрація іонів Na+ і виникає набряк гладком'язових клітин артеріол. Це має кілька наслідків:
а) потовщення стінки і зменшення просвіту артеріол;
б) збільшення чутливості гладком'язових клітин до дії ендогенних катехоламінів;
в) ушкодження і загибель клітин з наступним розвитком артеріосклерозу.
Усі наведені зміни викликають стійке збільшення загального периферичного опору і підвищення артеріального тиску.
28.29. Як класифікують артеріальну гіпотензію? Які гемодинамічні фактори можуть лежати в основі її розвитку?
Артеріальна гіпотензія (стійке зниження артеріального тиску) спостерігається частіше в осіб астенічної конституції і виявляється загальною адинамією, швидкою стомлюваністю, тахікардією, задишкою, запамороченням, головним болем, непритом-ностями і депресивним станом з періодичним підвищенням нервової збудливості.
Артеріальну гіпотензію класифікують у такий спосіб.
I. Фізіологічна (не супроводжується хворобливими симптомами).
II. Патологічна (з характерним симптомокомплексом):
1. Гостра.
2. Хронічна:
а) симптоматична (вторинна);
б) нейроциркуляторна дистонія гіпотензивного типу (первинна).
З огляду на те, що рівень артеріального тиску визначається величиною серцевого виштовху, об'ємом циркулюючої крові і тонусом резистивних судин, можливі три гемодинамічні форми артеріальної гіпотензії:
1) пов'язана з недостатністю скорочувальної функції серця;
2) викликана зменшенням об'єму циркулюючої крові;
3) така, що виникає внаслідок зниження тонусу резистивних судин.
28.30. Які причини і механізми розвитку хронічної артеріальної гіпотензії?
Симптоматична (вторинна) хронічна артеріальна гіпотензія є наслідком низки загальних соматичних гострих і хронічних захворювань серця (вади, міокардит, інфаркт міокарда), головного мозку (комоція), легень (крупозна пневмонія), печінки (гепатит, механічна жовтяниця), крові (анемія), ендокринних залоз, а також екзогенних інтоксикацій.
Стосовно нейроциркуляторної (первинної) артеріальної гіпотензії вважають, що її основним етіологічним і патогенетичним фактором, як і гіпертонічної хвороби, є перенапруження основних процесів кори великого мозку (збудження і гальмування). Однак, на відміну від первинної гіпертензії, спостерігається переважання гальмування і поширення його на підкіркові вегетативні утворення, зокрема на судиноруховий центр.
28.31. Які загальні і місцеві розлади гемодинаміки можуть бути пов'язані з первинними порушеннями функції ємнісних судин?
До ємнісних судин належать вени, що депонують кров з метою її розподілу і повернення до серця. Було відзначено, що в судинах ділянки низького тиску міститься 70-80 % загального об'єму крові. Про важливе функціональне значення венозного відділу свідчить хоча б той факт, що одномоментне зменшення його ємності всього лише на 3 % подвоює венозне повернення до серця, а при однакових за величиною змінах тиску в артеріальній і венозній системах об'єм останньої змінюється приблизно в ЗО разів більше, ніж артеріальної.
З урахуванням сказаного було зроблено висновок про те, що вже незначні порушення функції з боку ємнісних судин можуть призводити до істотних порушень загальної гемодинаміки. Такі розлади можуть виявлятися двома типами змін:
а) розвитком артеріальної гіпертензії при збільшенні тонусу гладких м'язів венозних судин, унаслідок чого збільшується діастолічний приплив крові до серця;
б) виникненням артеріальної гіпотензії— гострої (колапсу) або хронічної — при швидкому або тривалому збільшенні ємності венозної системи.
Місцеві розлади кровообігу, пов'язані з порушенням функції вен, призводять до розвитку венозної гіперемії (дав. розд. 13).
29 Патологічна фізіологія зовнішнього дихання
29.1. Що таке недостатність зовнішнього дихання?
Недостатність зовнішнього дихання — це патологічний стан, при якому система зовнішнього дихання не здатна забезпечити нормальний склад газів крові (газовий гомеостаз).
29.2. Як класифікують дихальну недостатність ?
I. За клінічним перебігом розрізняють гостру і хронічну недостатність дихання. Гостра недостатність розвивається протягом кількох днів, годин і навіть хвилин, її прикладом може бути асфіксія (див. запит. 29.16).
Хронічна недостатність розвивається протягом тривалого часу і є наслідком захворювань бронхів і легень (хронічна пневмонія, пневмосклероз, емфізема легень та ін.).
II. За вираженістю клінічних ознак недостатність дихання може бути компенсованою і декомпенсованою.
При компенсованій недостатності газовий склад крові ще не змінений (спрацьовують компенсаторні захисні механізми); при декомпенсованій - газовий гомеостаз порушений.
III. За патогенезом виділяють два різновиди: а) вентиляційну і б) паренхіматозну недостатність зовнішнього дихання.
29.3. Що таке вентиляційна недостатність дихання? Чим вона виявляє себе?
Вентиляційна недостатність дихання виникає внаслідок порушень обміну газів між атмосферним повітрям і альвеолами легень, тобто в результаті порушень легеневої вентиляції (гіповентиляції).
Для цього виду дихальної недостатності характерні такі порушення гомеостазу:
1) зменшення напруги кисню (р02) в артеріальній крові - гіпоксемія;
2) збільшення напруги вуглекислого газу (рС02) в артеріальній крові - гіперкапнія;
3) газовий ацидоз.
Для характеристики вентиляційної недостатності використовують показники легеневих об'ємів і ємностей (рис. 134).
29.4. Назвіть причини вентиляційної недостатності дихання.
І. Позалегеневі причини. Безпосередньо не пов'язані з порушеннями бронхів і
легень. До них відносять:
а) порушення функції дихального центру. Можуть бути наслідком прямої дії на
центральну нервову систему різних патогенних факторів або рефлекторних
впливів (на хемо-, барорецептори і т. д.). Недостатність дихання виникає
Рис. 134. Легеневі об'єми і ємності:
ДО — дихальний об'єм; РО^ —резервний об'єм вдиху; РО . —резервний об'єм видиху;
ЗОЛ-залишковий об'єм легень; ЖЄЛ- життєва ємність легень; Є^ - ємність вдиху;
ФЗЄ— функціональна залишкова ємність; ЗЄЛ—загальна ємність легень.
при зменшенні глибини, частоти дихання, при різних видах періодичного і термінального дихання;
б) порушення функції мотонейронів спинного мозку. Функція мотонейронів спинного мозку, що іннервують дихальні м'язи, може бути порушена при розвитку пухлини в спинному мозку, при сирингомієлії, поліомієліті. Характер і ступінь порушення зовнішнього дихання при цьому залежать від місця ушкодження спинного мозку (наприклад, при ураженні патологічним процесом верхньої шийної частини спинного мозку порушується робота діафрагми) і від кількості уражених мотонейронів;
в) порушення функції нервово-м 'язового апарату. Порушення вентиляції можуть виникати при ураженні нервів, що іннервують дихальні м'язи (запалення, авітаміноз, травма), при утрудненні передачі м'язам нервового імпульсу (міастенія, ботулізм, правець), при порушенні функції самих дихальних м'язів (міозит, дистрофія).
Із м'язів, які беруть участь в акті дихання, велике значення має діафрагма. Порушення роботи діафрагми може привести до значного розладу дихання, що буває, зокрема, при ураженні n. fremcus. У цьому випадку виникають парадоксальні рухи діафрагми: догори - при вдиху, вниз - при видиху. При клонічних судомах м'язів діафрагми з'являється гикавка, під час якої повітря втягується в легені;
г) порушення рухливості грудної клітки. Усі патологічні процеси, що обмежують рухливість грудної клітки, обмежують розтягання легень і, отже, впливають на альвеолярну вентиляцію. До них відносять уроджену або набуту деформацію ребер і хребетного стовпа, окостеніння реберних хрящів, зрощення листків плеври, асцит, метеоризм, велику надмірну масу тіла. Рухи грудної клітки можуть обмежуватися також різкими больовими відчуттями, що виникають під час дихання, наприклад, при міжреберній невралгії, запаленні і т. д.;
ґ) порушення цілості грудної клітки і плевральної порожнини. Цілість плевральної порожнини забезпечує створення постійного транспульмонального тиску, що підтримує легені в розправленому стані. Під час вдиху, коли об'єм грудної клітки збільшується, транспульмональний тиск зростає доти, доки не переборе еластичну тягу легень, унаслідок чого альвеоли розширюються. У тому випадку, коли цілість плевральної порожнини порушується і в негпотрапляє атмосферне повітря, транспульмональний тиск знижується, а легені спадаються (розвивається пневмоторакс).
Іноді, залежно від конкретних позалегеневих причин, розрізняють центральну форму дихальної недостатності (причини, позначені вище пунктами а, б), периферичну, або нервово-м 'язову (в) і торако-діафрагмальну, або парієтальну (г, ґ). II. Легеневі причини. Пов'язані з патологічними процесами в легенях і повітро-носних шляхах. До них відносять:
а) порушення прохідності дихальних шляхів (бронхіти, бронхіальна астма, злоякісні пухлини);
б) порушення еластичних властивостей легеневої тканини (емфізема, пневмосклероз);
в) зменшення кількості функціонуючих альвеол (пневмонія, набряк легень, ателектаз, пневмоторакс).
Форму дихальної недостатності, зумовлену легеневими причинами, часто називають легеневою.
29.5. Які виділяють патогенетичні варіанти вентиляційної недостатності дихання?
1. Дисрегуляторна недостатність (порушення центральної регуляції дихання).
2. Рестриктивна недостатність.
3. Обструктивна недостатність.
29.6. Чим можуть виявляти себе порушення центральної регуляції зовнішнього дихання?
При патології внаслідок дії рефлекторних, гуморальних або інших чинників на дихальний центр можуть змінюватися ритм, глибина і частота дихання, а також може виникати задишка. Ці зміни можуть бути проявом компенсаторних реакцій організму, спрямованих на підтримку сталості газового складу крові, або проявом порушень нормальної регуляції дихання, що ведуть до розладів альвеолярної вентиляції, до недостатності дихання.
Зміни центральної регуляції дихання можуть виявляти себе такими його типами. 1. Брадипное — рідке дихання. Механізм розвитку рідкого дихання полягає в зміні характеру нервової імпульсації, що йде від різних рецепторів до дихального центру, або в первинному порушенні діяльності самих дихальних нейронів. Рефлекторне зменшення частоти дихання може спостерігатися при підвищенні артеріального тиску (рефлекс із барорецепторів дуги аорти і каротидного синуса),
при гіпероксії (унаслідок періодичного збудження хеморецепторів, чутливих до зниження напруги кисню в артеріальній крові).
Глибоке рідке дихання може з'явитися при підвищенні опору руху повітря у верхніх дихальних шляхах — стенотичне дихання. У цьому випадку вдих і видих відбуваються повільніше, ніж звичайно. У встановленні такого типу дихання певну роль відіграють імпульси, що надходять у дихальний центр від міжреберних м'я-. зів, що працюють з підвищеним навантаженням. Крім того, має значення запізнювання в цьому випадку гальмівного рефлексу Герінга-Брейєра. Брадипное може розвиватися в результаті безпосередньої дії патогенних факторів на дихальний центр, що знижує збудливість дихальних нейронів. Пригнічення дихального центру можливе при тривалій і тяжкій гіпоксії (в умовах розрідженої атмосфери, при недостатності кровообігу та ін.), за умов впливу речовин з наркотичною дією, при деяких органічних ураженнях головного мозку (запалення, порушення мозкового кровообігу, набряк та ін.) і функціональних розладах центральної нервової системи (невроз, істеричні реакції). У всіх цих випадках рідке дихання може супроводжуватися зменшенням його глибини, що призводить до зниження альвеолярної вентиляції і розвитку недостатності дихання.
2. Поліпное (тахіпное) — часте поверхневе дихання. В основі розвитку поліпное лежить рефлекторна перебудова роботи дихального центру. У деяких тварин (наприклад, у собак) часте поверхневе дихання виникає при дії високої температури. У людини поліпное може спостерігатися при гарячці, функціональних порушеннях центральної нервової системи (істерія), ураженні легень (ателектаз, пневмонія, застійні явища).
Крім того, до розвитку поліпное може спричинятися біль, що локалізується у ділянках тіла, які беруть участь у дихальному акті (грудна клітка, черевна стінка, плевра). Біль призводить до обмеження глибини дихання і збільшення його частоти (щадне дихання).
Поліпное знижує ефективність дихання, тому що при цьому значно зменшується альвеолярна вентиляція і газообмін в основному відбувається у "мертвому" просторі.
3. Гіперппое — глибоке часте дихання. В умовах патології гіперпное розвивається при інтенсивній рефлекторній або гуморальній стимуляції дихального центру, наприклад, при зниженні парціального тиску кисню у вдихуваному повітрі або при підвищенні в ньому концентрації С02, при анемії, ацидозі і т. д.
Крайній ступінь збудження дихального центру виявляється диханням Куссмауля, яке найчастіше спостерігається у хворих у стані діабетичної коми. Воно являє собою гучне часте дихання, при якому після глибокого вдиху настає посилений видих з активною участю експіраторних м'язів.
4. Апное— тимчасова зупинка дихання. Тимчасова зупинка дихання може бути пов'язана зі зменшенням рефлекторної або безпосередньої хімічної стимуляції дихального центру. Наприклад, апное виникає у тварини або людини після пасивної гіпервентиляції під наркозом унаслідок зменшення в артеріальній крові напруги CO і припиняється зразу ж, як тільки вміст С02 нормалізується. При швидкому
підвищенні артеріального тиску, наприклад при введенні в кров адреналіну, також, спостерігається апное (рефлекс із барорецепторів). Апное, що часто повторюється і порушує звичайний ритм дихання, може бути пов'язане зі зниженням збудливості нейронів дихального центру (гіпоксія, інтоксикація, органічні ураження головного мозку).
5. Періодичне дихання (див. запит. 29.7).
6. Термінальне дихання (див. запит. 29.8).
7. Задишка (див. запит. 29.9).
29.7. Що таке періодичне дихання? Які відомі його форми?
Періодичним диханням називають таке порушення ритму дихання, при якому періоди дихання чергуються з періодами апное. Найчастіше бувають два типи періодичного дихання: дихання Чейна-Стокса і дихання Біота.
Дихання Чейна—Стокса характеризується наростанням амплітуди дихання до вираженого гіперпное, а потім зменшенням її до апное, після якого знову настає цикл дихальних рухів, що закінчуються також апное (рис. 135).
Рис. 135. Дихання Куссмауля і деякі види періодичного дихання
Циклічні зміни дихання у людини можуть супроводжуватися потьмаренням свідомості в період апное і його нормалізацією в період збільшення вентиляції. Артеріальний тиск при цьому також коливається, як правило, підвищуючись у фазі посилення дихання і знижуючись у фазі його ослаблення.
У більшості випадків дихання Чейна-Стокса є ознакою гіпоксії головного мозку. Воно може виникати при недостатності серця, захворюваннях мозку і його оболонок, уремії. Деякі лікарські препарати (наприклад, морфін) також можуть викликати дихання Чейна — Стокса. Його можна спостерігати і у здорових людей на великій висоті (особливо під час сну), у недоношених дітей, що, очевидно, пов'язане з недосконалістю нервових центрів.
Патогенез дихання Чейна-Стокса можна представити в такий спосіб. Клітини кори великого мозку і підкіркових утворень унаслідок гіпоксії пригнічуються — дихання зупиняється, свідомість зникає, пригнічується діяльність судинорухового центру. Однак хеморецептори при цьому все ще здатні реагувати на зміни вмісту газів
у крові. Різкого посилення імігульсації з хеморецепторів, поряд із прямою дією на центри високої концентрації вуглекислого газу і стимулами з барорецепторів унаслідок зниження артеріального тиску, виявляється достатньо для збудження дихального центру — дихання поновлюється. Поновлення дихання веде до оксигенації крові, що зменшує гіпоксію головного мозку і поліпшує функцію нейронів судинорухового центру. Дихання стає глибше, свідомість проясняється, підвищується артеріальний тиск, поліпшується наповнення серця. Вентиляція, що збільшується, веде до підвищення напруги кисню і зниження напруги С02 в артеріальній крові. Це у свою чергу призводить до ослаблення рефлекторної і хімічної стимуляції дихального центру, діяльність якого починає вгасати, - настає апное.
Дихання Біота відрізняється від дихання Чейна-Стокса тим, що дихальні рухи, які характеризуються постійною амплітудою, раптово припиняються, так само як і раптово починаються. Найчастіше дихання Біота спостерігається при менінгіті, енцефаліті та інших захворюваннях, що супроводжуються ушкодженням центральної нервової системи, особливо довгастого мозку.
29.8. Що таке термінальне дихання? Чим воно характеризується?
Термінальним називають дихання, що виникає в термінальних станах, тобто за умов, коли організм перебуває на межі життя і смерті. Найчастіше реєструють два типи термінального дихання: апнейстичне і гаспінг-дихання.
Апнейстичие дихання характеризується конвульсивним зусиллям вдихнути, що не припиняється, але зрідка переривається видихом. В експерименті його спостерігають після перетинання у тварини обох блукаючих нервів і мозкового стовбура між пневмотаксичним (у ростральній частині мосту) і апнейстичним (у середній і каудальній частинах мосту) центрами (рис. 136). Вважають, що апнейстичний центр має здатність збуджувати інспіраторні нейрони, які періодично гальмуються імпульсами із блукаючого нерва і пневмотаксичного центру. Перетинання зазначених структур призводить до постійної інспіраторної активності апнейстичного центру.
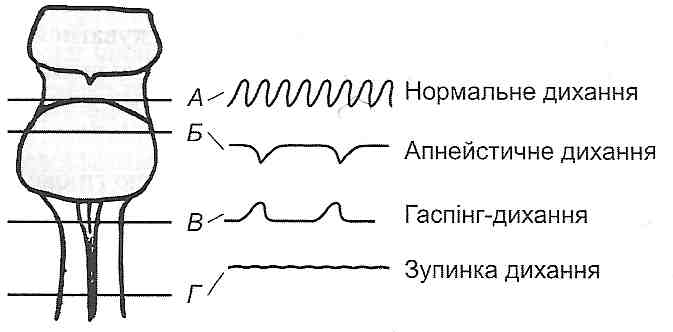
Рис. 136. Розвиток термінального дихання після перетинання стовбура мозку нарізних його рівнях (А, Б, В, Г)
Гаспінг-дихання - це поодинокі, рідкі вдихи, частота і амплітуда яких поступово зменшується аж до повної зупинки дихання. Такий вид дихання спостерігають
при агонії, наприклад у заключній стадії асфіксії. Звичайно характерні для гаспінг-дихання рідкі і низькоамплітуцні вдихи виникають після тимчасової зупинки дихання (претермінальної паузи). Поява їх, можливо, пов'язана зі збудженням клітин, що містяться в каудальній частині довгастого мозку, після вимикання функції вище розташованих відділів мозку.
29.9. Що таке задишка? Які механізми її розвитку?
Задишка (дисппое) - це відчуття нестачі повітря і пов'язана з цим потреба посилити дихання.
Відчуваючи нестачу повітря, людина не тільки мимовільно, але й свідомо збільшує активність дихальних рухів, прагнучи позбутися цього тяжкого відчуття, наявність якого і є найбільш істотною відмінністю диспное від інших видів порушення регуляції дихання (гіперпное, поліпное та ін.). Тому у людини, що втратила свідомість, задишки не буває.
Задишка виникає у тих випадках, коли впливи, що збуджують вдих, є сильнішими за впливи, що його пригнічують, а також у разі підвищення чутливості дихального центру до чинників, які стимулюють дихання. Найважливішими з цих впливів є такі.
1. Збудження рецепторів, що стимулюють центр вдиху, і які активуються при сильному зменшенні об'єму легеневих альвеол (сильнішому, ніж при максимальному видиху). При патології може виникнути постійна імпульсація від цих рецепторів. Наприклад, при застійних явищах у легенях (недостатність серця, пневмонія) переповнені кров'ю судини, що оточують альвеоли, здавлюють їх, ємність альвеол зменшується, що веде до збудження рецепторів спадіння.
2. Збудження рецепторів інтерстиціальної тканини легень (J-рецепторів). Усі патологічні процеси, що ведуть до застійних явищ у легенях (пневмонія, недостатність серця), можуть викликати тривале збудження J-рецепторів і підвищену стимуляцію дихальних нейронів.
3. Рефлекси з повітроносних шляхів. Можуть викликатися твердими частинками, парами хімічних сполук та іншими чинниками, що потрапили в повітроносні шляхи і подразнюють так звані іритантнірецептори (мають властивості одночасно ме-хано- і хеморецепторів), розташовані в субепітеліальному шарі трахеї, бронхів і бронхіол. Значне збудження іритантних рецепторів спостерігають при бронхітах, бронхопневмонії, бронхіальній астмі і хворобах, при яких у бронхах і альвеолах міститься слиз, ексудат або транссудат).
4. Рефлекси з барорецепторів аорти і сонної пазухи. Ці рефлекси долучаються до патогенезу задишки при крововтраті, шоку, колапсі. За умов зменшення артеріального тиску до рівня 70 мм рт. ст. і нижче, різко зменшується імпульсація від цих рецепторів, яка в нормі гальмує центр вдиху (через активацію центру видиху).
\ 5. Рефлекси з хеморецепторів аорти і сонної пазухи. При зниженні в крові напруги 02, підвищенні напруги С02 або збільшенні концентрації іонів водню відбувається посилене збудження рецепторів, розташованих в аортальному і каротидному тільцях, і, як наслідок, — посилене збудження центру вдиху. Цей механізм відіграє важливу роль у розвитку задишки при ацидозі, недостатності дихання, при анемії і т. д.
6. Безпосередня стимуляція нейронів дихального центру. У довгастому мозку є хеморецептори, вибірково чутливі до вуглекислого газу. Сильне збудження цих рецепторів при гіперкапнії також сприяє розвитку задишки.
7. Рефлекси з дихальних м 'язів. Відчуття нестачі кисню може виникнути при надмірному розтягненні міжреберних м'язів і сильному збудженні рецепторів розтягу, імпульсація від яких надходить у вищі відділи головного мозку. Цей механізм діє під час виконання тяжкої фізичної роботи, що потребує значної роботи інспіра-торних м'язів, при зменшенні еластичності легень, звуженні верхніх дихальних шляхів.
8. Стимуляція дихального центру продуктами власного метаболізму. Ідеться про накопичення вуглекислого газу, кислих продуктів обміну і зниження напруги кисню безпосередньо в нервових центрах унаслідок порушення мозкового кровообігу (спазм або тромбоз судин головного мозку, набряк мозку, колапс).
Дихання при задишці, як правило, часте і глибоке. Посилюється як вдих, так і видих, який за умов задишки носить активний характер і відбувається за участю експіраторних м'язів. Однак у деяких випадках може переважати або вдих, або видих, тоді ведуть мову про інспіраторну (посилений вдих) або експіраторну (посилений видих) задишку. Інспіраторну задишку спостерігають, наприклад, у першій стадії асфіксії, при загальному збудженні центральної нервової системи, при фізичному навантаженні у хворих з недостатністю кровообігу, при пневмотораксі. Експіраторна задишка буває рідше і виникає головним чином при бронхіальній астмі, емфіземі, коли при видиху збільшується опір потокові повітря в нижніх дихальних шляхах.
29.10. Що таке рестриктивна недостатність дихання? Які її причини?
PecmpuKmuenoio називають дихальну недостатність, пов'язану з обмеженням зовнішнього дихання (restrict - обмеження; рис. 137).
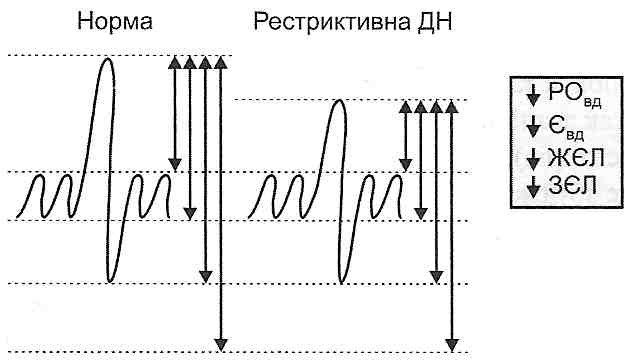
Рис. 137. Зміни показників вентиляції легень при рестриктивній дихальній недостатності (ДН)
Таке обмеження може бути обумовлене: 1) зменшенням дихальної поверхні легень, що буває після видалення сегмента, частки або цілої легені; при руйнуванні великих ділянок легень (туберкульоз); у результаті спадіння легеневої тканини (ателектаз, пневмоторакс);
2) збільшенням пружного опору легень — порушенням їхньої здатності розправлятися під час вдиху. Така ситуація закономірно виникає при:
а) зменшенні розтяжності легень у зв'язку із заміщенням еластичних структур легеневої тканини колагеновими (пневмосклероз);
б) збільшенні сили поверхневого натягу в альвеолах при порушеннях сурфак-танту (зменшенні утворення або посиленому руйнуванні).
29.11. Що таке пневмоторакс? Коли він виникає?
Пневмотораксом називають скупчення повітря в плевральній порожнині і підвищення тиску в ній. Повітря може потрапити в порожнину плеври при пораненнях, що проникають у грудну клітку, при розриванні емфізематозних альвеол на поверхні легені, розпаді легеневої тканини (туберкульоз, пухлина, абсцес). При цьому порожнина плеври може сполучатися з легенею та іншими повітроносними органами - стравоходом, шлунком, кишкою. Іноді повітря вводять у плевральну порожнину з лікувальною метою.
Якщо повітря, яке потрапило в плевральну порожнину, не сполучається з атмосферним повітрям, пневмоторакс називають закритим, якщо сполучається - відкритим. Нарешті, якщо особливості вхідного отвору в порожнині плеври допускають потрапляння повітря під час вдиху, але перешкоджають його виходу при видиху, пневмоторакс називають клапанним, або вентильним.
29.12. Що таке ателектаз?
Ателектаз — це патологічний процес, при якому припиняється вентиляція альвеол і вони спадаються внаслідок розсмоктування в них повітря. Факторами, що знижують транспульмональний тиск і ведуть до розвитку ателектазу, можуть бути підвищений тиск у плевральній порожнині (при пневмотораксі, скупченні ексудату), закупорка бронхів більшого або меншого діаметра, що завершується розсмоктуванням повітря у відповідній ділянці легені. Нарешті, у патогенезі ателектазу може мати значення дефіцит сурфактанту.
29.13. Що таке обструктивна недостатність дихання? Коли вона розвивається?
Обструктивною називають дихальну недостатність, яка виникає внаслідок збільшення аеродинамічного опору повітроносних шляхів. Основним фактором, що викликає таке збільшення, є зменшення радіуса повітроносних трубок (бронхів, бронхіол).
Причинами обструктивної недостатності дихання є:
1) спазм гладких м 'язів бронхів (бронхіальна астма);
2) набряк слизових оболонок (бронхіти, бронхіоліти); м
3) здавлювання бронхіол (емфізема легень).
29.14. Що таке емфізема легень? Чомупри цьому захворюванні розвивається обструктивна недостатність дихання?
Емфізема - це захворювання легеневої паренхіми, що супроводжується руйнуванням тонкої мережі легеневих капілярних судин і альвеолярних перегородок, а т*-
кож звуженням просвіту термінальних бронхіол. Механізм виникнення обструктивних явищ при емфіземі полягає в тому, що просвіт бронхіол, які мають м'які і тонкі стінки, підтримується транспульмональним тиском. Що більша еластичність легені, то більше розрідження необхідно створити в плевральній порожнині під час вдиху (і тим самим підвищити транспульмональний тиск), щоб перебороти еластичну тягу легень і розтягнути їх. Якщо ж легені втрачають свою еластичність, то вони розтягуються набагато легше, тобто при набагато меншому транспульмональному тиску. У результаті зменшується сила, що діє на стінки бронхіол зсередини і розправляє їх, - просвіт бронхіол зменшується внаслідок спадіння їхніх стінок.
Зменшення просвіту нижніх дихальних шляхів значно підвищує опір руху повітря і заважає рівномірному розподілу його в альвеолах. Особливо сильно порушується при цьому акт видиху. Пояснюється це тим, що при емфіземі під час вдиху транспульмональний тиск хоча і менший за нормальний, але все-таки достатній, щоб розправити стінки бронхіол. Під час видиху (який при емфіземі стає активним, тому що еластична тяга легень слабшає, а опір руху повітря збільшується) тиск у плевральній порожнині наростає і відповідно в міру видихання збільшується сила, що діє на бронхіоли ззовні. Стінки бронхіол поступово спадаються, і настає такий момент, коли подальший видих стає неможливим. Таким чином, при емфіземі стінки бронхіол відіграють роль клапана, який під час видиху закривається, і повітря опиняється неначе в пастці. Унаслідок цього альвеоли залишаються постійно роздутими, у них збільшується кількість залишкового повітря (рис. 138).
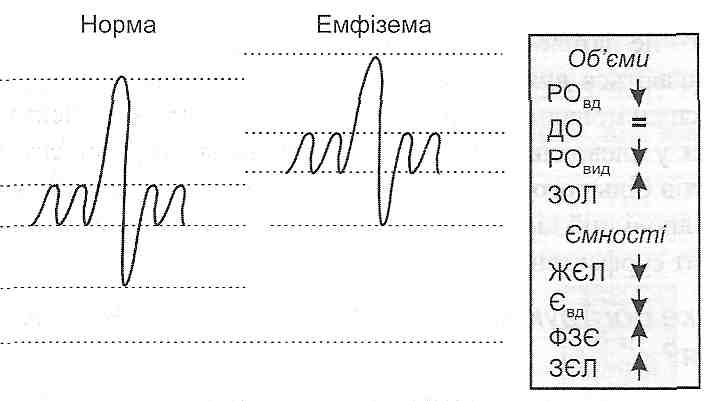
Рис. 138. Зміни показників вентиляції легень при емфіземі
29.15. Що таке бронхіальна астма? Як порушуються показники зовнішнього дихання при цьому захворюванні?
Бронхіальна астма — це алергічне захворювання, що характеризується повторними нападами експіраторної задишки, викликаної дифузним порушенням прохідності бронхів.
Під час тривалих нападів бронхіальної астми розвивається обструктивна недостатність дихання.
Для бронхіальної астми характерні такі зміни показників зовнішнього дихання (рис. 139): 1) зменшення резервного об'єму видиху; 2) зменшення життєвої ємності
легень; 3) збільшення залишкового об'єму легень; 4) зменшення об'єму форсованого видиху (проба Тифно; рис. 140).
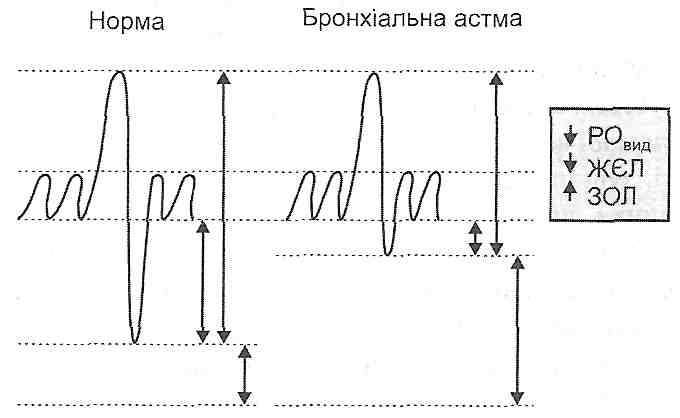
Рис. 139. Зміни показників вентиляції легень при бронхіальній астмі
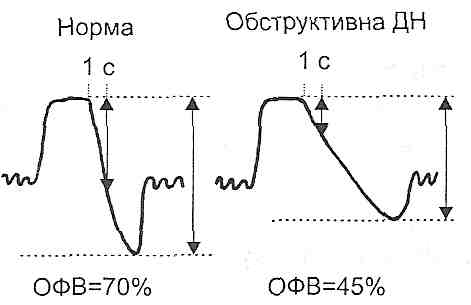
Рис. 140. Об'єм форсованого видиху (ОФВ; тест Тифно) при обструктивній дихальній недостатності (ДН)
29.16. Що таке асфіксія? Який ЇЇ патогенез?
Асфіксія (ядуха) - це загрозливий для життя стан, при якому гостра дихальна недостатність досягає такого ступеня, що у кров зовсім не надходить кисень, а з крові не виводиться вуглекислий газ.
Найчастіше асфіксія настає у разі здавлення дихальних шляхів (задушення), закупорки їхнього просвіту (сторонні предмети, запальний набряк), наявності рідини в дихальних шляхах і альвеолах (утоплення, набряк легень, потрапляння блювотних мас), у разі двостороннього пневмотораксу. Крім того, асфіксія може розвитися при сильному пригніченні дихального центру, порушенні проведення нервових імпульсів до дихальних м'язів, різкому обмеженні рухомості грудної клітки.
У перебігу асфіксії виділяють три періоди.
Перший період асфіксії характеризується швидким збільшенням глибини і частоти дихання з переважанням фази вдиху над фазою видиху. Розвивається загальне збудження, підвищується тонус симпатичної частини вегетативної нервової систе-
ми (розширюються зіниці, з'являється тахікардія, підвищується артеріальний тиск), можливі судоми.
У другому періоді частота дихання поступово зменшується при збереженій максимальній амплітуді дихальних рухів, посилюється фаза видиху. Переважає тонус парасимпатичної частини вегетативної нервової системи (зіниці звужуються, артеріальний тиск знижується, відзначається брадикардія).
В третьому періоді асфіксії спостерігають зменшення амплітуди дихання, його частоти і, нарешті, зупинку дихання. Артеріальний тиск значно знижується. Після короткочасної зупинки дихання, як правило, з'являється кілька рідких конвульсивних дихальних рухів (гаспінг-дихання), після яких настає параліч дихання.
Явища, що їх спостерігають при асфіксії, пов'язані спочатку з накопиченням в організмі вуглекислого газу. Діючи рефлекторно і безпосередньо на дихальний центр, С02 збуджує його, доводячи глибину і частоту дихання до максимально можливих величин. Крім того, дихання рефлекторно стимулюється і зниженням у крові напруги кисню. У міру збільшення в крові вмісту С02 підвищується і артеріальний тиск. Експерименти з вдиханням газових сумішей, що містять 10-20 % С02, показали, що таке підвищення пов'язане, по-перше, з рефлекторним впливом хеморецепторів на судиноруховий центр, по-друге, з посиленим викидом адреналіну в кров, по-третє, зі збільшенням хвилинного об'єму крові в результаті підвищення тонусу вен і збільшення надходження крові до серця при посиленні дихання.
При подальшому збільшенні концентрації С02 у крові починає виявлятися його наркотична дія, рН крові знижується до 6,8-6,5. Посилюється гіпоксемія і, відповідно, гіпоксія головного мозку. Це призводить, у свою чергу, до пригнічення дихання, зниження артеріального тиску. В результаті настає параліч дихання і зупинка серця.
29.17. Що таке паренхіматозна недостатність зовнішнього дихання? Які її причини й механізми розвитку?
Паренхіматозною називають недостатність дихання, що виникає як наслідок порушень газообміну між альвеолами легень і кров'ю.
її причинами є осередкові ураження легеневої паренхіми (ексудативні і проліфе-ративні запальні процеси), що призводять до порушень легеневого кровообігу.
Виділяють три основних механізми порушень газообміну між альвеолами і кров'ю:
1) порушення дифузії газів;
2) порушення легеневої перфузії (кровообігу); v
3) порушення загальних ірегіонарних вентиляційно-nepфузійних відношень.
У зв'язку з високим коефіцієнтом дифузії С02, що в 20-25 разів перевищує аналогічний показник для 02, перехід С02 із крові в альвеоли практично не страждає. Тому основною характеристикою паренхіматозної недостатності дихання є порушення надходження 02 з альвеол у кров, у результаті чого зменшується р02 крові, тобто розвивається гіпоксемія. Вона рефлекторно через хеморецептори синокаротидної
зони і дуги аорти викликає гіпервентиляцію легень, унаслідок чого може настати гіпокапнія і газовий алкалоз.
Таким чином, для паренхіматозної недостатності дихання характерні:
1) зменшення р02 артеріальної крові — гіпоксемія;
2) рС02 артеріальної крові не міняється або зменшується (гіпокапнія);
3) кислотно-основний стан не порушений або розвивається газовий алкалоз.
29.18. Назвіть причини порушення дифузії газів у легенях.
Дифузія газів через альвеоло-капілярну мембрану здійснюється відповідно до першого закону Фіка:
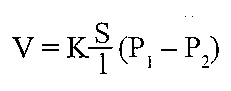
де V - кількість газу, що дифундує за одиницю часу; k - коефіцієнт дифузії; S -загальна площа, через яку відбувається дифузія; 1 - товщина мембрани; Р, і Р2 — парціальний тиск газів по обидві сторони мембрани.
З урахуванням цього можна виділити такі причини порушень дифузії газів у легенях:
1) зменшення коефіцієнта дифузії. Величина його залежить як від природи газу, так і від середовища, у якому відбувається дифузія. Практично має значення зменшення коефіцієнта дифузії кисню у зв'язку зі зміною властивостей легеневої тканини. При цьому перехід С02 із крові в альвеоли, як правило, не міняється, оскільки коефіцієнт його дифузії дуже високий (у 20-25 разів вищий, ніж кисню);
2) зменшення площі дифузії. Має місце при зменшенні дихальної поверхні легень;
3) збільшення товщини альвеоло-капілярноїмембрани;
4) зменшення різниці між парціальним тиском газів в альвеолярному повітрі та їх напругою в крові легеневих капілярів. Така ситуація виникає при всіх порушеннях вентиляції легень;
5) зменшення часу контакту крові з альвеолярним повітрям. Дифузія кисню порушується в тому випадку, якщо час контакту стає менше 0,3 с.
29.19. Назвіть причини порушень легеневої пєрфузії.
Порушення кровообігу в легенях (легеневої пєрфузії) можуть бути зумовлені такими причинами:
а) зменшенням тиску в правому шлуночку (недостатність правого серця, зменшення венозного повернення при крововтраті, шоку, колапсі);
6) збільшенням тиску в лівому передсерді (стеноз отвору двостулкового клапана, лі-вошлуночкова недостатність серця);
в) збільшенням опору судин малого кола кровообігу. Останнє може бути обумовлено рефлекторним збільшенням тонусу артеріол легень, збільшенням в'язкості крові, наявністю перешкод для руху крові (тромбоз, емболія).
29.20. Які причини і механізми розвитку гіпертензії малого кола кровообігу?
Гіпертензія малого кола кровообігу характеризується збільшенням тиску в легеневій артерії понад 25 мм рт. ст. її розвиток може бути обумовлений такими механізмами:
а) тривалий спазм артеріол легень. Найчастіше виникає в результаті зменшення парціального тиску кисню в альвеолярному повітрі, що буває при гіпоксичній гіпоксії (див. розд. 19) і порушеннях вентиляції легень;
б) гострий рефлекторний спазм легеневих артеріол. Розвивається при емболії судин легень, стенозі отвору двостулкового клапана. В останньому випадку вмикається рефлекс Китаєва: збільшення тиску в лівому передсерді і легеневих венах викликає збудження барорецепторів і спазм легеневих артеріол, що попереджає збільшення гідростатичного тиску в капілярах легень і розвиток набряку;
в) збільшення тиску повітря в бронхах і альвеолах. Викликає здавлення легеневих капілярів і, як наслідок, збільшення судинного опору в малому колі кровообігу. Буває у людей під час важких нападів кашлю. При цьому тиск у легеневій артерії може зростати до 250 мм рт. ст.;
г) облітерація легеневих судин (артеріол, капілярів, венул) унаслідок ураження їхніх стінок (наприклад, при емфіземі легень). В експерименті показано, що гіпертензія малого кола кровообігу виникає при вимиканні не менше 2/3 судинного русла. Отже, видалення однієї легені не призводить до розвитку цього синдрому;
ґ) збільшення хвилинного об'єму серця більше ніж у 3 рази;
д) порушення відтоку крові по легеневих венозних судинах (вади двостулкового клапана серця, недостатність лівого шлуночка, здавлювання легеневих вен);
є) збільшення в 'язкості крові (наприклад, при поліцитемії);
є) уроджені вади, пов'язані зі скиданням крові зліва направо (незарощення Боталло-вої протоки, дефекти міжшлуночкової перегородки). Залежно від того, на якій ділянці легеневих судин збільшується опір, розрізняють пре- і посткапілярну форму гіпертензії малого кола кровообігу.
29.21. Які механізми можуть лежати в основі розвитку набряку
легень?
Вихід рідини з кровоносних судин в інтерстиціальну тканину легень і альвеоли
може бути обумовлений такими механізмами.
1. Гідростатичний механізм - різке збільшення гідростатичного тиску в капілярах легень. Набряк розвивається, коли гідростатичний тиск стає вищим за 30 мм рт. ст. (у нормі 6-9 мм рт. ст.), тобто його величина стає більшою за онкотичний тиск крові. Така ситуація може виникати при гострій лівошлуночковій недостатності серця, обумовленій великим інфарктом міокарда, при стенозі отвору двостулкового клапана (кардіогенний механізм), при введенні великих кількостей (кілька літрів) крово- і плазмозамінних розчинів хворим з порушеним діурезом (гіперво-лемічний механізм).
2. Мембраногенний механізм — збільшення проникності легеневих капілярів. Буває при:
а) екзогенній інтоксикації (отруєння фосфорорганічними сполуками, наприклад, фосгеном);
б) ендогенній інтоксикації (уремія, печінкова недостатність);
в) алергічних реакціях І типу.
3. Онкотичний механізм — зменшення онкотичного тиску плазми крові. Відносно часто буває у хворих з нефротичними синдромом (див. розд. 32).
З урахуванням патогенезу розрізняють дві фази розвитку набряку легень.
I. Інтерстиціальний набряк - накопичення набрякової рідини в інтерстиціальній тканині легень. Клінічно виявляється нападами серцевої астми. Розвивається паренхіматозна недостатність дихання з явищами гіпоксемії.
II. Альвеолярний набряк — перехід набрякової рідини в альвеоли. При цьому порушується їх вентиляція — розвивається вентиляційна недостатність дихання з явищами гіпоксемії і гіперкапнії.
29.22. Якими порушеннями виявляє себе синдром емболії малого кола кровообігу?
Поява емболів у судинах легень викликає розвиток таких змін:
1) генеролізованш спазм артеріол усього малого кола кровообігу (а не тільки судин, де містяться емболи). Це викликає різку гіпертензію малого кола і розвиток гострої правопшуночкової недостатності серця (синдром гострого легеневого серця);
2) зменшення артеріального тиску у великому колі кровообігу. Пов'язане зі зменшенням хвилинного об'єму серця і зниженням тонусу артеріол у периферичних відділах (рефлекс Швічки—Паріна);
3) порушення зовнішнього дихання — розвиток паренхіматозної дихальної недостатності (див. запит. 29.17).
29.23. Які зміни загальних і місцевих вентиляційно-перфузійних відношень можуть спричинятися до розвитку недостатності зовнішнього дихання?
Вешпшяційно-перфузійне відношення для легень у цілому визначається як відношення хвилинного об'єму альвеолярної вентиляції (Va) до хвилинного об'єму крові (Q). У нормі:
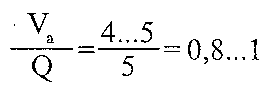
Якщо зазначене відношення більше одиниці, то це свідчить про збільшення функціонального мертвого простору, унаслідок чого ефективність вентиляції погіршується. Така ситуація виникає при гіпервентиляції, не підкріпленій збільшенням перфузії, або при нормальній вентиляції, але порушеному легеневому кровообігу.
Вентиляційно-перфузійне відношення менше 0,8 свідчить про так званий ефект шунтування, коли кров, не збагачена киснем, потрапляє в легеневі вени і потім у велике коло кровообігу. Це буває, коли величина легеневої перфузії значно перевищує величину альвеолярної вентиляції.
В умовах патології у зв'язку з нерівномірністю вентиляції і перфузії різних альвеол вентиляційно-перфузійні відношення в різних ділянках легень можуть бути різними. При цьому вентиляційно-перфузійне відношення для легень у цілому може бути в нормі, хоча і розвиваються ознаки дихальної недостатності. Вони обумовлені існуванням в уражених легенях трьох типів альвеол (рис. 141):
Рис. 141. Місцеві вентиляційно-перфузійні відношення:
1 — ефективний альвеолярний об 'єм;
2 — альвеолярний мертвий простір;
3 — альвеолярний артеріо-венозний шунт
1) альвеоли, які оптимально вентилюються і перфузуються (Va/Q = 0,8... 1). Вони утворюють ефективний альвеолярний об 'єм і становлять більшість альвеол здорових легень;
2) альвеоли, які вентилюються, але не перфузуються (V7Q >1). їхня сукупність становить альвеолярний мертвий простір^
3) альвеоли, які перфузуються, але не вентилюються (V/Q <0,8). З ними пов'язане артеріовенозне шунтування.
Збільшення кількості альвеол другого і третього типів може призводити до розвитку гіпоксемії. При цьому виділення С02 не порушується завдяки високому коефіцієнту його дифузії (розвивається паренхіматозна недостатність дихання).
29.24. Якими клінічними ознаками виявляє себе недостатність зовнішнього дихання?
1. Розвивається задишка (див. запит. 29.9).
2. Гіпоксемія (зменшення рО артеріальної крові) призводить до виникнення:
а) гіпоксії (дав. розд. 19);
б) ціанозу. Останній виникає, коли насичення гемоглобіну киснем стає нижче 80 % (синюшний відтінок шкіри і видимих слизових оболонок обумовлено відновленим гемоглобіном).
3. Гіперкапнія (збільшення рС02 артеріальної крові) викликає:
а) збудження дихального центру і задишку. Коли рС02 стає вище 90-100 мм рт. сх, збудження дихального центру змінюється його гальмуванням;
б) розширення мозкових судин і звуження судин м'язів, нирок;
в) зміщення кривої дисоціації оксигемоглобіну вправо;
г) газовий ацидоз.
4. Газовий ацидоз обумовлює:
а) порушення функцій білкових молекул (конформаційні зміни). Вони особливо небезпечні в центральній нервовій системі, з ураженням якої може бути пов'язаний розвиток дихальної коми;
б) збудження дихального центру;
в) збільшення проникності клітинних мембран, що викликає набряк і ушкодження клітин;
г) зміщення кривої дисоціації оксигемоглобіну вправо — ефект Бора.
ЗО. Патологічна фізіологія травлення
ЗО. 1. Що таке недостатність травлення?
Недостатність травлення - це патологічний стан, при якому травна система не забезпечує засвоєння поживних речовин, що надходять в організм. Наслідком цього є розвиток різного ступеня голодування (див. розд. 18).
30.2. Як класифікують недостатність травлення?
I. За клінічним перебігом виділяють гостру і хронічну недостатність травлення.
II. Відповідно до анатомічного принципу недостатність травлення може бути обумовлена порушеннями цього процесу:
а) у порожнині рота;
б) у шлунку;
в) у кишках.
III. Недостатність травлення може бути загальною (тотальною) і селективною (парціальною). При загальній недостатності порушено засвоєння всіх поживних речовин, при селективній - тільки окремих їхніх класів (наприклад, ліпідів, лактози, вітаміну В12 та ін.).
IV. За етіологією розрізняють спадково обумовлену (деякі види мальабсорбції) і набуту недостатність травлення. Остання може бути:
а) інфекційного походження;
б) обумовленою впливами фізичних факторів (наприклад, при гострій променевій хворобі);
в) пов'язаною із впливами хімічних агентів;
г) диерегуляторною; ґ) аліментарною.
V. Патофізіологічний принцип передбачає поділ недостатності травлення на три варіанти. Цс недостатність, обумовлена порушеннями:
а) рухової функції травної системи;
б) секреторної її функції;
в) процесів усмоктування.
30.3. Які причини можуть лежати в основі розвитку недостатності травлення?
І. Аліментарні (харчові) фактори:
а) приймання недоброякісної і грубої їжі;
б) сухоїдіння;
в) нерегулярне приймання їжі;
г) незбалансоване харчування (наприклад, зменшення вмісту вітамінів у раціоні);
ґ) зловживання алкоголем.
II. Фізичні фактори. Серед чинників цієї групи найбільше значення має іонізуюча радіація, яка уражує епітеліальні клітини травної трубки, що мають високу мітотичну активність.
При одноразовому загальному опроміненні організму дозою, що перевищує 10 Гр, розвивається так звана кишкова форма гострої променевої хвороби, що швидко завершується смертю (див. розд. 5).
III. Хімічні агенти. Є причиною розладів травлення при отруєннях неорганічними і органічними сполуками на виробництві і в побуті.
IV. Біологічні фактори:
а) бактерії (наприклад, холерний вібріон, збудники дизентерії, черевного тифу, паратифу та ін.);
б) бактеріальні токсини (наприклад, при сальмонельозах, стафілококовій інфекції);
в) віруси (наприклад, аденовіруси);
г) гельмінти.
V. Органічні ураження:
а) уроджені аномалії органів травлення;
б) післяопераційні стани;
в) пухлини травної системи.
VI. Порушення нервової й гуморальної регуляції. Розлади травлення можуть розвиватися при:
а) психоемоційних порушеннях (невротичні і неврозоподібні стани);
б) психічних захворюваннях (шизофренія, маніакально-депресивний синдром);
в) органічних захворюваннях центральної нервової системи (енцефаліти, ді-енцефаліт);
г) ураженнях периферичних структур вегетативної нервової системи; ґ) рефлекторних порушеннях (різні вісцеро-вісцеральні рефлекси).
Порушення гуморальної регуляції травлення можуть бути пов'язані з розладами синтезу і секреції гастроінтести-нальних гормонів (гастрину, секретину, холецистокінін-пан-креозиміну та ін.).
30.4. Які принципи використовують в експериментальному моделюванні недостатності травлення?
І. Принцип видалення. Полягає у хірургічному видаленні тих чи тих органів травної системи (шлунку, тонкої або товстої кишки). Найбільш фундаментальні дослідження в цьому напрямі було виконано Є. Лондоном. Він та його учні показали, що видалення різних відділів травного каналу призводить до розвитку різного ступеня недостатності травлення. Однак травна система має
досить високі компенсаторні можливості. Про це свідчить той факт, що життя тварин (собак) можливе навіть після поетапного видалення у них шлунку, всієї клубової і більшої частини порожньої кишки, а також майже всієї товстої кишки, за винятком її початкового відділу та прямої кишки. II. Принцип ізолювання. Набув широкого використання у фізіологічних і патофізіологічних дослідженнях завдяки роботам І. Павлова. Він, зокрема, запропонував методику ізольованого шлуночка. Крім того, часто застосовуються метод ізольованої петлі кишок, накладення анастомозів між різними відділами системи травлення. На відміну від методів видалення, ці методики моделювання дають можливість одержувати додаткову інформацію про характер і механізми змін в ізольованих ділянках травного каналу, про зміни нервової і гуморальної регуляції в цих умовах. III. Принцип відведення. Полягає у відведенні на різних рівнях травної системи їжі, що надходить в організм, та хімусу, а також у відведенні за межі травного каналу або в інші його відділи травних секретів. Ілюстрацією цього принципу можуть бути такі методики:
а) "удаване годування" (створення езофагостоми за І. Павловим) (рис. 142);
б) створення фістул кишок;
в) виведення назовні проток слинних залоз, панкреатичної протоки, загальної жовчної протоки.
Рис. 142. Езофаготомована собака з фістулою шлунку ("удаване годування ")
IV. Принцип екзогенних ушкоджень. При моделюванні патологічних процесів у травній системі використовують різні ушкоджувальні впливи: фізичні (висока і низька температури, іонізуюча радіація), хімічні (кислоти, луги, солі важких металів, органічні сполуки), біологічні (бактерії та їх токсини).
V. Принцип порушення нервової регуляції (див. запит. 30.5).
VI. Принцип порушення гуморальної регуляції (див. запит. 30.6).
ЗО. 5. За допомогою яких впливів на нервову систему моделюють розлади травлення?
1. Відтворення експериментальних неврозів— метод "зіткнення" умовних рефлексів (рос. "сшибка") за І. Павловим. За допомогою зіткнення процесів умовного збудження і умовного гальмування викликають позамежне гальмування в корі головного мозку. Наслідком цього є розгальмовування "підкірки", що виявляє себе у собак-ваготоніків підвищенням активності центрів парасимпатичної нервової системи. Останнє викликає збільшення секреторної і рухової активності органів травлення.
2. Збудження підкірковій структур центральної нервової системи. Для цього використовують або вживляння електродів з наступною електричною стимуляцією, або руйнування структур (наприклад, ядер блукаючого нерва).
3. Впливи на нервові провідники: перетинання блукаючого нерва та його гілок або їх електрична стимуляція.
4. Впливи на передачу збудження в парасимпатичних гангліях (наприклад, використання гангліоблокаторів).
5. Втручання в медіаторні механізми передавання збудження на ефекторні клітини (наприклад, застосування m-холіноміметиків або т-холінолітиків).
30.6. Які порушення гуморальної регуляції відтворюють з метою моделювання розладів травлення?
Основу гуморальної регуляції травлення становить секреція гастроінтести-нальних гормонів, до яких належать гастрин, секретин, холецистокінін-панкреози-мін, мотилін, гастроінгібіторний поліпептид (ГІП) та ін.
Для відтворення в експерименті різних типів порушень травлення використовують такі підходи:
а) введення в організм високих доз гастроінтестинальних гормонів або їхніх аналогів;
б) впливи, що активують утворення власних гастроінтестинальних гормонів (наприклад, застосування так званих сокогінних речовин);
в) пригнічення утворення і секреції гастроінтестинальних гормонів;
г) фармакологічна блокада чутливих до гастроінтестинальних гормонів рецепторів на поверхні ефекторних клітин.
ЗО. 7. Якими синдромами може виявляти себе недостатність травлення?
1. Голодування (див. розд. 18).
2. Диспепсичний синдром.
3. Зневоднення (див. розд. 23).
4. Порушення кислотно-основного стану (див. розд. 25).
5. Кишкова аутоінтоксикація (див. розд. 31).
6. Больовий синдром.
30.8. Якими ознаками може виявлятися диспепсичний синдром?
Диспепсичний синдром являє собою різні поєднання таких симптомів: а) анорек-сія; б) печія; в) відрижка; г) нудота; ґ) блювота; д) метеоризм; є) закрепи; є) пронос.
30.9. Що таке анорексія? Коли вона виникає?
Анорексія — це повна відсутність апетиту при об'єктивній потребі в харчуванні. Виділяють такі види анорексії:
а) інтоксикаційна - розвивається при гострих і хронічних отруєннях (наприклад, солями ртуті, лікарськими препаратами, бактеріальними токсинами);
б) диспепсична - виникає при захворюваннях органів травної системи, має найчастіше умовнорефлекторну природу;
в) нейродинамічна - розвивається в результаті реципрокного гальмування центру апетиту при перезбудженні окремих структур лімбічної системи (наприклад, больовий синдром при інфаркті міокарда, кольках, перитоніті);
г) невротична - пов'язана з надмірним збудженням кори головного мозку і сильними емоціями (особливо негативними);
ґ) психогенна— є результатом свідомого обмеження споживання їжі (наприклад, з метою схуднення або як результат нав'язливої ідеї при порушеннях психіки);
д) нейроендокринопатична - обумовлена органічними ураженнями центральної нервової системи (гіпоталамуса) і ендокринними захворюваннями (гіпофізарна кахексія, адисонова хвороба).
В основі розвитку анорексії можуть лежати два механізми:
1) зменшення збудливості харчового центру (інтоксикаційна, диспепсична, нейроен-докринопатична анорексія);
2) гальмування нейронів харчового центру (нейродинамічна, невротична, психогенна анорексія).
ЗО. 10. Що таке печія? Які її механізми?
Печія (pyrosis) - це відчуття жару по ходу стравоходу.
її розвиток пов'язаний з подразненням рецепторів стравоходу при закиданні вмісту шлунку у стравохід (рефлюкс). Це може бути обумовлено:
а) великою кількістю утворюваного шлункового соку;
б) функціональною недостатністю кардіального сфінктера.
30.11. Що таке відрижка? Які її механізми?
Відрижка (eructatio) - це раптове мимовільне виділення в порожнину рота газу зі шлунку або стравоходу, іноді з невеликими порціями вмісту шлунку.
Збільшення вмісту газів у шлунку може бути викликано двома причинами:
а) надходженням великої кількості газів з їжею і питвом (наприклад, газовані напої), заковтуванням повітря (аерофагія);
б) утворенням газів у самому шлунку, особливо при тривалій затримці там їжі (буває при виразковій хворобі, раку шлунку).
У результаті збільшення вмісту газів у шлунку збільшується внутрішньошлунко-вий тиск. Це рефлекторно викликає:
а) скорочення м'язів стінки шлунку;
б) спазм воротаря;
в) розслаблення м'язів стравохідно-шлункового сфінктера. Наслідком зазначених змін є витискання газів з порожнини шлунку у стравохід, глотку, а потім у порожнину рота.
ЗО. 12. Що таке нудота і блювота? Які їх механізми?
Hydoma (nausea) - це своєрідне тяжке відчуття в ділянці під грудьми, у грудях і порожнині рота, що нерідко передує блювоті і часто супроводжується загальною слабкістю, пітливістю, підвищенням слиновиділення, похолоданням кінцівок, блідістю шкіри, зниженням артеріального тиску, тобто ознаками активації парасимпатичної нервової системи. В основі нудоти лежить збудження блювотного центру, однак ще недостатнє для виникнення блювоти.
Блювота (vomitus) - це складнорефлекторний акт, що призводить до виверження вмісту шлунку назовні через рот. Виникає в результаті збудження блювотного центру, що міститься в довгастому мозку.
У механізмі блювоти виділяють ряд послідовних стадій.
1. Відбувається глибокий вдих з опущенням діафрагми, одночасно опускається надгортанник і піднімається гортань (перекривається вхід у гортань), піднімається м'яке піднебіння (перекривається вхід у носоглотку).
2. Скорочується воротар, тіло шлунку і стравохідно-шлунковий сфінктер розслаблюються, кардія підтягується догори, стравохід розширюється і коротшає.
3. Відбувається сильне скорочення діафрагми і м'язів черевного преса, у результаті чого підвищується внутрішньочеревний і внутрішньошлунковий тиск. Це у свою чергу викликає виверження вмісту шлунку через стравохід і порожнину рота назовні - виникає блювота.
Розрізняють такі патогенетичні варіанти блювоти:
а) центральна — пов'язана з підвищенням збудливості блювотного центру. Це буває при захворюваннях центральної нервової системи (менінгіти, енцефаліти, пухлини мозку), у разі надходження збуджувальних імпульсів з кори великих півкуль (умовнорефлекторна блювота) або рецепторів лабіринту (вестибулярна блювота);
б) гематогенно-токсична- обумовлена безпосередньою дією токсичних речовин, що містяться у крові, на хеморецептори структур блювотного центру. Це можуть бути екзогенні речовини (чадний газ, алкоголь, лікарські препарати, токсини бактерій) або токсичні продукти власного метаболізму, що накопичуються при уремії, печінковій недостатності, декомпенсованому цукровому діабеті та ін.;
в) вісцеральна (рефлекторна) — є результатом рефлексів, що виникають унаслідок подразнення різних рецепторних полів внутрішніх органів. Такі рефлексогенні зони є у шлунку, слизовій оболонці глотки, вінцевих судинах, очеревині, жовчних протоках та ін.
ЗО. 13. Що таке закрепи? Коли вони виникають?
Закрепи (obstipatio) — це уповільнене, утруднене або систематично недостатнє випорожнення кишок.
Виділяють два механізми розвитку закрепів - спастичний і атонічний. Перший обумовлений тривалим постійним скороченням гладких м'язів кишок (спазмом), другий - їх атонією.
До спастичних закрепів відносять: а) запальні — виникають унаслідок місцевих спастичних рефлексів зі зміненої слизової;
б) проктогенні - розвиваються при патології аноректальної ділянки;
в) механічні — виникають при непрохідності кишок;
г) токсичні - є результатом отруєнь свинцем, ртуттю, талієм.
Атонічними закрепами є:
а) аліментарні - розвиваються при надходженні їжі, що легко засвоюється і містить мало клітковини;
б) неврогенні - є результатом порушень нервової регуляції моторики кишок;
в) гіподинамічні - виникають у лежачих хворих, у старих людей, у людей з дуже низькою руховою активністю;
г) закрепи при аномаліях товстої кишки (наприклад, хвороба Гіршпрунга);
ґ) закрепи внаслідок порушень водно-сольового обміну (наприклад, при зневодненні).
ЗО. 14. Що таке метеоризм? Коли він виникає?
Метеоризм — це надмірне скупчення газів у травному каналі за рахунок їх підвищеного утворення або недостатнього виведення з кишок.
Джерелами газів у кишках є повітря, що його заковтують, процеси нейтралізації шлункового соку гідрокарбонатами (утворення вуглекислого газу), діяльність мікрофлори.
Надмірне утворення газів лежить в основі розвитку таких видів метеоризму:
а) аліментарного — розвивається при прийманні їжі, що містить багато клітковини, крохмалю (бобові, капуста, картопля);
б) при розладах травлення (ферментопатіях, порушеннях всмоктування, кишкових дисбактеріозах).
Порушення виведення газів становить сутність метеоризму:
а) механічного — розвивається в результаті порушення прохідності кишок (спазми, спайки, пухлини);
б) динамічного — виникає при розладах рухової функції кишок;
в) циркуляторного — є наслідком загальних і місцевих розладів кровообігу.
ЗО. 15. Що таке проноси? Чим вони можуть бути обумовлені?
Проноси (діарея) - це прискорене випорожнення кишок з виділенням розріджених, а в частині випадків і великих за об'ємом випорожнень.
Проноси виникають при порушенні нормальних співвідношень між секрецією і всмоктуванням рідини в кишках, при порушеннях моторики кишок.
Виділяють такі патогенетичні варіанти проносів:
а) осмотична діарея. Розвивається при збільшенні осмотичного тиску кишкового вмісту при прийманні всередину речовин, які погано або зовсім не всмоктуються (наприклад, проносних), а також при порушеннях травлення і всмоктування (синдроми мальдигестії й мальабсорбції);
б) секреторна діарея. Пов'язана з активацією секреції іонів електролітів (Na+, СІ"), що викликає посилену секрецію води в просвіт кишок (наприклад, при холері);
в) діарея, викликана гальмуванням активного транспорту іонів через клітинні мембрани в кишках (наприклад, уроджена хлордіарея - генетичний дефект усмоктування аніонів хлору у клубовій кишці);
г) діарея, обумовлена підвищенням проникності кишкової стінки (запальна); ґ) діарея при порушенні кишкової моторики. Може розвиватися як при посиленні, так і при ослабленні рухової активності кишок.
ЗО. 16. Що може бути причиною зневоднення організму при розладах функцій травлення?
Основними причинами зневоднення організму при розладах травлення можуть бути:
1) гіперсалівація — збільшення утворення й секреції слини;
2) нестримна блювота;
3) проноси.
При гіперсалівації розвивається гіперосмолярне зневоднення (слина гіпотонічна стосовно крові), а при нестримній блювоті і проносах — гіпоосмолярне (електролітів втрачається більше, якщо порівнювати з водою; докладно див. розд. 23).
Втрата рідини через травний канал може бути обумовлена специфічною дією деяких патогенних факторів. Так, установлено, що при холері зневоднення є результатом дії холерного екзотоксину на аденілатциклазу ентероцитів. При цьому відбувається стійке необоротне підвищення активності цього ферменту, внаслідок чого різко, у багато разів, збільшується секреція кишкового соку. Велика кількість кишкового секрету шляхом впливу на рецептори тонкої кишки викликає діарею.
ЗО. 17. Якими порушеннями кислотно-основного стану можуть виявляти себе розлади травлення?
При порушеннях функцій травної системи можуть розвиватися:
1) негазовш алкалоз. Є наслідком нестримної блювоти;
2) негазовш ацидоз. Виникає в результаті втрати великих кількостей гідрокарбонатів підшлункового соку і жовчі при діареї.
Докладну характеристику зазначених порушень представлено в розд. 25.
ЗО. 18. Чим обумовлюється кишкова аутоінтоксикація при \ порушеннях функцій травлення?
Кишкова аутоінтоксикація, як правило, пов'язана з дисбактеріозами і утворенням великих кількостей токсичних продуктів бродіння і гниття.
Дисбактеріоз - це порушення співвідношення між окремими видами кишкової мікрофлори. При цьому часто збільшується кількість бактерій, які викликають процеси гниття і бродіння. У результаті зростає утворення в кишках токсичних продуктів — сірководню, скатолу, індолу, фенолів, путресцину, кадавери-ну та ін. Якщо утворення цих продуктів перевищує функціональні можливості печінки щодо їх детоксикації, розвиваються ознаки печінкової недостатності (див. розд. 31).
Розвитку кишкової аутоінтоксикації сприяють зменшення перистальтики кишок (закрепи), зменшення секреції кишкового соку, кишкова непрохідність.
ЗО. 19. Які механізми можуть лежати в основі больового синдрому при ураженнях травної системи?
Біль часто супроводжує розвиток захворювань травного каналу. Він може мати різний характер залежно від причин і патогенезу.
Розрізняють такі механізми виникнення болю при ураженнях органів травлення:
а) спастичний механізм. Біль обумовлений спазмом гладкої мускулатури різних відділів травного каналу. Вважають, що в цьому випадку причиною болю є перетискання судин, що проходять у товщі стінки порожнистих органів, унаслідок чого розвивається ішемія. Остання викликає появу продуктів, що діють на больові нервові закінчення (див. розд. 34). У разі різкого сильного спазму, що виникає раптово, розвиваються болі за типом кольки',
б) гіпотонічний механізм. При зменшенні тонусу гладкої мускулатури (гіпотонії) болі виникають у результаті розтягнення стінки порожнистих органів (шлунку, кишок, жовчного міхура) їхнім вмістом. При цьому механічне розтягнення тканин обумовлює подразнення закладених у них нервових закінчень;
в) вплив біологічно активних речовин (гістаміну, серотоніну, кінінів, простагландинів та ін.) на нервові закінчення. Ці речовини утворюються і вивільнюються при ушкодженні клітин і запаленні (гастрити, дуоденіти, ентерити, коліти, холецистити). Особливо багато їх з'являється при гострому панкреатиті (див. запит. 30.49, 30.50).
ЗО. 20. Які порушення рухової, секреторної і всмоктувальної функцій травної системи можуть лежати в основі розладів травлення?
I. Порушення рухової (моторної) функції травного каналу:
1) порушення жування;
2) порушення ковтання — дисфагія;
3) шлункові дискінезії;
4) кишкові дискінезії;
5) дискінезії жовчного міхура і жовчних проток;
6) порушення дефекації.
II. Порушення секреторної функції травної системи:
а) гіперсекреторні стани:
1) гіперсалівація;
2) шлункова гіперсекреція;
3) панкреатична гіперсекреція;
4) гіперхолія;
б) гіпосекреторні стани:
1) гіпосалівація;
2) шлункова гіпосекреція;
3) панкреатична гіпосекреція;
4) ахолія.
III. Порушення всмоктувальної функції — синдром мальабсорбції.
30.21. Що може спричинятися до порушень жування? Яке значення для травлення мають ці порушення?
Причини порушень жування:
1) ураження зубів та їх відсутність (карієс, пародонтит);
2) ураження жувальних м'язів (міозит);
3) порушення іннервації жувальних м'язів (бульбарні паралічі, неврити);
4) ураження скронево-нижньощелепних суглобів;
5) травматичне ушкодження кісток (нижньої, верхньої щелепи);
6) ураження слизової оболонки порожнини рота і ясен (стоматит, гінгівіт);
7) ураження м'язів і слизової язика;
8) гіпосалівація.
Порушення жування мають такі наслідки:
а) зменшення рефлекторної секреції шлункового і підшлункового соків;
б) уповільнення травлення в шлунку;
в) травматизація слизової оболонки порожнини рота, стравоходу, шлунку;
г) відмова від споживання деяких продуктів харчування, що потрібні організму, але потребують пережовування.
30.22. Що таке карієс зубів? Які причини його розвитку?
Карієс зубів - це патологічний процес, що виявляє себе демінералізацією і про-гресуючим руйнуванням твердих тканин зуба з утворенням дефектів у вигляді порожнин.
Поширеність карієсу становить 80-90 %, а в деяких районах земної кулі - 100 %.
Етіологію карієсу нині остаточно не встановлено, хоча й доведено значення цілого ряду факторів у його розвитку. До таких відносять:
1. Мікроорганізми. Велике значення мають деякі штами стрептококів і лактобакте-рій. В експерименті доведено, що у тварин-гнотобіонтів (безмікробних тварин) карієс не виникає.
2. Надмірне споживання харчових продуктів, що легко ферментуються у порожнині рота бактеріями. Такими, зокрема, є вуглеводи (сахароза, глюкоза, фруктоза^ Запропоновано карієсогенні дієти (містять від 25 до 65 % сахарози), що викликають розвиток карієсу у щурів, хом'яків, мишей.
3. Недостатній вміст у раціоні деяких речовин, зокрема вітамінів групи В, кальцію, фосфору, мікроелементів (фтору, стронцію, молібдену).
4. Генетичні фактори, що впливають на закладку зубних зачатків. Отримано чисті лінії щурів, стійких і чутливих до розвитку карієсу.
5. Зменшення захисної і трофічної функції слини.
6. Загальні порушення фосфорно-кальцієвого обміну (гіпокальціємічні і гіпофосфа-темічні стани; див. розд. 24).
30.23. Як уявляють сьогодні патогенез карієсу?
У патогенезі карієсу провідне значення надають зубному нальоту (зубній бляшці) - скупченню мікроорганізмів та продуктів їх життєдіяльності на поверхні зубів.
Мікроорганізми зубного нальоту виділяють цілий ряд карієсогенних факторів. До них відносять:
а) органічні кислоти (молочна, піровиноградна та ін.), що викликають розчинення оксіапатиту емалі й дентину, внаслідок чого розвивається їх демінералізація;
б) речовини-хелатори (пептиди, амінокислоти). Вони утворюють комплекси з кальцієм, забираючи його від оксіапатитів і викликаючи цим демінералізацію твердих тканин зуба;
в) протеолітичні ферменти. Діючи на білкові компоненти зуба, вони руйнують ма-трикс емалі і дентину. Відбувається деструкція твердих тканин, порушується їх ремінералізація.
Інші етіологічні фактори карієсу (див. запит. 30.22) можуть втручатися в патогенез на різних його етапах. Так, зменшення захисних властивостей слини сприяє розвитку мікрофлори і утворенню зубного нальоту; вуглеводи їжі, адсорбуючись на поверхні зуба, є джерелом великої кількості карієсогенних органічних кислот; генетичні фактори (неповноцінність органічної матриці зуба) і дефіцит цілого ряду речовин сприяють порушенням ремінералізації емалі і дентину. Нарешті, загальні порушення фосфорно-кальцієвого обміну самі собою можуть обумовлювати демінералізацію твердих тканин зуба.
ЗО. 24. Що таке пародонтит? Які фактори можуть лежати в основі його розвитку?
Пародонтит — це дистрофічно-запальне ураження пародонта, тобто тканин, що оточують корінь зуба (періодонта, кісткової тканини зубної комірки, ясен, окістя). Хвороба виявляє себе резорбцією зубних комірок, гноєтечею з ясен, розхитуванням і випадінням зубів.
Серед численних факторів, що мають стосунок до етіології пародонтиту, виділяють такі.
1. Мікрофлора порожнини рота, з якою пов'язують виділення ферментів і біологічно активних речовин зубного нальоту і утворення зубного каменю, що порушує кровопостачання тканин пародонта.
2. Аліментарні порушення — неповноцінне харчування, дефіцит вітамінів, особливо С, Р і Е.
3. Нейрогенні фактори - емоційна перенапруга, що викликає стрес; порушення нервової трофіки.
4. Ендокринні розлади — гіпогонадизм, гіпотиреоз, гіпоінсулінізм, гіперпаратиреоз.
5. Аномалії прикусу, зменшення або надмірне збільшення навантаження на жувальний апарат.
30.25. Які причини і значення гіперсалівації?
Гіперсалівація - збільшення утворення і секреції слини — може бути обумовлена
такими причинами:
а) збудженням рецепторів порожнини рота, стравоходу і шлунку (рефлекторний механізм). Гіперсалівація супроводжує розвиток запальних процесів у порожнині рота (гінгівіти, стоматити, пульпіти, періодонтити), є реакцією надію бормашини;.
б) збудженням центру слиновиділення, що міститься в довгастому мозку (бульбарні паралічі);
в) подразненням вегетативних нервів, що іннервують слинні залози. При стимуляції парасимпатичних нервів виділяється багато рідкої слини, при активації симпатичних - мала кількість, але густої, в'язкої слини;
г) впливом т-холіноміметиків - фармакологічних препаратів, що активують т- хо-лінорецептори;
ґ) впливом гуморальних регуляторних факторів. Цей механізм лежить в основі так званої парадоксальної, або паралітичної, гіперсалівації. Вона починається через добу після повної денервації слинних залоз, досягає максимуму через 6-7 діб, з 15-ї доби починає зменшуватися і через 35-40 діб повністю зникає. У патогенезі парадоксальної гіперсалівації провідне значення має підвищення чутливості денервованих залоз до циркулюючих у крові гуморальних факторів (ацетилхоліну, гістаміну). При гіперсалівації виділяється від 5 до 20 л слини за добу (у нормі - 0,5-2 л). Це
призводить до:
1) зневоднення організму — розвивається гіперосмолярна гіпогідрія, оскільки слина гіпотонічна стосовно крові; ( 2) нейтралізації шлункового соку, що пов'язано зі слабколужним середовищем слини. Ця обставина викликає порушення травлення у шлунку.
30.26. Які причини і значення гіпосалівації?
Причинами зменшення утворення і виділення слини (гіпосалівації) можуть бути: І а) центральне гальмування секреції слинних залоз (страх, переляк, біль); [ б) дія m-холінолітиків — фармакологічних агентів, що блокують т-холінорецептори
периферичних тканин; І в) ушкодження секреторних клітин слинних залоз (запалення, пухлини);
г) порушення виведення секрету (закупорка проток слинних залоз каменями); І г) зневоднення організму. Наслідки гіпосалівації:
1) порушення жування, формування харчової грудки, ковтання, що спричиняється до розладів наступних етапів травлення;
2) травматизація слизової оболонки рота з розвитком її запалення (стоматиту);
3) активний розвиток мікроорганізмів. Він обумовлений зменшенням бактерицидної дії слини (мало лізоциму) і утворенням нальоту зі злущеного епітелію на поверхні язика (прекрасне поживне середовище для мікробів);
4) порушення трофічних впливів слини на зуби, що сприяє розвитку карієсу.
30.27. Що таке дисфагія? Назвіть її причини. Яке вона має значення?
Дисфагія - це порушення складного рефлекторного акту ковтання, що відбува-' ється в три фази: ротову (довільну), глоткову (швидку мимовільну) і стравохідну | (повільну мимовільну).
Причинами дисфагії можуть бути:
а) ураження рецепторів слизової порожнини рота (стоматити) і глотки (ангіни);
б) ураження чутливих аферентних і рухових еферентних нервових провідників, що беруть участь у здійсненні ковтальних рефлексів (волокна V, VII, IX, X, XII пар ] черепномозкових нервів);
в) ураження нервових центрів - у корі головного мозку (порушується довільна фаза ковтання) і центру ковтання, розташованого в ділянці дна IV шлуночка;
г) ураження м'язів язика, глотки і стравоходу. Порушення функції гладкої мускулатури стравоходу можуть виявлятися як спастичними станами (ідіопатичний дифузний спазм, ахалазія - спазм кардії), так і гіпотонічними (атонічними), для яких характерне порушення перистальтики стравоходу;
ґ) уроджені та набуті дефекти м'якого й твердого піднебіння;
д) механічні перешкоди (пухлини, рубці, здавлювання стравоходу ззовні).
При дисфагії вкрай утруднено приймання їжі, у результаті чого можуть розвиватися голодування і виснаження. Крім того, можливе потрапляння слини і часток їжі в дихальні шляхи, що викликає аспіраційну пневмонію, а іноді і гангрену легень.
30.28. Що таке шлункові дискінезії? Які існують їхні варіанти?
Шлунковими дискінезіями називають порушення рухової (моторної) функції шлунку.
Виділяють два варіанти шлункових дискінезій: гіпертонічний і гіпотонічний.
Для гіпертонічного варіанта характерне збільшення тонусу м'язів шлунку (гіпертонія) і посилення його перистальтики (гіперкінезія).
Гіпотонічний варіант, навпаки, характеризується гіпотонією і гіпокінезією.
30.29. Назвіть причини /'значення гіпертонічних дискінезій шлунку.
Причинами рухових розладів шлунку гіпертонічного типу можуть бути:
а) деякі харчові фактори (груба їжа, алкоголь);
б) підвищення шлункової секреції;
в) збільшення тонусу блукаючого нерва;
г) деякі гастроінтестинальні гормони, зокрема мотилін.
Гіпертонія і гіперкінезія шлунку спричинюються до:
1) тривалої затримки харчових мас у шлунку, що сприяє підвищенню шлункової секреції і розвитку виразок на слизовій оболонці;
2) розвитку антиперистальтики (зворотної перистальтики) шлунку, що обумовлює появу таких ознак диспепсії, як відрижка, нудота, блювота.
ЗО. ЗО. Що таке пілороспазм?
Шлороспазм — це спастичне скорочення пілоричної частини шлунку, одна з форм дискінезій шлунку гіпертонічного типу. Його спостерігають переважно у грудних дітей, частіше в перші тижні і місяці життя.
Вважають, що пілороспазм у дітей обумовлений функціональними розладами нервово-м'язового апарату пілоричної частини шлунку. Він буває головним чином у збудливих дітей, що зазнали внутрішньоутробної гіпоксії, що народилися в асфіксії, з ознаками пологової травми центральної нервової системи.
При пілороспазмі відзначають слабкий розвиток мускулатури кардіальної частини шлунку і більш виражений її розвиток у ділянці воротаря. Це сприяє легкому виникненню блювоти і зригувань.
30.31. Назвіть причини і значення гіпотонічних дискінезій шлунку.
Зменшення рухової (моторної) активності шлунку може бути обумовлено:
а) аліментарними факторами (жирна їжа);
б) зменшенням шлункової секреції (гіпоацидні гастрити);
в) зменшенням тонусу блукаючого нерва;
г) дією гастроінтестинальних гормонів, що пригнічують моторику шлунку, - гастро-інгібіторного пептиду, секретину та ін.;
ґ) видаленням пілоричної частини шлунку;
д) загальним ослабленням організму, виснаженням, гастроптозом (опущенням шлунку).
При гіпотонічних дискінезіях зменшується час перебування їжі в шлунку, що веде до порушення її перетравлювання. Дія неперетравлених компонентів їжі на рецептори слизової оболонки кишок викликає підвищення їхньої перистальтики і проноси.
30.32. Назвіть типи патологічної шлункової секреції.
Порушення шлункової секреції в експерименті виявляють шляхом визначення кількості шлункового соку і його складу після механічного, а потім хімічного впливу. Розрізняють такі типи патологічної шлункової секреції (рис. 143):
Рис. 143. Типи шлункової секреції
1) збудливий тип. Характеризується збільшенням секреції шлункового соку як на механічні, так і на хімічні стимули;
2) астенічний тип. Виявляється збільшенням секреції шлункового соку на механічні впливи і зменшенням - на хімічні;
3) інертний тип. Шлункова секреція зменшена при дії механічних стимулів і зростає при хімічній стимуляції;
4) гальмівний тип. Характеризується зменшенням секреції шлункового соку в умовах як механічної, так і хімічної стимуляції.
... ...
У людини залежно від кількості шлункового соку і його якісних характеристик J виділяють шлункову гіпер- і гіпосекрецію.
30.33. Чим виявляє себе шлункова гіперсекреція? Яке її значення?
Для шлунковоїгіперсекреціїхарактерно:
1) збільшення кількості шлункового соку як після приймання їжі, так і натще;
2) гіперацидітас і гіперхлоргідрія — відповідно збільшення загальної кислотності і вмісту вільної соляної кислоти в шлунковому соку;
3) збільшення перетравлювальної здатності шлункового соку.
Порушення травлення, пов'язані зі шлунковою гіперсекрецією, обумовлені тривалою затримкою вмісту у шлунку (воротар закритий, тому що нейтралізація дуже кислого хімусу, що надходить у дванадцятипалу кишку, вимагає багато часу). Ця обставина має такі наслідки:
1) у кишки надходить мало хімусу, що призводить до зменшення перистальтики кишок і розвитку закрепів;
2) у шлунку посилюються процеси бродіння і утворення газів. Це викликає появу відрижки й печії;
3) збільшується рухова активність шлунку — розвивається гіпертонус і гіперкінезія його гладкої мускулатури.
Утворення великої кількості активного шлункового соку є важливим чинником* що сприяє розвитку виразок у шлунку і дванадцятипалій кишці.
ЗО. 34. Як можна моделювати шлункову гіперсекрецію в експерименті?
Збільшення утворення і секреції шлункового соку моделюють за допомогою таких впливів.
I. Вплив на нервові механізми регуляції шлункової секреції:
а) збудження рецепторів слизової оболонки шлунку (введення в шлунок со-когінних екстрактів, алкоголю);
б) зменшення гальмівного впливу кори головного мозку на центри блукаючого нерва - метод "зіткнення" за І.Павловим (див. запит. 30.5);
в) електричне подразнення центрів блукаючого нерва або його еферентних волокон, що іннервують шлунок;
г) введення т-холіноміметиків.
II. Вплив на гуморальні механізми регуляції шлункової секреції:
а) введення гістаміну або фармакологічних агентів, що стимулюють його утворення в слизовій шлунку;
б) введення гастрину;
в) введення фармакологічних препаратів — стимуляторів Н2-рецепторів (ацетилсаліцилової кислоти, індометацину);
г) пригнічення утворення простагландинів, які в нормі гальмують секрецію соляної кислоти. З цією метою вводять глюкокортикоїди, ацетилсаліцилову кислоту.
30.35. Що таке виразкова хвороба? Яка її етіологія?
Виразкова хвороба — це хронічне рецидивуюче захворювання, що характеризується утворенням виразки в шлунку або дванадцятипалій кишці.
Етіологію виразкової хвороби донині остаточно не з'ясовано. Вважають, що у виникненні виразок шлунку і дванадцятипалої кишки мають значення такі фактори ризику.
1. Тривала психоемоційна перенапруга, як правило, негативного характеру (негативні емоції, конфліктні ситуації, почуття постійної тривоги, перевтома і т. п.), яка часто повторюється.
2. Стрес (див. розд. 33).
3. Спадкова схильність. Значення цього фактора підтверджує відносно висока (40-60 %) частота захворювання у батьків і родичів хворих, особливо молодого віку. Установлено, що у хворих з обтяженою спадковістю в слизовій оболонці шлунку у 1,5-2 рази більше парієтальних клітин (клітин, що продукують соляну кислоту), ніж у здорових. Ознаками генетичної схильності є також 0 (І) група крові, яку часто виявляють у хворих на виразку, дефіцит с^-антитрипсину і фукоглікопротеїдів.
4. Неправильне харчування - їжа всухом'ятку, нерегулярне приймання їжі, вживання грубої або гострої їжі, погане її пережовування, їда поспіхом, відсутність зубів, недостатній вміст у харчових продуктах білків і вітамінів.
5. Хронічний гастрит і дуоденіт з підвищеною секреторною активністю залоз слизової оболонки.
6. Мікробний фактор - Helicobacter (Campylobacter) pylori.
7. Шкідливі звички — куріння, уживання алкоголю.
30.36. Які існують теорії патогенезу виразкової хвороби?
1. Судинна теорія (Р. Вірхов). Відповідно до цієї теорії, виразки в шлунку і дванадцятипалій кишці виникають у результаті порушень кровопостачання їхніх стінок.
2. Механічна теорія (Л. Ашофф). Виникнення виразки пов'язано з травмуванням грубою їжею слизової оболонки шлунку в ділянці малої його кривизни.
3. Запальна теорія (Г. Конечний). В основі утворення виразок лежать запальні зміни слизової оболонки (гастрит та ерозії).
4. Пептична теорія (Е. Рігель). Пояснює виникнення виразки перетравлювальною дією шлункового соку на ділянки слизової, що найбільше зазнають впливу протеолітичних ферментів.
5. Нервово-вегетативна теорія (Г. Бергман). її прихильники вважають, що причиною утворення виразки є гіперсекреція шлункового соку, гіпермоторика шлунку і судинні порушення в ньому в осіб з конституціонально обумовленим переважанням тонусу парасимпатичної нервової системи.
6. Нервово-рефлекторна теорія (І. Греков, М. Стражеско). Пояснює виникнення виразки рефлекторними впливами на шлунок, наприклад, при хронічному апендициті, коліті, жовчнокам'яній хворобі.
7. Кортико-вісцеральна теорія (К. Биков, І. Курцин). Підкреслює провідну роль порушень умовнорефлекторної діяльності головного мозку (неврозів) в утворенні виразок.
8. Інфекційна теорія (Б. Маршали). Виразка у шлунку є результатом інфікування бактерією Helicobacter pylori (див. запит. 30.38.). Нині формується концепція, відповідно до якої утворення виразки в шлунку і дванадцятипалій кишці відбувається в результаті змін у співвідношенні факторів "агресії" і "захисту". До "факторів агресії" відносять підвищення кислотності і ферментативної активності шлункового соку в умовах порушення моторики шлунку і дванадцятипалої кишки, Helicobacter pylori. Зменшення захисних властивостей обумовлено зниженням продукції слизу, уповільненням процесів фізіологічної регенерації поверхневого епітелію, порушеннями місцевого кровообігу і нервової трофіки слизової оболонки.
30.37. Які виділяють патогенетичні варіанти виразки шлунку?
1. Екзогенна виразка.
2. Пептична виразка.
3. Трофічна виразка..
4. Гіпорегенераторна виразка.
30.38. Що таке екзогенна виразка шлунку? Що може бути її причиною?
Екзогенною називають виразку, що виникає в результаті безпосередньої дії на слизову оболонку шлунку ушкоджувальних факторів зовнішнього середовища.
В експерименті множинні виразки шлунку екзогенного походження моделюють за допомогою:
а) фізичних впливів - уведення в шлунок роздробленого скла (механічне ушкодження), окропу, рідкого азоту і т. п.;
б) хімічних впливів - уведення в шлунок сильних кислот і лугів.
У розвитку екзогенної виразки в людини найбільше значення мають мікробні фактори, зокрема Helicobacter pylori. Цей збудник потрапляє в шлунок через рот, занурюється в захисний шар слизу і прикріплюється до апікальної поверхні клітин епітелію. Найактивніше він заселяє вихідний відділ шлунку і цибулину дванадцятипалої кишки. Проникнення збудника в слизову викликає локальну імунну відповідь — антитіла, що утворилися, взаємодіють з поверхневими антигенами бактерій. Це у свою чергу спричиняє вихід у слизову поліморфноядерних лейкоцитів (нейтрофілів) - розвивається запалення (набряк, гіперемія), утворюються спочатку ерозії, які згодом переходять у виразку.
30.39. Що таке пептична виразка шлунку? Як її моделюють в експерименті?
Пептичною називають виразку, що виникає в результаті перетравлювальної дії шлункового соку на слизову оболонку шлунку і дванадцятипалої кишки.
Розрізняють два патогенетичних варіанти пептичної виразки: 1) виразка, що утворюється в результаті підвищення "агресивності" шлункового соку (збільшення кількості соку, його кислотності і перетравлювальної сили). В експерименті може виникати при моделюванні шлункової гіперсекреції (див. запит. 30.34);
2) виразка, обумовлена зменшенням захисних властивостей слизової оболонки. За цих умов шлунковий сік, що має нормальну "агресивність", чинить перетравлю-вальну дію на змінену слизову. Існує два підходи до моделювання в експерименті пептичної виразки цього виду:
а) порушення утворення слизу (введення глюкокортикоїдів, саліцилатів);
б) руйнування слизового бар'єра речовинами-детергентами (введення в шлунок жовчних кислот, відтворення дуодено-гастрального рефлюксу).
30.40. Що таке трофічні виразки шлунку? Як їх відтворюють в експерименті?
Трофічними називають виразки, які виникають у результаті порушення живлення слизової оболонки шлунку. До трофічних виразок відносять:
а) судинні виразки. Утворюються внаслідок порушень кровопостачання стінки шлунку. В експерименті їх моделюють перев'язуванням артерій, що живлять шлунок, введенням у шлункові артерії атофану — речовини, яка викликає склерозування стінки і облітерацію просвіту судин; введенням великих доз катехоламінів, що спричиняють спазм артерій та ішемію слизової шлунку;
б) нейрогенні виразки. Є результатом порушень нервової трофіки і розвитку нейро-дистрофічного процесу. В експерименті моделюють перетинанням нервів, що ін-нервують стінку шлунку.
30.41. Що таке гіпорегенераторні виразки шлунку? Чим вони можуть бути зумовлені?
Гіпорегенераторні виразки шлунку виникають у результаті порушення регенерації епітелію слизової оболонки. У нормі покривний епітелій і залозистий апарат слизової шлунку повністю оновлюється через кожні 5 діб. Якщо процеси регенерації з якихось причин порушуються, то спочатку утворюються поверхневі дефекти — ерозії, а потім і глибші — виразки.
Розвиток гіпорегенераторних виразок може бути зумовлений:
а) дією високих доз глюкокортикоїдів, що пригнічують біосинтез білка і клітинний поділ;
б) білковим голодуванням;
в) гіповітамінозами;
г) дією отрут — інгібіторів біосинтезу білків.
30.42. Чим виявляє себе шлункова гіпосекреція? Яке її значення?
Для шлунковоїгіпосекреціїхарактерні:
1) зменшення кількості шлункового соку натще і після приймання їжі;
2) зменшена або нульова кислотність шлункового соку (гіпо- або онацидітас), зменшення вмісту в ньому або повна відсутність вільної соляної кислоти (гіпо- або ахлоргідрія);
3) зменшення перетравлювальної здатності шлункового соку аж до ахілії (повного припинення утворення соляної кислоти і ферментів).
Зменшення шлункової секреції спричиняється до розладів травлення у всьому травному каналі. Це обумовлено тим, що в умовах недостатнього утворення шлунко- і вого соку воротар постійно відкритий і вміст шлунку швидко переходить у дванадця- І типалу кишку, де середовище внаслідок відсутності надходження зі шлунку соляної кислоти постійно лужне. Ця обставина гальмує утворення секретину, внаслідок чого зменшується секреція підшлункового соку й порушуються процеси порожнинного травлення в кишках. Недостатньо перетравлені компоненти їжі подразнюють рецептори слизової оболонки кишок, що призводить до посилення їхньої перистальтики, -розвиваються проноси.
Крім того, внаслідок відсутності соляної кислоти в шлунку посилено розмножується мікрофлора. З цим пов'язана активація процесів гниття й бродіння в шлунку і поява таких диспепсичних симптомів, як відрижка, обкладений язик та ін.
30.43. Як в експерименті можна моделювати шлункову гіпосекрецію?
І. Вплив на нервові механізми регуляції шлункової секреції:
а) блокада рецепторів слизової оболонки шлунку (застосування речовин, що викликають місцеву анестезію);
б) перетинання блукаючого нерва або його гілок (селективна ваготомія);
в) використання гангліоблокаторів (п-холінолітиків);
г) застосування ш-холінолітиків.
її. Вплив на гуморальні механізми регуляції утворення і секреції шлункового соку:
а) блокада Н -рецепторів (гістамінових), наприклад, циметидином;
б) застосування антагоністів гастрину - секретину, гастроінгібіторного пептиду та ін.
30.44. Які причини панкреатичної гіперсекреції? Чим вона може виявлятися?
Збільшення утворення і секреції підшлункового соку (панкреатична гіперсе-креція) може бути обумовлене:
1) підвищенням тонусу парасимпатичної нервової системи (блукаючого нерва);
2) збільшенням утворення і секреції гастроінтестинальних гормонів, що стимулюють секрецію води і гідрокарбонатів у складі підшлункового соку (секретин) і підвищують вміст у ньому травних ферментів (холецистокінін-панкреозимін).
Збільшення панкреатичної секреції поліпшує процеси порожнинного травлення, однак за деяких умов може сприяти розвитку гострого панкреатиту.
30.45. Що таке гострий панкреатит? Яка його етіологія?
Гострий панкреатит - це запалення підшлункової залози, що характеризується гострим перебігом.
Його виникнення і розвиток можуть бути обумовлені такими етіологічними факторами:
1) приймання великої кількості жирної їжі;
2) зловживання алкоголем і переїдання, що його супроводжує;
3) жовчні камені та поліпи протоки підшлункової залози;
4) механічне ушкодження підшлункової залози при травмах і хірургічних втручаннях;
5) інфекційні агенти (віруси епідемічного паротиту, коксакі, бактеріальна інфекція);
6) інтоксикації, у тому числі дія деяких лікарських засобів (імунодепресанти, тіази-ди та ін.).
30.46. Що є головною ланкою патогенезу гострого панкреатиту? Які розрізняють патогенетичні варіанти цього захворювання?
Провідним механізмом розвитку гострого панкреатиту будь-якої етіології є передчасна активація ферментів підшлункового соку в протоках підшлункової залози. Найбільше значення має активація трипсину, тобто утворення його із трипсиногену під дією активних протеолітичних ферментів клітинного (лізосомного) або кишкового походження. Особливістю активації ферментів у протоках підшлункової залози є те, що цей процес має лавиноподібний характер і, виникнувши в одному місці залози, швидко охоплює всі її протоки. Це обумовлено аутокаталітичними реакціями, під час яких молекули активних ензимів (трипсину, хімотрипсину) перетворюють неактивні ферменти (трипсиноген, хімотрипсиноген) в активні.
Таким чином, для ініціювання гострого панкреатиту потрібна "іскра", якою можуть стати навіть поодинокі молекули активних протеаз, що потрапили в протоки залози з ушкоджених клітин або кишок.
Залежно від джерела таких протеаз і механізмів їх потрапляння в протоки підшлункової залози розрізняють три патогенетичних варіанти гострого панкреатиту.
I. Первинно-альтеративний.
II. Гіпертензивний.
III. Рефлюксний.
ЗО. 4 7. Чим характеризується первинно-альтеративний варіант розвитку гострого панкреатиту?
Первинно-альтеративний варіант гострого панкреатиту розвивається в результаті ушкодження тканини підшлункової залози різними патогенними факторами. Це може бути механічне ушкодження (травма, операційні втручання), вплив хімічних агентів (отруєння лугами, кислотами) і біологічних чинників (віруси, бактерії); ішемічне або алергічне ушкодження.
Ініціаторами передчасної активації ферментів підшлункового соку у всіх цих випадках є лізосомні ферменти, що вивільняються з ушкоджених клітин залозистої тканини.
30.48. Що лежить в основі розвитку гіпертензивного варіанта гострого панкреатиту?
Гіпертензивний варіант гострого панкреатиту розвивається в результаті підвищення тиску панкреатичного соку в протоках підшлункової залози.
Залежно від причин виникнення гіпертензії розрізняють два різновиди цього варіанта:
1) гіперсекреторний панкреатит. Причиною підвищення тиску в протоках залози є різке збільшення секреції підшлункового соку (жирна їжа, алкоголь, що стимулю-
ють утворення секретину і холецистокінін-панкреозиміну), коли панкреатичні протоки не в змозі швидко пропустити весь сік, що утворився (відносна недостатність^ проток). У цьому випадку мають значення анатомо-фізіологічні особливості вивід-: них проток залози, тому панкреатит у такій формі може виникати не в усіх людей; 2) обтураційний панкреатит. Гіпертензія в протоках підшлункової залози виникає при нормальному рівні секреції, але при наявності перешкод (обтурації) відтоку соку (камені, здавлювання проток ззовні та ін.). Підвищений тиск соку є причиною ушкодження клітин, що вистилають протоки залози. Це призводить до виходу лізосомних ферментів і активації трипсиногену з наступною реалізацією аутокаталітичного механізму (див. запит. 30.46).
30.49. Які умови викликають рефлюксний варіант розвитку гострого панкреатиту?
Рефлюксний варіант гострого панкреатиту виникає в результаті закидання в протоки підшлункової залози вмісту дванадцятипалої кишки або жовчі.
Дуодено-шнкреатичний рефлюкс виникає при збільшенні внутрішньодуоде-нального тиску (кишкова непрохідність) або розслабленні сфінктера Одді. Ініціатором передчасної активації ферментів підшлункової залози в цьому випадку є енте-рокіназа - протеолітичний фермент, що утворюється клітинами слизової оболонки дванадцятипалої кишки.
Умовою виникнення жовчно-панкреатичного рефлюксу є наявність спільної кінцевої ділянки панкреатичної і загальної жовчної проток (спільна ампула). При цьому жовчні кислоти, що мають детергентну дію, при потраплянні в протоки підшлункової залози ушкоджують їхні клітини. З останніх виходять лізосомні ферменти, які і запускають процес активації панкреатичних ензимів. Крім того, жовчні кислоти можуть активувати ліпазу підшлункового соку, хоча є дані, що панкреатична ліпаза не потребує активації.
30.50. Опишіть патогенез місцевих змін у підшлунковій залозі при гострому панкреатиті.
Передчасна активація ферментів панкреатичного соку в протоках підшлункової залози викликає самоперетравлювання залозистої тканини. Так, під дією трипсину й хімотрипсину відбувається гідролітичне розщеплення тканинних білків і білкових компонентів клітин. Еластаза спричинює гідроліз еластину — складової частини базальних мембран судин. Фосфоліпаза, розщеплюючи фосфоліпіди клітинних мембран, обумовлює порушення їх бар'єрної функції, а ліпаза, гідролізуючи тригліцериди, викликає розвиток некрозу жирової тканини. Із фосфоліпідів мембран вивільняється арахідонова кислота, з якої утворюються простагландини. З крові в тканину залози надходить калікреїноген і а2-глобуліни. Під дією трипсину і хімотрипсину відбувається активація калікреїну і в кінцевому підсумку утворюються кініни.
Активні ферменти підшлункового соку, простагландини, кініни - усі ці фактори викликають вторинну альтерацію тканини підшлункової залози, підвищення проникності судин з розвитком набряку, геморагій; виникнення болю. Розвивається гостре запалення, що має ряд відмітних ознак. По-перше, воно є генеролізованим, тобто охо-
плює всю тканину залози; по-друге, надто сильною є альтерація (іноді розвивається повний некроз залози), по-третє, дуже характерні судинні зміни і пов'язана з ними ексудація (набряк, геморагії).
ЗО. 51. Які механізми лежать в основі розвитку панкреатичного шоку?
Панкреатичний шок є важким загальним проявом гострого панкреатиту і характеризується порушеннями загальної гемодинаміки (падінням артеріального тиску) та генералізованими розладами мікроциркуляції.
У патогенезі панкреатичного шоку мають значення два механізми.
I. Больовий механізм. Причиною різкого гострого оперізуючого болю, що виникає при панкреатиті, з одного боку, є набряк підшлункової залози (тиск на сонячне сплетіння), а з другого - дія активних травних ферментів (трипсин, фосфоліпаза та ін.) та біологічно активних речовин (кініни, простагландини) на нервові закінчення залози. Інтенсивний біль викликає спочатку збудження, а потім позамежне гальмування в життєво важливих центрах головного мозку. Це веде до пригнічення зовнішнього дихання і порушення системного кровообігу — розвивається больовий шок (див. розд. 12).
II. Гуморальний механізм. Він обумовлений ферментемією — надходженням у кров активних панкреатичних ферментів. Ферменти, що потрапили в кров, інак-тивуються природними інгібіторами протеаз (aj-інгібітор протеаз, антитромбін III, а2-макроглобулін, а2-антиплазмін, інтер-а-інгібітор трипсину та ін.). Однак якщо потужність інгібіторів недостатня і всі ферменти, що надійшли, не можуть бути інактивовані, то наступною подією є активація біохімічних систем крові. її зумовлює головним чином трипсин. При цьому активуються калікреїн-кінінова система, системи зсідання крові і фібринолізу. Кініни, що утворюються, викликають генералізоване розширення судин, яке веде до зменшення загального периферичного опору, з другого ж боку вони підвищують проникність судин, унаслідок чого рідка частина крові переходить у тканини (плазморагії) і, отже, зменшується об'єм циркулюючої крові. Зазначені зміни гемодинаміки обумовлюють падіння артеріального тиску. Активація системи зсідання і фібринолітичної системи є причиною розвитку ДВЗ-синдрому і пов'язаних з ним розладів мікроциркуляції (див. розд. 26.3).
30.52. Якими синдромами виявляє себе панкреатичний шок?
1. Гостра артеріальна гіпотензія (колапс). Обумовлена розширенням судин (зменшується загальний периферичний опір) і падінням об'єму циркулюючої крові (зменшується хвилинний об'єм серця).
2. ДВЗ-синдром. Пов'язаний з активацією системи зсідання крові і фібринолітичної системи, а також з підвищенням проникності кровоносних судин.
3.- Гіпоксичний синдром. Обумовлений:
а) циркуляторною гіпоксією — порушеннями загальної гемодинаміки (падіння артеріального тиску) і мікроциркуляції (ДВЗ-синдром);
б) дихальною гіпоксією—пригніченням діяльності дихального центру (позамежне гальмування життєво важливих центрів при сильному больовому синдромі);
в) гемічною гіпоксією, що виникає в результаті гемолізу еритроцитів, обумовленого панкреатичною фосфоліпазою. 4. Інтоксикаційний синдром. Пов'язаний з надходженням у кров продуктів аутолі-тичного розпаду тканини підшлункової залози.
30.53. Які причини викликають розвиток панкреатичної гіпосекреції? Яке її значення?
Причинами зменшення утворення і секреції підшлункового соку (панкреатичної гіпосекреції) можуть бути:
а) нейрогенне гальмування зовнішньосекреторної функції підшлункової залози, (зменшення тонусу блукаючого нерва, отруєння атропіном та ін.);
б) дуоденіт - запалення слизової дванадцятипалої кишки, унаслідок чого зменшується утворення стимуляторів панкреатичної секреції - секретину і холецистокі-1 нін-панкреозиміну;
в) порушення виведення підшлункового соку (закупорка проток, їх здавлювання);
г) зменшення кількості секреторних клітин (руйнування, хронічні панкреатити);
ґ) спадково обумовлена недостатність ентерокінази, внаслідок чого порушується початкова активація протеолітичних ферментів підшлункового соку (трипсину, хімотрипсину). Головним наслідком панкреатичної гіпосекреції є порушення порожнинного
травлення в кишках — розвиток синдрому мальдигестії.
30.54. Що таке синдром мальдигестії? Чим він виявляє себе?
Синдромом мальдигестії позначають порушення порожнинного травлення, обумовлені недостатнім надходженням в кишки травних ферментів, зокрема при панкреатичній гіпосекреції.
Цей синдром виявляє себе:
1) порушенням перетравлювання жирів (відсутність ліпази, фосфоліпаз). Не засво- | юється 60-80 % жиру, він виводиться з калом— з'являється симптом стеаторег\ (жир у калі);
2) розладами всмоктування жиророзчинних вітамінів — розвиваються ознаки гіповітамінозів А, Е, К;
3) порушенням перетравлювання білків (відсутність травних протеаз). Не засвою- | ється до 30—40 % харчового білка. У калі з'являється велика кількість м 'язових j волокон;
4) порушенням перетравлювання вуглеводів (відсутність амілази);
5) порушенням розщеплення нуклеїнових кислот (відсутність нуклеаз).
30.55. Що таке кишкові дискінєзії? Які існують їхні варіанти?
Кишковими дискінезіями називають порушення рухової (моторної) функції кишок.
Виділяють два варіанти кишкових дискінезій: гіперкінетичний і гіпокінетичний. \ Для першого характерно посилення перистальтики кишок, ритмічної сегментації і маятникоподібних рухів, що виявляється розвитком проносів (діареї).
Другий, навпаки, характеризується послабленням рухової активності кишок, унаслідок чого виникають закрепи.
30.56. Назвіть причини і значення гіперкінетичних дискінезій кишок.
Причинами кишкових дискінезій гіперкінетичного типу можуть бути:
а) підвищення збудливості рецепторів кишок до адекватних подразників під час розвитку запалення слизової оболонки кишок (ентерити, коліти);
б) дія на рецептори кишок незвичайних, патологічних подразників — неперетравленої їжі (наприклад, при ахілії), продуктів гниття й бродіння, токсичних речовин і т. п.;
в) збільшення збудливості центрів блукаючого нерва;
г) збільшення утворення деяких гастроінтестинальних гормонів, що посилюють перистальтику кишок, наприклад, мотиліну.
Наслідками кишкових дискінезій гіперкінетичного типу є:
а) розлади травлення (перетравлювання, всмоктування);
б) зневоднення;
в) видільний негазовий ацидоз (втрата гідрокарбонатів).
30.57. Чим виявляють себе гіпокінетичні дискінезії кишок? Які причини їх розвитку і значення?
Кишкові дискінезії гіпокінетичного типу виявляються зменшенням перистальтики кишок, що призводить до виникнення закрепів. Залежно від механізмів розвитку виділяють два види закрепів: спастичні й атонічні.
Спастичні закрепи виникають у результаті стійкого тривалого тонічного скорочення гладкої мускулатури кишок (спазму) і можуть бути обумовлені вісцеро-вісце-ральними рефлексами або дією токсичних факторів (наприклад, отруєння свинцем).
Причиною розвитку атонічних закрепів, пов'язаних зі зменшенням скорочувальної функції гладких м'язів кишок, можуть бути:
а) бідне харчування, низький вміст клітковини в харчових продуктах;
б) надмірне перетравлювання їжі в шлунку (наприклад, при шлунковій гіперсекреції);
в) вікові зміни рецепторного апарату кишок в осіб старечого віку, а також структурні зміни кишкової стінки при ожирінні;
г) зменшення тонусу блукаючого нерва;
ґ) порушення внутрішньокишкової іннервації, наприклад, при хворобі Гіршпрун-га- відсутність гангліонарних клітин ауербахового сплетіння в сигмоподібній і прямій кишках. Кишкові дискінезії гіпокінетичного типу призводять до:
1) розвитку кишкової аутоінтоксикації;
2) виникнення метеоризму;
3) утворення калових каменів;
4) у крайніх випадках може розвиватися кишкова непрохідність.
30.58. Що таке кишкова непрохідність? Як її класифікують?
Непрохідність кишок (ileus) - це захворювання, що характеризується порушенням проходження хімусу по кишках. Причиною цього можуть бути або обтурація, або здавлювання кишок, або порушення їхніх функцій.
Кишкову непрохідність класифікують у такий спосіб.
I. Механічна:
а) обтураційна. Розвивається внаслідок закупорки просвіту кишки пухлиною, каловими каменями, клубком гельмінтів;
в) странгуляційна. Є результатом здавлювання кишки ззовні (заворот, защемлення в грижових воротах, утворення вузлів). Особливістю цього виду непрохідності є здавлювання судин брижі, що порушує живлення стінки кишки аж до некрозу.
II. Динамічна:
а) спастична. Обумовлена спастичним скороченням гладких м'язів кишок;
б) паралітична. Розвивається внаслідок глибокого пригнічення рухової функції кишок.
ЗО. 59. Якими змінами в організмі виявляє себе непрохідність кишок? Який їх патогенез?
I. Больовий синдром. Розвивається внаслідок спастичного скорочення гладких м'язів, некрозу стінки кишки, розтягнення її рідиною. У результаті тривалого інтенсивного болю можуть з'являтися ознаки больового шоку (падіння артеріального тиску, розлади зовнішнього дихання), обумовлені позамежним гальмуванням судинорухового і дихального центрів після їх перезбудження.
II. Зневоднення. В умовах кишкової непрохідності відбувається накопичення великої кількості рідини в кишках вище місця перешкоди. Причиною цього є:
а) збільшення секреції травних залоз у відповідь на розтягування стінки кишки рідиною і газами;
б) перехід рідини з кровоносних судин у кишки (транссудація) внаслідок застою крові і збільшення проникності стінок судин;
в) порушення процесів всмоктування води.
Накопичення рідини в кишках призводить до збільшення внутрішньокишково-jo тиску, що обумовлює подразнення великої кількості рецепторів і виникнення блювоти. Що вище перешкода в кишках, то інтенсивніша блювота і більш виражене зневоднення. Ш. Порушення обороту травних ферментів. Збільшення внутрішньокишкового тиску призводить до закидання вмісту кишок (у тому числі і травних соків, ен-терокінази) у протоки підшлункової залози. Відбувається передчасна активація ферментів підшлункового соку, унаслідок чого розвивається гострий панкреатит з явищами панкреатичного шоку (див. запит. 30.49 і 30.54).
IV. Порушення кислотно-основного стану. Якщо при нестримній блювоті втрачаються в більшій мірі хлориди — розвивається негазовий алкалоз, якщо гідрокарбонати - негазовий ацидоз (див. розд. 25).
V. Кишкова аутоінтоксикація. Особливо виражена при низькій кишковій непрохідності. Виявляється ознаками недостатності печінки (див. розд. 31).
VI. Гострий перитоніт (запалення очеревини). Обумовлений мікробами, які проникають через некротизовану стінку кишки в порожнину очеревини. Перитоніт є чинником, що викликає і посилює біль та больовий шок, а також, зумовлю-
ючи рефлекторну блювоту, сприяє розвитку зневоднення, порушень кислотно-основного стану і розладів системної гемодинаміки.
VII. Розлади загального кровообігу і мікроциркуляції. Порушення системної гемодинаміки (падіння артеріального тиску, зменшення хвилинного об'єму серця, зменшення загального периферичного опору) є проявом больового і панкреатичного шоку, а також зневоднення. Згущення крові (гемоконцентрація) призводить до розладів мікроциркуляції.
VIII. Гіпоксія. Обумовлена розладами кровообігу і зовнішнього дихання.
IX. Порушення функції життєво важливих органів (нирок, серця, головного мозку). Зумовлені розвитком гіпоксії та інтоксикації.
30.60. Які причини викликають порушення дефекації? Чим можуть виявлятися такі розлади?
Причинами порушень випорожнення кишок (дефекації) можуть бути: . а) випадання впливу кори головного мозку на спинномозковий центр дефекації (переляк, страх);
б) ушкодження центру дефекації в попереково-крижовому відділі спинного мозку;
в) ураження периферичних нервів: ші. pelvici, hypogastrici;
г) розлади функції м'язів, що беруть участь у дефекації.
Порушення випорожнення кишок можуть виявлятися:
1) мимовільною дефекацією;
2) відсутністю позивів на дефекацію або, навпаки, частими помилковими позивами;
3) нетриманням калових мас.
30.61. Що таке синдром мальабсорбції? Чим можуть бути обумовлені порушення всмоктування?
Синдромом мальабсорбції називають симптомокомплекс, що виникає в результаті порушень усмоктування речовин у кишках.
Порушення всмоктування в кишках можуть бути обумовлені розладами, що виникають на трьох рівнях. Тому виділяють:
1) передентероцитарні порушення. Розвиваються внаслідок розладів процесів травлення, що передують усмоктуванню; ; 2) ентероцитарні. Виникають у результаті порушення діяльності епітеліальних клітин слизової кишок (ентероцитів); 3) постентероцитарні. Є наслідком порушення процесів, які забезпечують надходження речовин, що всмокталися, у внутрішнє середовище організму (кров, лімфу).
30.62. Які передентероцитарні порушення можуть лежати в основі розладів усмоктування в кишках?
1. Порушення рухової функції травного каналу.
2. Порушення порожнинного травлення (синдром мальдигестії). За походженням вони можуть бути гастрогенними, панкреатогенними, гепатогенними, ентероген-ними, дисрегуляторними, ятрогенними (пов'язаними з тривалим застосуванням антибіотиків та інших лікарських препаратів).
З. Порушення пристінкового травлення. Найчастіше зумовлені розладами утворення і вмонтовування ферментів у плазматичну мембрану мікроворсинок ентероцитів.
30.63. Що таке інтестинальні ферментопатії? Чим вони можуть виявлятися?
Інтестинальні ферментопатії — це спадково обумовлені порушення синтезу травних ферментів мікроворсинок, що забезпечують процеси пристінкового (мембранного) травлення.
Серед інтестинальних ферментопатій найчастіше бувають непереносність дисахаридів (лактози, сахарози, трегалози) і недостатність пептидаз (глютенова хвороба).
Непереносність дисахаридів обумовлена недостатністю дисахаридаз. У деяких регіонах Африки, Азії і Південної Америки 90 % населення не переносить молока і його не споживає. У представників зазначених етнічних груп нема ферменту лактази, що розщеплює лактозу молока. Через це нерозщеплена лактоза, накопичуючись на поверхні слизової, подразнює рецептори кишок і викликає профузні проноси.
Глютенову хворобу зумовлює недостатність пептидаз, вона характеризується не-переносністю продуктів, виготовлених зі злаків. Поширена в Європі, зокрема в Голландії. Глютен — білкова частина клейковини деяких злаків (пшениці, рису, ячменю, вівса) - складається з нешкідливого глютеїну і гліадину, який токсично діє на слизову тонкої кишки (викликає ентерит, проноси). При недостатності пептидаз гліадин не розщеплюється і чинить свою токсичну дію.
ЗО. 64. Які ентероцитарні порушення можуть обумовлювати синдром мальабсорбції?
Причинами мальабсорбції можуть бути такі ентероцитарні порушення:
1) зменшення площі всмоктування (стан після резекції кишки, атрофія ворсинок і мікроворсинок);
2) спадково обумовлені і набуті порушення утворення білків — переносників моносахаридів (непереносність глюкози, галактози, фруктози), амінокислот (триптофан-мальабсорбція), іонів кальцію (гіповітаміноз D);
3) порушення функціонування іонних насосів ентероцитів (транспорт моносахаридів і амінокислот пов'язаний з роботою Na-K-насосів);
4) дефіцит енергії (всмоктування більшості речовин — процес енергозалежний);
5) порушення формування в ентероцитах комплексів, що транспортуються (хіломі-кронів, ліпопротеїдів).
30.65. Які постентероцитарні розлади викликають порушення всмоктування речовин у кишках?
Причинами мальабсорбції можуть бути такі постентероцитарні порушення: 1) порушення кровообігу в стінках кишок. Можуть бути зумовлені розладами загальної гемодинаміки в системі ворітної вени і місцевими порушеннями (ішемія, венозна гіперемія, тромбоз, емболія, реакції судин при запаленні);
2) порушення лімфовідтоку. Крім загальних розладів лімфообігу, вони можуть бути пов'язані з порушеннями скорочення ворсинок кишкової стінки. Таке скорочення в нормі здійснюється завдяки місцевим рефлексам за участю підслизового нервового сплетіння (сплетіння Месснера) і речовини віллікініну, що її утворює слизова оболонка тонкої кишки.
31. Патологічна фізіологія печінки
31.1. Які фактори можуть бути причиною уражень печінки?
Ураження печінки можуть викликати:
1) фізичні фактори - іонізуюче випромінювання, механічна травма;
2) хімічні агенти, що мають токсичну (гепатотропну) дію. Вони можуть бути як екзогенного походження (алкоголь, промислові отрути - чотирихлористий вуглець, фосфорорганічні сполуки, хлороформ, миш'як; лікарські препарати - ПАСК-на-трій, сульфаніламіди, цитостатики, деякі антибіотики; рослинні отрути - афлаток-син, мускарин, алкалоїди геліотропа), так і ендогенного (продукти розпаду тканин при опіках, некрозах; токсикоз вагітних);
3) інфекційні агенти- віруси (вірусного гепатиту, інфекційного мононуклеозу), збудники туберкульозу, сифілісу, найпростіші (лямблії, амеби), гриби (актиноміцети), гельмінти (ехінокок, аскариди);
4) аліментарні фактори - білкове, вітамінне голодування, дуже жирна їжа;
5) алергічні реакції'на введення вакцин, сироваток, харчових продуктів і лікарських препаратів;
6) порушення кровообігу в печінці місцевого (ішемія, венозна гіперемія, тромбоз, емболія) і загального (при недостатності кровообігу) характеру;
7) ендокринні і обмінні порушення в організмі (цукровий діабет, гіпертиреоз, ожиріння);
8) пухлини (гепатоцелюлярний рак) та їхні метастази в печінку (рак шлунку, легень, молочної залози, лейкозні проліферати);
9) генетичні дефекти обміну речовин (спадкові ферментопатії), уроджені вади розвитку печінки.
31.2. Що таке недостатність печінки? Як її класифікують?
Недостатність печінки— цс патологічний стан, при якому діяльність цього органа не здатна забезпечити сталість внутрішнього середовища організму відповідно до його вимог.
Класифікують її в такий спосіб. І. Недостатність печінки може бути відносною і абсолютною.
Відносна виникає при первинному підвищенні навантаження на печінку, коли вимоги організму щодо підтримання гомеостазу перевищують функціональні можливості печінки.
Абсолютна недостатність розвивається при первинному ураженні печінки, внаслідок чого зменшуються її функціональні можливості і вона не здатна забезпечувати сталість внутрішнього середовища навіть у звичайних умовах. Відносна недостатність печінки згодом може переходити в абсолютну. Послідовність подій у цьому випадку така: підвищення навантаження на печінку → від-
носна недостатність печінки → порушення сталості внутрішнього середовища → вторинне ураження печінки → абсолютна печінкова недостатність.
II. Залежно від причин ушкодження гепатоцитів абсолютна недостатність печінки може бути:
а) печінково-клітинною',
б) холестатичною;
в) печінково-судинною.
III. Залежно від кількості функцій, які порушуються при ураженнях печінки, недостатність цього органа може бути тотальною (порушуються всі види функцій печінки) і парціальною (страждає одна або кілька функцій).
IV. За клінічним перебігом недостатність печінки може бути гострою і хронічною.
31.3. У яких випадках розвивається печінково-клітинна недостатність печінки? Які її причини? Як її моделюють в експерименті?
Печінково-клітинна недостатність розвивається внаслідок прямого ушкодження гепатоцитів патогенними агентами.
Причинами її виникнення можуть бути фізичні фактори (іонізуюча радіація), гепатотропні отрути (чотирихлористий вуглець, токсини блідої поганки, алкалоїди геліотропа), віруси інфекційного і сироваткового гепатиту. Останні викликають ураження печінки внаслідок прямої цитотоксичної дії (розмножуються в гепатоцитах) і утворення аутоантитіл на змінені вірусом власні білки печінкових клітин (аутоімун-ний механізм ушкодження).
В експерименті на тваринах печінково-клітинну недостатність моделюють повним або частковим видаленням печінки, а також за допомогою отрут, що їх вводять в організм (чотирихлористого вуглецю, хлороформу, тринітротолуолу та ін.).
31.4. Що є причиною розвитку холестатичноїнедостатності печінки? Як її відтворюють в експерименті?
Холестшпична недостатність розвивається внаслідок первинних розладів жовчоутворення і жовчовиділення. Найчастішою причиною її розвитку є механічна жовтяниця (див. запит. 31.25).
Ушкодження гепатоцитів в умовах тривалого холестазу (порушення виведення жовчі) обумовлено принаймні двома причинами:
а) механічною дією жовчі на печінкові клітини (здавлення, розрив мембран);
б) роз'єднанням окиснення і фосфорування (дія білірубіну і жовчних кислот на мі-тохондрії), у результаті чого виникає дефіцит АТФ, а потім і дегенеративні зміни в гепатоцитах.
В експерименті холестатичну недостатність моделюють перев'язуванням жовчовивідних проток.
31.5. У яких випадках розвивається печінково-судинна недостатність печінки? Як її можна моделювати в експерименті?
Печінково-судинна недостатність розвивається в результаті первинних порушень кровообігу в печінці. При цьому основним механізмом ушкодження гепатоцитів є гіпоксія.
Найчастішими причинами печінково-судинної недостатності є портальна гіпер- \ тензія та ішемія печінки.
В експерименті розлади кровообігу в печінці моделюють за допомогою таких методів:
а) накладання порто-кавальних анастомозів (фістула Екка, фістула Екка-Павлова);
б) перев'язування ворітної вени;
в) перев'язування печінкових вен;
г) накладання лігатури на печінкову артерію.
31.6. Що таке цироз печінки? Які його патогенетичні варіанти виділяють?
Цироз печінки - це хронічне прогресуюче захворювання, що характеризується розростанням сполучної тканини, патологічною регенерацією тканини печінки і перебудовою структури органа. Ця хвороба виявляє себе ознаками печінкової недостатності. Цироз є наслідком необоротного ушкодження великої кількості печінкових клітин.
Залежно від причин такого ушкодження розрізняють три патогенетичні варіанти цирозу печінки:
а) постнекротичний. Виявляється ознаками печінково-клітинної недостатності печінки;
б) біліарний. Супроводжується холестатичною недостатністю печінки;
в) портальний. Є структурною основою печінково-судинної недостатності печінки.
31.7. Порушеннями яких функцій печінки може виявляти себе її недостатність ?
При недостатності печінки можуть порушуватися такі її функції.
I. Метаболічні — участь печінки у вуглеводному, жировому, білковому обміні, обміні вітамінів, гормонів і біологічно активних речовин.
II. Захисні — фагоцитарна і антитоксична функції печінки.
III. Екскреторні — утворення і виділення жовчі. Секреція жовчі обумовлює видільну і травну функції печінки.
IV. Гемодинамічні - участь печінки у підтриманні системного кровообігу.
31.8. Які порушення вуглеводного обміну можуть розвиватися при ураженнях печінки?
Участь печінки у вуглеводному обміні полягає головним чином у підтримуванні сталості концентрації глюкози в плазмі крові. Це досягається завдяки тому, що в печінці відбувається депонування глюкози у вигляді глікогену (на глікоген припадає близько 20 % маси печінки).
Можливі два принципово різні стани, при яких порушується підтримування печінкою сталості концентрації глюкози крові. І. Зменшення вмісту глікогену в печінці. До цього можуть спричинятися: а) ненадходження глюкози з кишок у печінку (голодування);
б) порушення перетворення харчових моносахаридів (фруктози, галактози) у глюкозу, що характерно для спадково обумовлених захворювань — фрукто-земії і галактоземії;
в) порушення глюконеогенезу (наприклад, при гіпофункції кори надниркових залоз);
г) порушення синтезу глікогену з глюкози, обумовлене зменшенням активності ферментів глікогенезу (наприклад, спадково обумовлене захворювання — аглікогеноз);
ґ) дефіцит АТФ, необхідного для транспорту глюкози в гепатоцити і реакцій біосинтезу глікогену. її. Порушення процесів вивільнення глюкози з глікогену і надходження її в кров. Такі порушення лежать в основі спадкових захворювань, що одержали назву глікогенозів.
Оскільки утворення глюкози з глікогену в гепатоцитах може відбуватися двома шляхами (фосфоролітичним і гідролітичним), то можливі дві групи глікогенозів.
1. Глікогенози, при яких порушується фосфоролітичне розщеплення глікогену. До них, зокрема, відносять дефіцит кінази фосфорилази, фосфорилази (хвороба Гер-ша), глюкозо-6-фосфатази (хвороба Гірке).
2. Глікогенози з розладами гідролітичного розщеплення глікогену в лізосомах гепа-тоцитів. У цю групу, зокрема, входять хвороба Помпе, хвороба Форбса-Корі, хвороба Андерсена.
Морфологічно глікогенози виявляються значним збільшенням вмісту глікогену в гепатоцитах. Однак цей глікоген не може бути джерелом глюкози крові.
Основним проявом порушень вуглеводної функції печінки є розвиток печінкової гіпоглікемії, що у важких випадках може призводити до гіпоглікемічної коми (див. розд. 20).
31.9. Які розлади жирового обміну можуть виникати при ураженнях печінки?
1. Порушення перетравлювання і всмоктування жирів у тонкій кишці при патології жовчоутворення і жовчовиділення (див. запит. 31.29);
2. Порушення синтезу тригліцеридів, фосфоліпідів, холестеролу, а також утворення ліпопротеїдів плазми крові (ЛПДНГ і ЛПВГ);
3. Надмірне утворення кетонових тіл (див. розд. 21);
4. Жирова дистрофія (жирова інфільтрація, жировий гепатоз) печінки.
31.10. Що таке жирова дистрофія печінки? Який її патогенез?
Жирова дистрофія печінки — це збільшення у кілька разів проти норми вмісту тригліцеридів у печінкових клітинах, унаслідок чого розвивається дифузне або осередкове ожиріння печінки.
Найчастішими причинами жирової дистрофії печінки є ожиріння, цукровий діабет, алкоголь, хронічні інфекції та інтоксикації, гепатотропні отрути, аліментарні фактори (білкове голодування, дефіцит у їжі так званих ліпотропних речовин — холіну, метіоніну).
У патогенезі жирової дистрофії печінки виділяють два механізми (рис. 144).
Рис. 144. Механізми розвитку жирової дистрофії печінки
I. Надмірне утворення тригліперидів з жирових кислот. Це може бути зумовлено:
а) збільшеним надходженням жирових кислот у печінку (гіперліпацидемія);
б) порушенням окиснення жирових кислот у гепатоцитах (гіпоксія, дефіцит ] коферментів, переважне використання для енергетичних потреб інших сполук, зокрема алкоголю).
II. Недостатнє виведення тригліцеридів з печінки в кров у складі ЛХІДНГ (ліпо- І протеїдів дуже низької густини). Причинами цього можуть бути:
а) порушення синтезу основних компонентів ліпопротеїдних часток (білкової частини — апопротеїну, фосфоліпідів);
б) порушення формування міцел ліпопротеїдів;
в) розлади процесу секреції ліпопротеїдів.
31.11. Які розлади білкового обміну можуть виникати при ураженнях печінки?
Ураження печінки можуть виявляти себе такими розладами білкового обміну:
1) порушеннями біосинтезу білків, у тому числі і білків плазми крові (розлади біло-ксинтетичної функції печінки);
2) порушеннями перетворення амінокислот (дезамінування, трансамінування, де-карбоксилювання);
3) порушенням утворення сечовини (див. розд. 22).
31.12. Що може бути причиною порушень білоксинтетичної функції печінки? Чим виявляють себе такі порушення?
Білоксинтетична функція печінки порушується при: 1) змінах структури генів, що несуть інформацію про будову відповідних білків (наприклад, афібриногенемія);
2) порушеннях процесів транскрипції, тобто синтезу інформаційної РНК на матриці ДНК (наприклад, дія токсинів блідої поганки, антибіотика актиноміцину D);
3) розладах процесів транскрипції, тобто зчитування інформації з інформаційної РНК у рибосомах (наприклад, дія антибіотика пуроміцину);
4) зменшенні кількості амінокислот, необхідних для біосинтезу білків (наприклад, білково-енергетична недостатність, дефіцит у їжі незамінних амінокислот);
5) дефіциті АТФ, енергія якого використовується в біосинтетичних процесах (гіпоксія, голодування, гіповітамінози В] В2, РР, дія інгібіторів ферментів, роз'єднання окиснення й фосфорування).
У печінці синтезується переважна більшість білків плазми крові: всі альбуміни, 75-90 % а-глобуліців, близько 50 % р-глобулінів. Тому найяскравіші прояви порушень білоксинтетичної функції печінки пов'язані з розладами утворення саме білків плазми крові. Такими проявами є гіпопротеїнемічний і геморагічний синдроми.
Гіпопротеїнемічний синдром виникає в результаті зменшення концентрації альбумінів у плазмі крові. Це призводить до зниження онкотичного тиску плазми, наслідком чого може бути розвиток набряків.
Геморагічний синдром (підвищена кровоточивість) є наслідком порушень синтезу білків - факторів зсідання крові (фібриногену, протромбіну, проконвертину, про-акцелерину).
31.13. Які порушення обміну вітамінів можуть розвиватися при ураженнях печінки?
При ураженнях печінки можуть розвиватися такі розлади обміну вітамінів:
1) порушення утворення біологічно активних форм вітамінів з вітамінів-попередни-ків (наприклад, тіамінпірофосфату з вітаміну В,);
2) порушення депонування вітамінів у печінці (наприклад, вітаміну В12);
3) розлади всмоктування жиророзчинних вітамінів у результаті порушень функції утворення жовчі в печінці.
Клінічно всі зазначені розлади виявляють себе розвитком ознак відповідних гіповітамінозів.
31.14. Як порушується обмін мікроелементів при ураженнях печінки?
При захворюваннях печінки порушуються: а) депонування заліза (у формі феритину), міді, цинку, кобальту, молібдену, марганцю та ін.;
6) синтез транспортних білків, що забезпечують перенесення мікроелементів в організмі (трансферину, церулоплазміну);
в) екскреція мікроелементів з жовчю.
31.15. Які порушення обміну гормонів і біологічно активних речовин можуть супроводжувати розвиток недостатності печінки?
У печінці відбувається інактивація багатьох гормонів і біологічно активних речовин.
Інактивації зазнають усі стероїдні гормони (глюкокортикоїди, мінералокортико-їди, жіночі й чоловічі статеві гормони), тиреоїдні гормони, інсулін. При порушенні функції гепатоцитів зменшується інтенсивність перетворення зазначених гормонів, тому їхня концентрація в крові зростає і з'являються ознаки гіперфункції відповідних ендокринних залоз (вторинного гіперальдостеронізму, гіпертиреозу та ін.; див. розд. 33). Подібні ознаки можуть бути пов'язані і з порушенням синтезу в печінці транспортних білків (наприклад, транскортину), що зв'язують вільні гормони.
Печінка є органом, де відбувається руйнування багатьох біологічно активних речовин, і зокрема — біогенних амінів: катехоламінів, гістаміну, серотоніну. У зв'язку з цим у разі порушення функції гепатоцитів концентрація зазначених біологічно активних речовин у крові може зростати. У результаті розвиваються ознаки активації симпатоадреналової системи (підвищення артеріального тиску, тахікардія та ін.), підвищеного утворення гістаміну (свербіж шкіри, розвиток виразок у травному каналі).
31.16. Чим можуть виявляти себе порушення захисної (бар'єрної) функції печінки?
Захисна функція печінки складається з фагоцитарної й антитоксичної функцій.
Фагоцитарну функцію здійснюють особливі зірчасті ендотеліальні клітини (клітини Купфера), що належать до системи мононуклеарних фагоцитів. Можливі розлади фагоцитарної функції цих клітин описано в розд. 8.
Антитоксична функція властива гепатоцитам. Вона полягає в інактивації кінцевих продуктів обміну речовин (сечовиноутворення) і екзогенних речовин, що надходять з кишок (фенол, крезол, індол, скатол, аміни), а також з навколишнього середовища. Детоксикація різних речовин здійснюється за допомогою різних хімічних реакцій (синтезу, окиснення, відновлення, гідролізу, метилювання, кон'югації), що відбуваються в ендоплазматичному ретикулумі, мітохондріях, пероксисомах і цитоплазмі печінкових клітин.
Порушення антитоксичної функції печінки виявляються ознаками інтоксикації, що уражає насамперед центральну нервову систему. Тому комплекс змін, що виникають, одержав назву синдрому гепатоцеребральної недостатності.
31.17. Які фактори можуть викликати розвиток розладів антитоксичної функції печінки?
До порушень антитоксичної функції печінки можуть спричинятися:
1) зменшення кількості функціонуючих гепатоцитів (наприклад, при масивних не- ' крозах печінки). Так, установлено, що синтез сечовини з аміаку порушується при ураженні 80 % і більше паренхіми печінки;
2) зменшення активності ферментів, що беруть участь у реакціях детоксикації. Воно може бути зумовлено генетичними дефектами відповідних ферментів (наприклад, спадкове порушення ферментів синтезу сечовини) або набутими порушеннями їх утворення (розлади білоксинтетичної функції печінки);
3) зменшення концентрації речовин, що беруть участь в інактивації токсичних продуктів (кисню, глюкуронової і сірчаної кислот, гліцину, таурину, цистеїну та ін.);
4) дефіцит АТФ. При цьому можуть порушуватися процеси надходження й виведення з гепатоцитів токсичних речовин, а також реакції синтезу й кон'югації, які вимагають витрат енергії.
31.18. У чому сутність синдрому гепатоцеребральної недостатності? Чим він може виявлятися?
Синдром гепатоцеребральної недостатності виникає в результаті порушень антитоксичної функції печінки і виявляється комплексом психічних і невротичних розладів аж до втрати свідомості й розвитку коматозного стану.
Залежно від ступеня порушень антитоксичної функції печінки зазначений синдром може виявлятися:
а) емоційно-психічними розладами: емоційною нестійкістю (почергові зміни ейфорії й депресії), безсонням уночі й сонливістю вдень, головним болем, запамороченнями;
б) вираженими порушеннями свідомості - розвитком ступору (сонливість, сплутаність свідомості);
в) печінковою комою.
В основі розвитку синдрому гепатоцеребральної недостатності лежить накопичення в крові так званих церебротоксичнихречовин.
31.19. Які речовини, що накопичуються в крові при недостатності печінки, є церебротоксичними?
1. Аміак. В організмі існує два джерела надходження аміаку в кров: а) всі периферичні тканини, де відбуваються процеси дезамінування амінокислот; б) кишки, де утворення аміаку обумовлене процесами гниття, тобто діяльністю кишкової мікрофлори.
2. Продукти окиснення амінокислот у кишках. Такими є фенол, індол, скатол, аміни (путресцин, кадаверин); сполуки сірки, що утворюються при окисненні метіоніну (диметилсульфід, метилмеркаптан). З останніми, зокрема, пов'язують появу специфічного симптому печінкової коми — печінкового запаху.
3. Похідні молочної і піровиноградної кислот - ацетоїн, 2,3-бутиленгліколь. Причиною появи цих речовин є порушення реакцій циклу Кребса в гепатоцитах при їхньому ушкодженні.
4. Низькомолекулярні жирові кислоти: масляна, валеріанова, капронова. Вони з'являються внаслідок порушень окиснення жирових кислот в ушкоджених гепатоцитах.
31.20. Які виділяють патогенетичні варіанти печінкової коми?
Печінкова кома — це патологічний стан, що виникає в результаті важких порушень антитоксичної функції печінки і виявляється втратою свідомості, випадінням рефлексів на внутрішні і зовнішні подразники, розладами життєво важливих функцій організму (кровообігу, дихання).
Причиною розвитку печінкової коми є накопичення в крові церебротоксичних речовин (див. запит. 31.19).
Залежно від джерела й механізмів надходження в кров зазначених речовин виділяють два патогенетичних варіанти печінкової коми.
І. Ендогенна (печінково-клітинна, або розпадна) печінкова кома. У цьому випадку поява церебротоксичних речовин у крові пов'язана з порушеннями антитоксичної функції печінки при ушкодженні і загибелі гепатоцитів. її. Екзогенна (портокавальна, або шунтова) печінкова кома. її розвиток обумов- 1 лений тим, що церебротоксичні речовини потрапляють у системний кровообіг з кишок через ворітну вену і портокавальні анастомози, минаючи печінку. При цьому антитоксична функція печінки може істотно не страждати.
. 31.21. Який патогенез печінкової коми?
Центральне місце в патогенезі печінкової коми належить надходженню в кров церебротоксичних речовин. Про це, зокрема, свідчить той факт, що у хворих у стані | печінкової коми в багато разів збільшується вміст зазначених сполук у крові. Крім J того, в експерименті показано, що введення тваринам церебротоксичних речовин су- ] проводжується розвитком ознак печінкової коми.
Механізми дії церебротоксичних сполук на центральну нервову систему досить І складні. Велике значення в патогенезі печінкової коми можуть мати такі зміни.
1. Порушення синаптіїчної передачі. Цілий ряд церебротоксичних речовин (низь-комолекулярні жирові кислоти, тирамін та ін.) є несправжніми нейромедіатора-ми. Вони, накопичуючись в тканинах головного мозку, або заміщають нормальні медіатори нервової системи, або порушують їх утворення з попередників. Такі зміни призводять до порушень передавання нервових імпульсів і, як наслідок, | до порушень міжнейронних взаємодій та розладів інтегративних функцій цен- ] тральної нервової системи. Крім того, у тканинах головного мозку зростає вміст ГАМК - гальмівного медіатора центральної нервової системи. Збільшення концентрації цієї речовини обумовлене її утворенням у кишках під дією мікрофлори, а також порушеннями процесів інактивації в печінці.
2. Порушення енергетичного обміну, що призводять до дефіциту АТФ. Провідна роль у розвитку таких порушень належить аміаку (рис. 145). Накопичуючись у великих кількостях, аміак зв'язується з глютаміновою і а-кетоглютаровою кислотами, перетворюючи їх в кінцевому підсумку в глютамін. Оскільки а-кетоглю-тарова кислота є одним із центральних метаболітів циклу Кребса, зв'язування її з аміаком веде до порушення функціонування цього метаболічного шляху і, як наслідок, до порушення реакцій ресинтезу АТФ.
Зменшення вмісту АТФ у нервових клітинах призводить до розладів процесів активного транспорту катіонів, порушення генерації нервових імпульсів, змін величини мембранного потенціалу.
3. Порушення функції клітинних мембран у результаті прямої дії церебротоксичних речовин. Такий їхній вплив виявляється насамперед розладами роботи Na-К-насосів, унаслідок чого падає мембранний потенціал і стають неможливими ] генерація і проведення нервових імпульсів.
Рис. 145. Церебротоксична дія аміаку
4. Розвиток метаболічного ацидозу і пов'язаних з ним порушень обміну електролітів. Ацидоз при печінковій комі обумовлений церебротоксичними речовинами — кислотами (піровиноградною і молочною кислотою, амінокислотами та їхніми похідними, низькомолекулярними жировими кислотами).
31.22. Якими синдромами можуть виявляти себе порушення екскреторної функції печінки ?
Сутність екскреторної функції печінки полягає в утворенні і виділенні жовчі. Завдяки цьому здійснюється:
1) виведення з організму продуктів метаболізму (білірубіну) і надлишку деяких речовин (холестеролу);
2) участь печінки в процесах травлення (емульгування, перетравлювання і всмоктування жирів).
Порушення екскреторної функції печінки можуть виявляти себе такими синдромами: жовтяниця, холемічний синдром, ахолічний синдром.
31.23. Як у нормі відбувається обмін жовчних пігментів?.
Основний жовчний пігмент - білірубін — являє собою кінцевий продукт обміну гема. Головним джерелом білірубіну крові є гемоглобін еритроцитів.
Утворення білірубіну з гема відбувається в клітинах системи мононуклеарних фагоцитів (макрофагах селезінки, червоного кісткового мозку, клітинах Купфера в печінці), де фагоцитовані еритроцити зазнають гемолізу. Білірубін, що утворився,
нерозчинний у воді, тому його транспорт у печінку здійснюється у зв'язаному з білками вигляді. Такий білірубін одержав назву непрямого, оскільки дає реакцію з діа-зореактивом Ерліха тільки після попереднього осадження білків. Непрямий білірубін з крові надходить у печінку, де відбуваються три процеси, важливі з погляду обміну жовчних пігментів:
1) захоплення гепатоцитами зв 'язаного з білками (непрямого) білірубіну. Його забезпечують специфічні білкові рецептори плазматичної мембрани печінкових клітин;
2) кон'югація білірубіну з глюкуроновою кислотою, внаслідок чого утворюються глюкуроніди білірубіну (прямий білірубін);
3) екскреція прямого білірубіну у складі жовчі.
Усі зазначені процеси (захоплення, кон'югація і екскреція) вимагають витраті енергії АТФ.
Екскретований з жовчю білірубін у жовчному міхурі і тонкій кишці під дією ферментів мікрофлори перетворюється в уробіліноген. Основна кількість уробіліноге-ну в товстій кишці далі перетворюється в стеркобіліноген, що виводиться з калом і частково з сечею, всмоктуючись у кров у ділянці нижнього і середнього гемороїдальних сплетінь прямої кишки.
Друга, набагато менша частина уробіліногену бере участь у так званому печінково-кишковому кругообігу — всмоктується в тонкій кишці, потрапляє в печінку, частково зазнає окиснення, а частково знову надходить у жовчовивідні шляхи і у кишки.
31.24. Що таке жовтяниця? Які існують її види?
Жовтяниця (icterus) — це синдром, обумовлений збільшенням рівня білірубіну в крові, що виявляється жовтим забарвленням шкіри і слизових оболонок. Виділяють три види жовтяниці.
1. Гемолітична (надпечінкова) жовтяниця. Виникає в результаті гемолізу еритроцитів і збільшеного утворення білірубіну в клітинах системи мононуклеарних фагоцитів.
2. Паренхіматозна (печінкова) жовтяниця. її розвиток пов'язаний з ураженням печінки.
3. Механічна (рбтураційна, або підпечінкова) жовтяниця. Виникає в результаті порушення відтоку жовчі по жовчовивідних шляхах.
31.25. Який механізм розвитку гемолітичної жовтяниці? Які зміни пігментного обміну характерні для цього виду жовтяниці?
Гемолітична жовтяниця розвивається внаслідок гемолізу еритроцитів (див. розд. 26.1). Посилений фагоцитоз еритроцитів або самого гемоглобіну, що вивільнився зі зруйнованих еритроцитів, спричиняється до утворення у фагоцитах великої кількості білірубіну, який, зв'язуючись з білками, надходить у кров, а потім і в печінку. Гепатоцити при цьому зазнають підвищеного навантаження, перетворюючи великі кількості непрямого білірубіну в прямий і екскретуючи останній у складі жовчі. У цьому пояснення високого вмісту стеркобіліногену в калі (гіперхолічний кал) і в сечі. У зв'язку з тим, що непрямий білірубін не фільтрується в нирках (бо зв'язаний з білками), його нема в сечі.
З урахуванням цього основними ознаками порушення пігментного обміну при гемолітичній жовтяниці є:
а) збільшення вмісту непрямого білірубіну в крові;
б) збільшення вмісту стеркобіліногену в калі (гіперхолічний кал);
в) збільшення вмісту стеркобіліногену в сечі;
г) поява в сечі уробіліногену (у зв'язку з тим, що печінка не в змозі окислити великі кількості цієї речовини, яка надходить із кишок).
31.26. Які існують різновиди паренхіматозної (печінкової) жовтяниці? Як змінюється обмін жовчних пігментів при кожному з них?
В основі розвитку паренхіматозної (печінкової) жовтяниці лежать ізольовані або комбіновані порушення захоплення, кон'югації і екскреції білірубіну клітинами печінки.
Виділяють такі різновиди паренхіматозної жовтяниці:
1. Печінково-клітинна жовтяниця. Характеризується порушеннями всіх трьох процесів, що відбуваються в гепатоцитах: захоплення, кон'югації і екскреції білірубіну. Виникає при ушкодженні гепатоцитів (наприклад, вірусний гепатит), при дефіциті АТФ. При цьому внаслідок загибелі печінкових клітин утворюються сполучення між жовчними і кровоносними капілярами. У результаті жовч потрапляє в кров (холемія), а разом з нею і прямий білірубін. У клітинах, які не загинули, але ушкоджені, порушується захоплювання непрямого білірубіну й екскреція жовчі. Вона починає виділятися не тільки в жовчні капіляри, але й у кров. При цьому зменшується надходження жовчі в кишки (гіпохолія).
У зв'язку із зазначеними порушеннями виникають такі зміни показників пігментного обміну:
а) збільшення вмісту в крові непрямого білірубіну (порушується його захоплювання гепатоцитами);
б) збільшення вмісту в крові прямого білірубіну (результат надходження жовчі У кров);
в) зменшення вмісту стеркобіліногену в калі (гіпохолічний кал);
г) поява в сечі білірубіну;
ґ) зменшення або повна відсутність стеркобіліногену в сечі. Крім того, у крові і сечі виявляють жовчні кислоти (холалемія і холалурія).
2. Печінкові жовтяниці з ізольованими порушеннями процесів, що забезпечують виведення білірубіну з організму. Вони можуть бути обумовлені: ^
а) порушеннями захоплення непрямого білірубіну (синдром Жільбера — спадково обумовлений дефіцит рецепторів до білок-білірубінового комплексу). Виявляється збільшенням вмісту непрямого білірубіну в крові і відсутністю стеркобіліногену в калі і сечі;
б) розладами кон 'югації білірубіну (фізіологічна жовтяниця новонароджених, синдром Кріглера—Найяра). Вони найчастіше пов'язані з набутим або спадковим дефіцитом ферменту глюкуронілтрансферази. Виявляються збільшенням вмісту непрямого білірубіну в крові і зменшенням вмісту стеркобіліногену в калі і сечі;
в) порушеннями екскреції білірубіну (синдром Дабіна-Джонсона, синдроїЛ Ротора). їх причиною є дефекти (найчастіше спадкові) систем транспорту білірубіну і жовчі з гепатоцитів у жовчні капіляри. Виявляють себе збільшенням вмісту прямого білірубіну в крові, появою в крові й сечі жовчних кислот, білірубінурією, зменшенням вмісту або повною відсутністю стерко-біліногену в калі і сечі.
31.27. Які причини і патогенез механічної жовтяниці? Дайте характеристику порушень пігментного обміну при цьому виді
жовтяниці.
Механічна (підпечінкова) жовтяниця розвивається в результаті механічної пе- І решкодй^відтоку жовчі. Це може бути:
1) здавлення жовчовивідних шляхів ззовні (пухлина головки підшлункової залози, дія рубця);
2) їх закупорка каменем, гельмінтами, густою жовчю.
Механічне перешкоджання відтоку жовчі призводить до застою й підвищення тиску жовчі, розширення й розриву жовчних капілярів і надходження жовчі в кров як прямо, так і через лімфатичні шляхи.
У зв'язку з цим виникають такі зміни показників обміну жовчних пігментів:
а) збільшується вміст у крові прямого білірубіну (гіпербілірубінемія);
б) у крові з'являються жовчні кислоти (холалемія);
в) збільшується вміст у крові холестеролу (гіперхолестеролемія). З'являються моди- j фіковані ліпопротеїди (ліпопротеїд X), що мають атерогенні властивості;
г) у сечі з'являється білірубін (білірубінурія), унаслідок чого вона набуває темного забарвлення ("колір пива"), крім того в ній виявляють жовчні кислоти (холалурія). Із сечі зникає стеркобіліноген;
ґ) у калі нема стеркобіліногену (безбарвний кал).
Зазначені зміни пігментного обміну зумовлюють розвиток двох важливих клінічних синдромів, характерних для механічної жовтяниці: холемічного і ахолічного.
31.28. Що таке холємічний синдром? У яких випадках він виникає? Чим виявляє себе?
Холємічний синдром (синдром холестазу) обумовлений надходженням компонентів жовчі (жовчних кислот, прямого білірубіну, холестеролу) у кров у зв'язку з порушенням формування і відтоку жовчі.
Він закономірно виникає при механічній жовтяниці, а також деяких формах печінкової жовтяниці (печінково-клітинній, печінковій жовтяниці, обумовленій порушеннями екскреції жовчі).
Походження основних проявів синдрому: 1. Поява в крові жовчних кислот - холалемія. Цим зумовлені такі порушення:
а) розлади діяльності центральної нервової системи, що виникають як наслідок загальнотоксичної дії жовчних кислот (загальна астенія, дратівливість, що змінюється депресією; сонливість удень і безсоння вночі; головні болі, стомлюваність);
б) артеріальна гіпотензія, брадикардія. їхній розвиток обумовлений підвищенням тонусу блукаючого нерва і прямою дією жовчних кислот на синусно-передсердний вузол та кровоносні судини;
в) свербіж шкіри, що виникає в результаті подразнення нервових закінчень жовчними кислотами;
г) множинні ушкодження і загибель клітин, обумовлені детергентною дією жовчних кислот. Цим, зокрема, пояснюють гемоліз еритроцитів, запалення і некрози в різних органах і тканинах (печінковий некроз, перитоніт, гострий панкреатит та ін.);
ґ) поява жовчних кислот у сечі (холалурія).
2. Надходження у кров білірубіну. Ця обставина викликає появу жовтого забарвлення шкіри і слизових оболонок, тобто власне жовтяницю.
3. Збільшення вмісту в крові холестеролу. Це з невідомих поки що причин зумовлює появу аномального ліпопротеїду X, що має атерогенну дію.
31.29. Що таке ахолічний синдром? Чим він виявляється?
Ахолічним називають синдром, обумовлений ненадходженням жовчі в кишки у зв'язку з порушеннями її формування і відтоку. Для цього синдрому характерні:
1. Розлади перетравлювання і всмоктування жирів. Обумовлені порушенням процесів емульгування жирів, зменшенням активності панкреатичної ліпази, що активується жовчю; порушенням утворення міцел, що всмоктуються в тонкій кишці. Наслідком зазначених змін є:
а) поява жиру в калі — стеаторея;
б) розлади всмоктування жиророзчинних вітамінів, у результаті чого розвиваються гіповітамінози А, Е, К;
в) зменшення надходження в організм ненасичених жирових кислот, необхідних для побудови фосфоліпІдів клітинних мембран.
2. Порушення рухової функції кишок — ослаблення перистальтики і зменшення тонусу кишок (закрепи).
3. Посилення процесів гниття і реакцій бродіння в кишках у результаті зменшення бактерицидної дії жовчі. Це призводить до збільшення навантаження на антитоксичні системи печінки.
4. Зміни з боку калу - знебарвлення, стеаторея.
31.30. Що таке дисхолія? Які її причини? Який механізм виникнення жовчних каменів ?
Дисхолія — це порушення фізико-хімічних властивостей жовчі, внаслідок чого вона набуває літогенних властивостей, тобто здатності утворювати камені (конкременти) у жовчному міхурі й жовчних протоках. Результатом цього є розвиток жов-чноком 'яної хвороби.
Дисхолія і жовчні камені виникають унаслідок взаємодії багатьох чинників, серед яких: а) спадкова схильність;
б) нераціональне харчування;
в) порушення обміну речовин;
г) інфекційно-запальні процеси в жовчному міхурі і жовчних протоках; ґ) застій жовчі (холестаз).
Одним з основних механізмів виникнення літогенної жовчі є зниження холато-холестеролового і лецитин-холестеролового індексів (відношення жовчних кислот і лецитину до холестеролу жовчі). Це може бути зумовлено зменшенням печінково-кишкового кругообігу жовчних кислот при патології кишок і зміні їхньої мікрофлори, пригніченням синтезу жовчних кислот у печінці, прискоренням їх всмоктування слизовою оболонкою запаленого жовчного міхура, зменшенням вмісту лецитину і збільшенням синтезу холестеролу. При зменшенні концентрації жовчних кислот і лецитину, що забезпечують завислий стан холестеролу, останній випадає в осад і започатковує утворення холестеролових каменів.
Інфекція, застій жовчі також сприяють процесу утворення каменів, тому що супроводжуються зміною властивостей жовчі — зміщенням рН у кислий бік, зниженням розчинності солей, випаданням їх в осад, коагуляцією білків із клітин, що розпадаються. Крім холестеролових, утворюються пігментні (при гемолізі еритроцитів), вапняні і складні камені (наприклад, холестеролово-пігментно-вапняні). Камені обумовлюють порушення жовчовиділення і розвиток механічної жовтяниці.
31.31. Які причини, механізми розвитку і значення дискінезій жовчного міхура і жовчних проток?
Існує три варіанти порушень скоротливої функції жовчного міхура і жовчних проток (дискінезій).
1. Гіпертонічний (гіперкінетичний). Виявляється підвищенням тонусу гладких м'язів жовчного міхура і міхурової протоки, а також спазмом сфінктера Одді. Причиною його розвитку може бути підвищення тонусу блукаючого нерва або збільшення секреції холецистокінін-панкреозиміну.
2. Гіпотонічний (гіпокінетичний). Характеризується зменшенням тонусу жовчного міхура й міхурової протоки, сфінктер Одді постійно розслаблений. Виникає при зменшенні тонусу блукаючого нерва або при пригніченні утворення холецистокінін-панкреозиміну.
3. Змішаний варіант. Виявляє себе різноспрямованими змінами тонусу жовчного міхура й міхурової протоки, з одного боку, і сфінктера Одді —з другого. При цьому можливі два типи змін:
а) спазм сфінктера Одді і зменшення тонусу жовчного міхура і міхурової протоки;
б) розслаблення сфінктера Одді і підвищення тонусу жовчного міхура і міхурової протоки.
Порушення скоротливої функції жовчного міхура і жовчних проток спричиняється до розвитку больового синдрому, розладів жовчовиділення, а отже, і травлення.
31.32. Які функції печінки відносять до гемодинамічних? Чим виявляють себе розлади цих функцій?
Гемодинамічними називають функції печінки, які забезпечують її участь у здійсненні системного кровообігу. До них відносять:
а) колекторну функцію. Печінка збирає через систему ворітної вени кров з великого басейну- від органів черевної порожнини. Через печінку проходить 30-35 % хвилинного об'єму крові, що становить 1,5—1,8 л/хв;
б) депонування крові. У печінці може міститися до 700 мл крові, тимчасово виведеної з кровообігу. При необхідності (наприклад, після крововтрати) ця кров може бути мобілізована;
в) участь у підтриманні тонусу кровоносних судин через синтез білків, що є попередниками біологічно активних речовин — регуляторів артеріального тиску. Ідеться, зокрема, про синтез ангіотензиногену, з якого утворюється ангіотензин II.
Порушення гемодинамічних функцій печінки виявляють себе розвитком синдрому портальної гіпертензії.
31.33. Що таке синдром портальної гіпертензії? Які його причини? Чим він виявляється?
Синдром портальної гіпертензії розвивається в результаті порушення відтоку крові з органів черевної порожнини по судинах системи ворітної вени.
Залежно від того, де знаходиться перешкода відтоку крові, виділяють такі форми портальної гіпертензії:
1) підпечінкову — перешкода в стовбурі або великих гілках ворітної вени (емболи, здавлення пухлиною);
2) внутрішньопечінкову — перешкода в самій печінці (тривалий спазм гладком'язо-вих сфінктерів синусоїдів; здавлення дрібних печінкових вен вузлами регенеруючих гепатоцитів при ушкодженні печінки, її цирозі);
3) надпечінкову — перешкода локалізована у позаорганних відділах печінкових вен або в нижній порожнистій вені проксимальніше місця впадіння в неї печінкових вен. Сюди ж відносять портальну гіпертензію, що виникає при збільшенні тиску в системі нижньої порожнистої вени в умовах недостатності правого шлуночка серця.
Основні прояви синдрому портальної гіпертензії. 1. Здійснення колатерального кровообігу як результат розкриття портокавальних анастомозів. Це зумовлює розвиток таких ознак:
а) варикозне розширення вен стравоходу і кардіальної частини шлунку;
б) шлунково-кишкові кровотечі, причина яких ушкодження варикозно розширених вен;
в) розширення підшкірних вен передньої грудної і черевної стінки ("голова медузи");
г) скидання крові з ворітної вени в порожнисті в обхід печінки, що викликає інтоксикацію, а у важких випадках — розвиток екзогенної (портокавальної, або шунтової) печінкової коми.
2. Гепато-лієиальний синдром. Його важливими складовими є спленомегалія і пі перспленізм.
Спленомегалія — це збільшення розмірів селезінки. Вона виникає в результаті за-] стою крові.
Гіперспленізм — збільшення функціональної активності селезінки- виявляється посиленим руйнуванням формених елементів крові. Характеризується анемією, лейкопенією і тромбоцитопенією. В основі цього явища лежить збільшення фаго-І цитарної активності макрофагів селезінки в умовах уповільнення циркуляції кровя
3. Асцит (див. запит. 31.34).
4. Гепато-ренальний синдром. Виявляє себе порушеннями фільтраційної здатності ниркових клубочків при збереженні функцій канальцевого епітелію. Причиною цього, цілком імовірно, є зменшення тканинного кровообігу у зв'язку зі зменшен-І ням об'єму циркулюючої крові і зміною тонусу кровоносних судин, що спостері-1 гається при розладах гемодинамічних функцій печінки.
З1.34. Які механізми розвитку асциту?
Асцитом називають значне скупчення вільної рідини (як правило, транссудату) у черевній порожнині.
Причинами асциту можуть бути:
а) портальна гіпертензія різного походження;
б) набряки при хронічній недостатності серця, захворюваннях нирок, аліментарнім дистрофії;
в) порушення відтоку лімфи грудною протокою (її поранення, здавлення);
г) ураження очеревини пухлинним або туберкульозним процесом (асцит-перитоніт).
Асцитична рідина за своїм характером буває звичайно серозною, значно рідше -геморагічною.
У патогенезі асциту мають значення такі механізми:
1) гідростатичний. Пов'язаний з підвищенням тиску крові в капілярах судин ворітної системи;
2) онкотичний. Обумовлений зменшенням білоксинтетичної функції печінки, внаслідок чого розвивається гіпопротеїнемія й падає онкотичний тиск крові;
3) затримка натрію в організмі. Пов'язана зі збільшенням вмісту альдостерону в крові. Це у свою чергу обумовлено активацією ренін-ангіотензинної системи (застій крові в судинах ворітної системи → зменшення венозного повернення -* падіння хвилинного об'єму серця → гіпоксія нирок → вивільнення реніну). Крімj того, у зв'язку з розладами метаболічних функцій печінки може порушуватися! інактивація альдостерону, що рівнозначно його гіперпродукції;
4) лімфогенниймеханізм. У зв'язку з порушенням лімфовідтоку відбувається перехід багатої білками лімфи в черевну порожнину. Це викликає підвищення онкотичного тиску рідини черевної порожнини з наступним виходом у неї води із кровонос-і них судин та інтерстиціального простору.
31.35. Які порушення в системі крові можуть розвиватися при ураженнях печінки?
При ураженнях печінки часто розвиваються зміни, що торкаються як фізико-хі-мічних властивостей, так і клітинного складу крові.
У результаті порушень білоксинтетичної функції печінки розвивається гіпопро-теїнемія, знижується онкотичний тиск крові (гіпоонкія), зменшується співвідношення альбумінів і глобулінів (альбуміно-глобуліновий коефіцієнт), що виявляється збільшенням JJIOE.
Зміни клітинного складу крові виявляють себе анемією, лейкопенією і тромбоцитопенією.
Розвиток анемії може бути пов'язаний з різними патогенетичними механізмами: порушенням еритропоезу (зменшення депонування в печінці ціанокобаламіну, фолієвої кислоти, заліза), гемолізом еритроцитів (гіперспленізм, детергентна дія жовчних кислот при холемічному синдромі), крововтратою (геморагічний синдром).
Лейкопенія і тромбоцитопенія, так само як і анемія, можуть бути обумовлені дефіцитом деяких речовин, необхідних для кровотворення (ціанокобаламіну, фолієвої кислоти) і руйнуванням формених елементів крові макрофагами при гіперспленізмі.
Ураження печінки часто супроводжуються геморагічним діатезом — коагулопа-тіями. В основі їх розвитку лежать порушення синтезу в печінці протромбіну, факторів V, VII, IX, X, фібриногену; порушення всмоктування вітаміну К при гіпо- і ахолії. У разі тромбоцитопенії приєднуються розлади судинно-тромбоцитарного гемостазу.
320 Патологічна фізіологія нирок JT:
32.1. Якими порушеннями гомеостазу можуть виявляти себе ураження нирок?
Оскільки основною функцією нирок є гомеостатична, тобто підтримання сталості внутрішнього середовища, то при ураженні цих органів виникають насамперед порушення гомеостазу.
З розладами екскреторної функції нирок пов'язані:
1) порушення водного гомеостазу — зміни об'єму позаклітинної рідини (гіпер- і гіпо-гідрія);
2) порушення осмотичного гомеостазу - гіпер- і гіпоосмія;
3) порушення балансу електролітів у позаклітинній рідині (дисіонія);
4) порушення кислотно-основного стану (найчастіше негазовий ацидоз);
5) зміни хімічного складу плазми крові, що виявляють себе, з одного боку, накопиченням кінцевих продуктів метаболізму (азотемія), з другого — втратою необхід- і них організму хімічних сполук (гіпопротеїнемія, гіпоаміноцидемія, гіпоглікемія).
Розлади інкреторних функцій нирок можуть обумовлювати розвиток:
1) артеріальної гіпертензії;
2) анемії;
3) порушень фосфорно-кальцієвого обміну — ниркової остеодистрофії.
32.2. Які процеси в нирках можуть порушуватися за умов ураження цих органів?
У нирках відбувається дві групи процесів, що забезпечують підтримання гомеостазу, - сечоутворення (екскреторна функція) і вивільнення в кров гормонів, ферментів, біологічно активних сполук (інкреторні функції).
При ураженні нирок розлади їх екскреторної функції можуть бути обумовлені порушеннями:
1) клубочкоеої (гломерулярної) фільтрації;
2) канальцевої реабсорбції;
3) канальцевої секреції.
Розлади інкреторних функцій нирок можуть виявлятися порушеннями:
1) секреції реніну юкстагломерулярним апаратом нирок, а також ниркових депресорних факторів;
2) вивільнення еритропоетинів та інгібіторів еритропоезу;
3) утворення гормонально активної форми вітаміну D.
32.3. Що таке недостатність нирок? Як її класифікують?
Недостатність нирок - це патологічний стан, для якого характерно порушення сталості внутрішнього середовища організму внаслідок нездатності нирок здійснювати свої гомеостатичні функції.
Ниркову недостатність класифікують у такий спосіб.
I. За клінічним перебігом розрізняють гостру і хронічну ниркову недостатність.
II. Залежно від причин розвитку недостатність нирок може бути преренальною, ренальною, постренальною і аренальною.
III. Залежно від обсягу порушених функцій ниркова недостатність може бути тотальною (порушено всі функції) і парціальною (порушено лише окремі функції).
IV. За механізмами розвитку розрізняють недостатність нирок:
1) пов'язану з первинним ураженням клубочків - гломерулярну;
2) пов'язану з первинним ураженням канальців — тубулярну.
32.4. Які причини гострої ниркової недостатності? Які стадії виділяють у її розвитку?
Гостра ниркова недостатність характеризується швидким виникненням і зна& І чними порушеннями екскреторної функції нирок.
Етіологія гострої недостатності нирок (ГНН) пов'язана з дією внутрішньо- і позаниркових факторів.
Внутрішньониркові фактори ГНН. гострий гломерулонефрит, пієлонефрит, тромбоз і емболія ниркових судин, видалення однієї-єдиної нирки (аренальна ГНН).
Позаниркові фактори ГНН.
а) шок і колапс;
б) гемолітичні та міолітичні стани (переливання несумісної крові, масивне роздавлювання тканин, опіки);
в) зневоднення організму;
г) екзогенна й ендогенна інтоксикації (солями важких металів, оцтовою кислотою, хлороформом, грибною й зміїною отрутами, при токсикозі вагітних, діабетичній комі);
ґ) алергічні стани;
д) порушення виділення сечі внаслідок непрохідності сечоводів або сечівника.
Патогенез ГНН може бути пов'язаний з трьома групами чинників:
1) порушенням кровообігу в нирках (преренальні фактори). Найчастіше ГНН розвивається внаслідок тимчасової ішемії нирок, обумовленої гіповолемією, спазмом аферентних артеріол, ДВЗ-синдромом. Наслідком цього є значне зниження фільг траційного тиску й клубочкової фільтрації, припинення діяльності певної кількості нефронів. Якщо порушення тканинного кровообігу нетривале, то ГНН є оборотним станом (функціональна фаза ГНН). Затяжна ішемія викликає необоротні структурні зміни клубочків і канальців, що відповідає структурній фазі ГНН;
2) прямим ушкодженням структур клубочків і канальців (ренальні фактори). Цим механізмом зумовлюється розвиток ГНН при дії нефротоксичних отрут і деяких інфекційних агентів;
3) порушенням відтоку сечі (постренальні фактори). Ця обставина викликає змен-
Ішення клубочкової фільтрації аж до повного її припинення у зв'язку зі збільшенням тиску первинної сечі в капсулі ниркових клубочків. У клінічному перебігу ГНН виділяють чотири стадії: 1) початкову; 2) ояіго-, анурії; 3) поліурії; 4) видужання.
Найбільш характерні і значні порушення спостерігають на стадії оліго-, анурії:
а) різке зменшення (аж до повного припинення) діурезу з розвитком ознак водного отруєння організму - набряк головного мозку, штерстиціальний набряк легень та ін.;
б) тяжкі порушення діяльності системи кровообігу — зменшення скоротливої функції серця, порушення ритму у вигляді екстрасистолії, брадикардії, блокади; артеріальна гіпотензія з наступним переходом у гіпертензію;
в) розлади зовнішнього дихання за типом Куссмауля;
г) значні порушення функцій нервової системи - від головного болю, блювоти до арефлексії, порушень свідомості, судом, коми.
Більша частина хворих з ГНН гине на висоті цієї стадії. При сприятливому перебігу захворювання, а головне - при проведенні ефективних терапевтичних заходів, через 5—10 діб настає перехід у стадію відновлення діурезу й поліурії. Підвищення клубочкової фільтрації спочатку має в своїй основі відновлення цього процесу в не-фронах, що залишилися, а потім (через кілька місяців) збільшення кількості функціонуючих нефронів.
32.5. Які етіологія і патогенез хронічної недостатності нирок?
Етіологічними факторами хронічної недостатності нирок (ХНН) є хронічні прогресуючі захворювання нирок запальної (хронічний гломерулонефрит, хронічний пієлонефрит та ін.), судинної (гіпертонічна хвороба, стеноз ниркової артерії) та метаболічної (діабетичний гломерулосклероз, амілоїдоз, подагра) природи.
У патогенезі ХНН виділяють такі стадії: 1) початкову, 2) ранню поліуричну, 3) пізню олігуричну і 4) термінальну.
ХНН розвивається в результаті одночасного або поступового зменшення маси діючих нефронів (МДН).
Початкові ознаки ХНН з'являються при зменшенні МДН до 50-30 % від вихідної кількості нефронів, клінічно виражена картина розвивається при зниженні МДН до 30—10 % і величини клубочкової фільтрації нижче 20 % (якщо порівнювати з нормою). Дальше зменшення МДН і клубочкової фільтрації (нижче 10 %) веде до розвитку термінальної стадії недостатності нирок —уремії.
32.6. Що таке швидкість клубочкової фільтрації? Як її визначають? Як вона змінюється при різних видах ниркової недостатності?
Швидкість клубочкової фільтрації (ШКФ) - це об'єм плазми крові, що фільтрується в ниркові канальці за одиницю часу.
ШКФ визначають за кліренсом інуліну. Кліренс інуліну — це об'єм плазми, що повністю очищається від цієї речовини нирками за 1 хв.
![]()
де Сіп - кліренс інуліну; Uin - концентрація інуліну в сечі; Ріп - концентрація інуліну в плазмі; V - діурез за 1 хв.
У нормі Сіп, а отже, і ШКФ дорівнюють 100-140 мл/хв.
Зменшення ШКФ є основним показником розвитку недостатності нирок.
Так, при ГНН ШКФ швидко зменшується від 100-140 до 10-1 мл/хв.
Для початкової стадії ХНН характерне падіння ШКФ від 100-140 до 30 мл/хв. для ранньої поліуричної- від 30 до 10 мл/хв. для пізньої олігуричної - від 10 до 5 мл/хв. для термінальної - нижче 5 мл/хв.
32.7. Які механізми можуть лежати в основі порушень ниркових функцій?
1. Преренальні — порушення кровопостачання нирок (рис. 146).
2. Ренальнг — порушення функції клубочків (клубочкової фільтрації) і ниркових ка-нальців (канальцевої реабсорбції й секреції). у •'
3. Постренальні - порушення, що виникають на шляху відтоку сечі.
4. Аренальні - порушення, обумовлені відсутністю нирок.
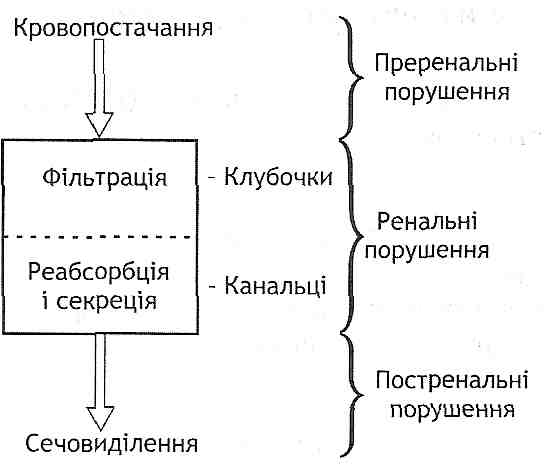
Рис. 146. Механізми порушень ниркових функцій
32.8. У чому-сутність преренальних порушень функцій нирок?
Преренальними називають порушення ниркових функцій, обумовлені розладами кровообігу в нирках.
Інтенсивність ниркового кровообігу в нормі дуже висока (близько 1300 мл/хв. або 25 % хвилинного об'єму крові в стані спокою), що обумовлено його специфічною функцією, тобто участю в здійсненні фільтрації і реабсорбції. Оскільки
![]()
де Qn, - об'ємна швидкість ниркового кровообігу; Ра - тиск на початку, а Рвеп -наприкінці системи перфузії ниркових судин; R - опір ниркових судин; п — в'язкість крові; 1 - довжина судин; г - радіус судин, то зменшення інтенсивності ниркового кровообігу може бути обумовлене:
1) зменшенням артеріального тиску (Р ) нижче 80 мм рт. ст. (не спрацьовує механізм міогенної ауторегуляції - механізм Бейліса). Це спостерігають при всіх видах [ шоку й колапсу;
2) збільшенням венозного тиску (РВСІІ). Причиною цього можуть бути загальні порушення (наприклад, правошлуночкова недостатність серця, що веде до збільшення центрального і периферичного венозного тиску) і місцеві розлади - венозна гіперемія (наприклад, при інтерстиціальному запаленні ниркової тканини);
3) ішемією нирок (зменшенням радіуса судин), що буває при атеросклерозі і артеріальній гіпертензії;
4) збільшенням в'язкості крові (наприклад, при ДВЗ-синдромі).
Усі зазначені порушення спричиняються до зменшення фільтраційного тиску в ниркових клубочках - це виявляє себе зменшенням швидкості клубочкової фільтрації, а отже, ознаками недостатності нирок.
32.9. Чим можуть бути обумовлені порушення клубочкової фільтрації?
Зменшення швидкості клубочкової фільтрації (ШКФ) є основним показником розвитку недостатності нирок. Оскільки
ШКФ = ЕФТ-К.
Ф
де ЕФТ - ефективний фільтраційний тиск; К - коефіцієнт фільтрації, то можна виділити дві групи механізмів порушення клубочкової фільтрації.
I. Зменшення ЕФТ. Оскільки
ЕФТ=Р -(Р + Р),
де Р — гідростатичний тиск у капілярах клубочків; Ро - онкотичний тиск крові; Р — гідростатичний тиск у капсулі клубочків — так званий тканинний тиск, то зменшення ШКФ може бути обумовлено:
1) зменшенням гідростатичного тиску в капілярах клубочків (Рк) унаслідок загальних і місцевих розладів кровообігу (див. запит. 32.8);
2) збільшенням онкотичного тиску крові (Р^, що буває, наприклад, при зневодненні;
3) збільшенням тканинного тиску в нирках (Рт). Причиною цього можуть бути перешкоди відтоку фільтрату або сечі при ушкодженні канальців (закупорка канальців некротичними масами і циліндрами), при інтерстиціальному запаленні (здавлення канальців набряковою рідиною), при порушеннях прохідності сечоводів і сечовивідних шляхів (камені, стриктури, здавлення пухлиною).
II. Зменшення коефіцієнта фільтрації (Кф). Воно може бути обумовлено:
1) зменшенням загальної площі фільтрації, що, у свою чергу, залежить від кількості діючих нефронів;
2) зменшенням проникності стінки клубочкового фільтра, що спостерігається при потовщенні мембрани (наприклад, при діабетичній нефропатії), скле-розуванні клубочків (наслідок гломерулонефриту), забиванні nop фільтра білками (гемоглобіном, міоглобіном відповідно при гемолізі еритроцитів і роздавлюванні м'язової тканини).
32.10. Які фактори можуть викликати збільшення клубочкової фільтрації?
Збільшення фільтрації відбувається під впливом таких чинників.
1. Підвищення гідростатичного тиску у капілярах клубочків, що буває:
а) у разі збільшення об'єму внутрішньосудинного сектора води у зв'язку з прийманням великої кількості рідини, з розсмоктуванням набряків, транссудатів і ексудатів;
б) при зменшенні тонусу привідних;
в) підвищенні тонусу вивідних артеріол.
2. Зниження онкотичного тиску крові, що відбувається у разі перерозподілу білкових фракцій крові у сторону переважання грубодисперсних глобулінів з низьким онкотичним тиском (при гепатиті, цирозі печінки).
32.11. Які механізми можуть лежати в основі порушень функцій ниркових канальців?
1. Ушкодження клітин канальцевого епітелію. Може бути обумовлено ішемією, не-фротропними отрутами, дією фізичних (радіація) і біологічних (інфекція) факторів. При ушкодженні канальців для порушення ниркових функцій мають значення:
а) вихід фільтрату через ушкоджені канальці в інтерстицій, що веде до збільшення тканинного тиску і зменшення клубочкової фільтрації;
б) обтурація канальців некротичними масами і циліндрами, що також зумовлює зменшення ефективного фільтраційного тиску.
2. Зменшення активності ферментів і транспортних білків, що беруть участь у процесах реабсорбції і секреції. Це можуть бути спадково обумовлені дефекти систем транспорту глюкози (ренальна глюкозурія), амінокислот (аміноацидурія), фосфатів (фосфатний нирковий діабет); складні поєднані порушення реабсорбції глюкози, амінокислот, гідрокарбонату і фосфатів (синдром Фанконі). Можливі й набуті розлади транспортних систем, що забезпечують реабсорбцію і секрецію. Наприклад, при отруєнні флоридзином, що пригнічує гексокіназу і глюкозо-6-фосфатазу, розвивається ренальна глюкозурія.
3. Порушення енергозабезпечення. Закономірно виникають при гіпоксії, голодуванні, гіповітамінозах, зменшенні активності ферментів енергетичного обміну і виявляються дефіцитом АТФ. При цьому страждають усі енергозалежні механізми реабсорбції і секреції (первинний і вторинний активний трансмембранний транспорт, ендо- і екзоцитоз).
4. Надлишок речовин, що реабсорбуються. Він зумовлює функціональне перевантаження систем зворотного транспорту. Цей механізм стосується так званих по-рогових речовин, до яких належать глюкоза і гідрокарбонати. Якщо концентрація
цих сполук у крові перевищує пороговий рівень (для глюкози- 10 ммоль/л, для гідрокарбонатів - 27 ммоль/л), то їхній надлишок виводиться із сечею - розвивається глюкозурія (при цукровому діабеті), сеча стає лужною (при алкалозах).
5. Порушення гуморальної регуляції процесів факультативної реабсорбції. Пов'язані зі змінами вмісту в крові альдостерону, передсердного натрійуретично-го гормону, вазопресину, паратирину.
32.12. У чомусутність постренальних порушень ниркових функцій?
Постреішльними називають порушення, що виникають на шляхах виведення сечі. Вони можуть бути обумовлені:
а) обтурацією сечовивідних шляхів (камені) і б) здавленням ззовні — компресією, що виникає як наслідок стриктур сечоводів, аденоми простати й інших пухлин.
Зазначені причини, які перешкоджають відтоку сечі, .викликають підвищення тиску рідини в капсулі клубочків, що призводить до зменшення ефективного фільтраційного тиску і зменшення швидкості клубочкової фільтрації.
32.13. Які кількісні і якісні зміни сечі можуть наставати при ураженнях нирок?
Кількісні зміни: 1) оліго- і анурія; 2) поліурія; 3) ніктурія; 4) гіпо- та ізостенурія. Якісні зміни: 1) протеїнурія; 2) гематурія; 3) циліндрурія; 4) лейкоцитурія (піурія).
32.14. Що таке оліго- і анурія? У яких випадках виникає олігурія? Які її наслідки?
В умовах нормальної життєдіяльності з організму щодоби має виводитися із сечею 700 мосм осмотично активних речовин. Щоб вивести таку їх кількість при максимально можливій осмоляльності сечі (1000 мосм/кг), необхідно як мінімум 700 мл сечі на добу. Такий добовий об'єм сечі одержав назву облігшпного діурезу, або облігатного об'єму.
Олігурія - це зменшення добового діурезу нижче облігатного об'єму, тобто менше 700 мл/добу.
Анурія — це повна відсутність діурезу.
Причиною олігурії є порушення клубочкової фільтрації. Олігурія виникає тоді, коли під дією преренальних, ренальних і постренальних факторів швидкість клубочкової фільтрації стає нижче 10 мл/хв.
Олігурія призводить до:
1) збільшення об'єму позаклітинної рідини — гіпергідрії (див. розд. 23);
2) накопичення в організмі осмотично активних речовин. Зокрема, розвиваються гі-пернатріємія, гіперкаліємія;
3) накопичення в крові кінцевих продуктів обміну речовин — азотемії.
32.15. Що таке поліурія? Які механізми її розвитку?
Поліурія — це збільшення добового діурезу понад 1,8 л. У людини максимально можливий діурез за умови, що він не осмотичний, дорівнює 25 л/добу, що становить 15 % об'єму профільтрованої води.
Причинами поліурії можуть бути позаниркові (психогенна полідипсія, порушення водно-сольового обміну та його регуляції, наприклад, нецукровий діабет) і ниркові (поліурична стадія гострої і хронічної недостатності нирок) фактори.
Залежно від механізмів розвитку виділяють такі види поліурії.
1. Водний діурез. Обумовлений зменшенням факультативної реабсорбції води. Виникає при водному навантаженні, нецукровому діабеті. Сеча при такій поліурії гіпотонічна, тобто містить мало осмотично активних речовин.
2. Осмотичний діурез (салурез). Пов'язаний зі збільшенням вмісту в сечі нереаб-сорбованих осмотично активних речовин, що спричиняється до вторинного порушення реабсорбції води. Поліурія цього типу розвивається при:
а) порушенні реабсорбції електролітів;
б) збільшенні вмісту в первинній сечі так званих порогових речовин (наприклад, глюкози при цукровому діабеті);
в) дії екзогенних речовин, які погано реабсорбуються (манітол) або порушують реабсорбцію електролітів (салуретики).
В умовах максимального осмотичного діурезу виділення сечі може досягати 40 % величини клубочкової фільтрації.
3. Гіпертензивний діурез. Розвивається при артеріальної гіпертензії, коли збільшується швидкість руху крові в прямих судинах мозкового шару нирок (ці судини йдуть паралельно колінам петлі Генле). При цьому збільшується конвекційний транспорт речовин; саме цей транспорт, а не дифузія стає провідним. Наслідком посилення конвекційного транспорту є "вимивання" натрію, хлору, сечовини з інтерстицію. Це веде до зменшення осмотичного тиску позаклітинної рідини, у результаті зменшується реабсорбція води в нисхідній ділянці петлі Генле і розвивається поліурія.
32.16. Що таке ніктурія? Коли вона виникає?
Ніктурія — це патологічна ознака, сутність якої полягає у переважанні нічної частини діурезу над денною.
У нормі 60-80 % добової кількості сечі виділяється в період з 8 до 20 год., тобто відношення нічного діурезу до денного становить 1:2.
При ніктурії нічна порція сечі може більше ніж удвічі перевищувати денну.
Залежно від причин виділяють:
1) серцеву ніктурію— розвивається при серцевій недостатності. Удень у іворих збільшуються навантаження на серце й приймання води, що веде до застою крові й затримки води в тканинах (набряки). Уночі в горизонтальному положенні поліпшується венозний відтік і зменшується навантаження на серце. Це викликає виділення передсердного натрійуретичного гормону, збільшення діурезу і зменшення набряків;
2) ниркова ніктурія — характерна для ураження нирок. її пояснюють поліпшенням уночі порушеного тканинного кровообігу. У результаті прискорюється рух крові по судинах нирок, розвивається гіпертензивний діурез.
32.17. Що таке гіпо- та ізостенурія?
Гіпостенурія виникає при зменшенні здатності нирок концентрувати сечу. Вона характеризується зменшенням відносної густини сечі до 1012-1006, причому зміни цієї густини протягом доби незначні (рис. 148).
Рис. 148. Добові коливання густини сечі
в нормі, при гіпо- та ізостенурії:
І - норма; II - слабке обмеження; III
- гіпостенурія; IV - ізостенурія
Поєднання гшостенурії з поліурією свідчить про ушкодження канальців при відносно достатній функції клубочків. Якщо гіпостенурія виникає на тлі олігурії, то це ознака ушкодження всіх структур нефронів (канальців і клубочків).
При повній втраті нирками здатності концентрувати і розводити сечу розвивається ізостенурія, при якій відносна густина сечі дорівнює густині фільтрату, тобто 1010, і не міняється протягом доби (монотонний діурез).
Ізостенурія є ознакою дуже важких порушень, при яких ниркові канальці, по суті, перетворюються у звичайні трубки, що проводять фільтрат у ниркові миски.
32.18. Які механізми протеїнурії?
Протеїнурія — це виділення білка із сечею. В основі її розвитку можуть лежати такі механізми:
1) збільшення проникності клубочкового фільтра у зв'язку з ураженням базальної мембрани (клубочкова протеїнурія);
2) зменшення канальцевої реабсорбції білка, що профільтрувався (канальцева про-теїнурія);
3) патологічне надходження білка у просвіт канальців з ушкоджених клітин тубуляр-ного епітелію або з перитубулярної лімфатичної рідини (секреторна протеїнурія).
Протеїнурія може бути селективною, коли в сечі визначають тільки низькомолекулярні білки, і неселективною, для якої характерна поява в сечі як низько-, так і високомолекулярних білків.
За ступенем селективності розрізняють нефротичний тип протеїнурії (у сечі тільки альбуміни або альбуміни+а-глобуліни) і нефритичний тип (у сечі визначаються всі класи білків плазми крові — альбуміни, а-, р- і у-глобуліни).
32.19. Що може бути причиною гематурії, лейкоцитурії, циліндрурії?
Гематурія — поява еритроцитів у сечі. Може бути обумовлена:
1) ушкодженням клубочкового фільтра і надходженням еритроцитів у первинну сечу. При цьому в кінцевій сечі виявляють "вилужені" еритроцити;
2) ушкодженням сечовивідних шляхів.
Лейкоцитурія - поява в сечі лейкоцитів понад 5 у полі зору. Лейкоцитурію, при якій виявляють дуже велику кількість лейкоцитів у сечі, у тому числі й зруйнованих, називають піурією. Основна причина лейкоцитурії — запальні процеси в нирковій тканині і сечовивідних шляхах.
Цііліидрурія — поява в сечі циліндрів. Циліндри являють собою зліпки ниркових канальців. Вони утворюються при ушкодженні епітелію канальців і складаються з осадженого білка і загиблих клітин. Залежно від будови розрізняють гіалінові, зернисті і епітеліальні циліндри.
32.20. Що таке уремічний синдром і уремічна кома? Який їх патогенез?
Уремічний синдром супроводжує розвиток гострої і хронічної недостатності нирок і розвивається внаслідок порушення клубочкової фільтрації. Перші його ознаки з'являються, коли швидкість клубочкової фільтрації (ІИКФ) стає нижче 50 мл/хв, а виражені клінічні симптоми - при падінні ШКФ нижче 10 мл/хв.
Основу уремічного синдрому становить інтоксикація, обумовлена накопиченням у крові кінцевих продуктів обміну речовин, які в нормі виводяться із сечею. Нині відомо понад 200 речовин, здатних викликати інтоксикацію при недостатності нирок. Найбільше значення мають:
а) сечовина;
б) сечова кислота;
в) похідні гуанідину — креатин, креатинін, метилгуанідин, диметилгуанідин та ін.;
г) ароматичні сполуки - феноли, індол, ароматичні аміни; ґ) аліфатичні аміни;
д) низькомолекулярні пептиди.
Ознакою накопичення зазначених токсичних речовин є азотемія - збільшення вмісту в крові залишкового азоту.
Клінічні прояви уремічного синдрому пов'язані з ураженням практично всіх органів і систем. Однак на перший план виступають ознаки токсичного ураження центральної нервової системи: анорексія, нудота, блювота, психічні порушення, ураження периферичних нервів.
Украй важким проявом інтоксикації є розвиток уремічної коми - повної втрати свідомості зі зникненням рефлексів на зовнішні і внутрішні подразники, пригніченням життєво важливих функцій. Крім інтоксикації, в патогенезі уремічного синдрому мають значення порушення обміну води, електролітів, розлади кислотно-основного стану. -?
32.21. Які механізми можуть обумовлювати розвиток набряків при ураженнях нирок?
Патогенетично розрізняють три типи набряків, що розвиваються при різних ураженнях нирок.
1. Набряки при гострій і хронічній недостатності нирок. Основний механізм їх розвитку — гідростатичний (гіперволемічний) (див. розд. 23). Зменшення швидкості клубочкової фільтрації, характерне для ниркової недостатності, призводить до затримки натрію й води в організмі (позитивний водний баланс) і, як наслідок, до гіперволемії. Остання, будучи причиною збільшення гідростатичного тиску в капілярах, викликає розвиток набряків за механізмом Старлінга.
2. Нефротичні набряки. Основний механізм їх розвитку — онкотичний (гіпопроте-гнемічний). Порушення клубочкового фільтра при нефрозі викликають масивну протеїнурію, у результаті якої розвивається гіпопротеїнемія і падає онкотичний тиск крові. Це, у свою чергу, за механізмом Старлінга викликає перехід води з судин у тканини - розвиваються набряки.
3. Нефритичні набряки. Розвиваються при гострому і хронічному гломерулонефриті. Патогенез цих набряків складний і в своїй основі має такі механізми:
а) запалення клубочків → застій крові в судинах нирок → гіпоксія юкстагло-мерулярного апарату → активація ренін-ангіотензинної системи → секреція альдостерону → затримка натрію в організмі і підвищення осмотичного тиску крові → секреція антидіуретичного гормону → затримка води → гі-перволемія → набряки;
б) запалення клубочків → порушення ниркового кровообігу → зменшення швидкості клубочкової фільтрації → затримка натрію й води в організмі → гіперволемія → набряки;
в) запалення клубочків → збільшення проникності ниркового фільтра →про-теїнурія → гіпопротеїнемія → набряки.
32.22. Які порушення кислотно-основного стану можуть розвиватися при ураженнях нирок?
Найчастіше розвивається негазовий ацидоз. Залежно від сутності патологічних процесів у нирках можливі три його варіанти.
1. Клубочковий ацидоз (нирковий азотемічний ацидоз). Виникає внаслідок розвитку ниркової недостатності при зменшенні швидкості клубочкової фільтрації ниж-
че 25 мл/хв. Він є метаболічним ацидозом зі збільшеною аніонною різницею (див. розд. 25). Його розвиток обумовлений накопиченням іонів водню, що утворюються ендогенно, в основному у формі сірчаної, фосфорної, сечової та інших кислот.
2. Проксимальний нирковий канальцевий ацидоз. Є наслідком первинних порушень реабсорбції гідрокарбонату в проксимальних звивистих канальцях не-фронів. Його відносять до видільних ацидозів з нормальною аніонною різницею (гіперхлоремічний).
3. Дистальний нирковий канальцевий ацидоз. Обумовлений первинними порушеннями процесів ацидогенезу в дистальних звивистих канальцях, де відбувається відтитровування буферів (збереження гідрокарбонату) і підкислення (аци-дифікація) сечі. Є метаболічним ацидозом з нормальною аніонною різницею (гіперхлоремічний). Виявляється нездатністю витримувати ендогенне навантаження іонами водню. При цьому рН сечі не досягає значень нижче 6.
Зміни функції нирок іноді можуть спричинятися до розвитку негазового гіпо-каліємічного алкалозу. Це, зокрема, буває при гіперальдостеронізмі. Альдостерон, посилюючи секрецію іонів калію в дистальних звивистих канальцях, викликає гіпо-каліємію, яка, у свою чергу, обумовлює виникнення алкалозу (див. розд. 25).
32.23. Які порушення фосфорно-кальцієвого обміну характерні для недостатності нирок? Який їх патогенез?
Порушення фосфорно-кальцієвого обміну при недостатності нирок виявляються розвитком ниркової остеодистрофії (остеопатії).
Ниркова остеодистрофія — це комплекс дистрофічних порушень у кістках, що виникають унаслідок розладів фосфорно-кальцієвого обміну при ураженнях нирок. Клінічно вона виявляє себе двома групами змін.
I. Дистрофічні зміни в кістках:
а) резорбція кісткової тканини (фіброзно-кістозний остеїт) — у кістках утворюються порожнини, які заповнюються фіброзною тканиною. Є наслідком гіперпаратиреозу;
б) остеомаляція - розм'якшення кісток, їх деформація, болі в кістках. У дітей порушується окостеніння хрящів. Є наслідком гіпокальціємії;
в) остеопороз — зменшення щільності кісткової тканини без деформації кісток;
г) остеосклероз — збільшення щільності кісткової тканини.
II. Обвапнеїшя м'яких тканин - кальцифікація (див. розд. 24).
У патогенезі ниркової остеодистрофії мають значення такі механізми:
а) хронічна недостатність нирок (ХНН) → зменшення екскреції фосфатів → гіперфосфатемія → гіпокальціємія (див. розд. 24) → збільшення продукції паратирину → резорбція кісткової тканини, її демінералізація;
б) ХНН → порушення перетворення 25-оксивітаміну D у його гормонально активну форму- 1,25-діоксивітамін D → зменшення всмоктування іонів Са2+ у кишках → гіпокальціємія → див. вище;
в) ХНН → клубочковий ацидоз → обмін іонів Са2+ і Na+ на іони Н+ крові (буферна функція кісткової тканини) → демінералізація кісток.
32.24. Які види артеріальної гіпертензії можуть виникати при ураженнях нирок? Який їх патогенез?
Артеріальна гіпертензія є одним з проявів хронічної недостатності нирок (ХНН). Виділяють такі її види.
1. Гіпертензія, залежна від об'єму. Цей вид буває у 80-90 % хворих з ХНН. Гї патогенез пояснюють таким чином: ХНН → зменшення швидкості клубочкової фільтрації → порушення екскреції натрію і води → гіперволемія → збільшення хвилинного об'єму серця → збільшення артеріального тиску.
2. Гіпертензія з високим рівнем реніну (високоренінова). Цей вид виявляють в.І 5-10 % хворих з ХНН. Основний її механізм: ураження нирок → порушення ниркового кровообігу → гіпоксія юкстагломерулярного апарату → вивільнення реніну → активація ренін-ангіотензинної системи → далі див. розд. 28.
3. Гіпертензія, не залежна від об'єму. Буває в 5-10 % хворих з ХНН. Характеризується нормальним об'ємом циркулюючої крові й нормальним рівнем реніну в крові. Вважають, що розвиток цього виду гіпертензії пов'язаний з порушенням депресорних функцій нирок, а саме — зі зменшенням утворення ниркових простагландинів, зокрема ПГ Е2, зменшенням активності ниркової калікреїн-кінінової системи, пригніченням утворення нейтрального ліпіду, що має гіпотензивну дію.
32.25. Які механізми обумовлюють розвиток анемії, що супроводжує недостатність нирок?
Ознаки анемії у хворих з хронічною недостатністю нирок з'являються при зменшенні швидкості клубочкової фільтрації нижче 40 мл/хв. У її патогенезі мають значення такі механізми.
1. Пригнічення еритропоезу:
а) порушення регуляції еритропоезу, обумовлене пригніченням утворення ниркових еритропоетинів і посиленим продукуванням інгібіторів кровотворення;
б) ушкодження кровотворних клітин уремічними токсинами;
в) дефіцит заліза, що розвивається як наслідок хронічних крововтрат при захворюваннях нирок і втрат трансферину в результаті протеїнурії.
2. Крововтрати. Можуть бути пов'язані з гематурією, з частим утворенням виразок у шлунку і кишках хворих з ураженням нирок, з розвитком геморагічного діатезу, і
3. Гемоліз еритроцитів, обумовлений уремічними токсинами.
32.26. Якими порушеннями гемостазу може виявляти себе недостатність нирок?
І. ТрОхМбофілічні порушення- гіиеркоагуляція. В основі їх розвитку можуть бути:
а) зменшення активності фібринолітичної системи внаслідок дефіциту урокі-нази — активатора плазміногену;
б) зменшення активності антикоагулянтної системи, що пов'язано з порушеннями синтезу і депонування гепарину в нирках;
в) втрата із сечею внаслідок протеїнурії антитромбіну III— основного природного антикоагулянта. її. Геморагічний діатез. Може бути обумовлений розладами як судинно-тромбо-цитарного гемостазу (тромбоцитопенія внаслідок мієлотоксичної дії уремічних токсинів), так і коагуляційного (втрата із сечею факторів зсідання крові при протеїнурії).
32.27. Що таке нефротичний синдром?Який його патогенез?
Нефропшчним називають синдром, що виникає при різноманітних ураженнях нирок і виявляється масивною протеїнурією, гіпопротеїнемією, розвитком набряків, гіперліпгдемією.
Нефротичний синдром за походженням поділяють на первинний і вторинний.
Первинний нефротичний синдром не пов'язаний з яким-небудь попереднім захворюванням нирок. Найчастіше в основі його виникнення є наявність генетично обумовлених дефектів обміну речовин (ліпоїдній нефроз) або трансплацентарне перенесення специфічних протиниркових антитіл від матері до плода (уроджений сімейний нефроз).
Вторинний нефротичний синдром обумовлений деякими захворюваннями нирок (гломерулонефрит) або інших органів (нефропатія вагітних, цукровий діабет, амілоїдоз, системний червоний вовчак, сироваткова хвороба, стафілококовий сепсис та ін.). Його спостерігають також при отруєнні солями важких металів, при великих опіках, радіаційному ураженні, при відторгненні ниркового трансплантата, при застосуванні деяких лікарських препаратів (сульфаніламіди, пеніцилін, кортикостероїди).
У патогенезі нефротичного синдрому мають значення два механізми.
I. Імунний механізм. У його основі лежать імунокомплексні реакції (реакції ПІ типу за Кумбсом і Джеллом), які спричиняються до ушкодження базальної мембрани клубочків. Джерелами антигенів можуть бути екзогенні фактори: бактеріальні, вірусні, паразитарні, лікарські, харчові, сполуки важких металів та ін. Ендогенними антигенами можуть бути ДНК, денатуровані нуклеопротеїди, білки пухлинного походження, тиреоглобулін. Антитіла до зазначених антигенів переважно являють собою імуноглобуліни класу Ig M.
Ураження клубочків нирок пов'язують з відкладанням на поверхні або в самій мембрані капілярних судин амілоїду, гліко- і ліпопротеїдів, фібриногену, з активацією гуморальних і клітинних ланок запальної реакції. Внаслідок цього втрачається структурна цілісність базальної мембрани, змінюється її склад і фізико-хімічні властивості, різко підвищується проникність для плазмових білків.
II. Метаболічний механізм і пов'язані з ним фізико-хімічні зміни. Підвищення проникності клубочкового фільтра може бути пов'язане зі зменшенням негативного електричного заряду стінки капілярів клубочків (зумовленого аніонами внутрішньої поверхні судин), зникненням сіалопротеїну, який у нормі тонким шаром покриває ендотелій капілярів і його відростки. У місцях, де втрата аніонів і сіа-лопротеїнів максимальна, накопичуються поліморфноядерні лейкоцити, лізосомні ферменти яких безпосередньо ушкоджують базальну мембрану капілярних судин.
32.28. Які наслідки для організму може мати втрата білків із сечею
при нефротичному синдромі?
Масивна протеїнурія, що її спостерігають при нефротичному синдромі, спричиняється до зменшення вмісту в крові майже всіх функціонально важливих білків. Цим і пояснюють широкий спектр порушень, що виникають в організмі:
Білки, що втрачаються 3 сечею |
Наслідки зменшення вмісту білка в крові |
|
Альбуміни |
|
Ппоонкія, набряки |
Антитромбін III |
|
Схильність до тромбозів і тромбоемболій |
Фактори зсідання крові |
|
Геморагічний діатез |
Компоненти комплементу |
|
Зменшення резистентності до інфекцій |
Імуноглобуліни (Ig) |
|
Те ж саме |
Ліпопротеїди високої густини |
(ЛПВГ) |
Прискорений розвиток атеросклерозу |
Білки, що транспортують мікроелементи |
Дефіцит Fe, Cu, Zn |
|
Білки, що транспортують гормони |
Ендокринні порушення |
|
32.29. Що таке гломерулонефрит? Які етіологія й патогенез гострого гломерулонефриту?
Гломерулонефрит - це двостороннє дифузне захворювання нирок алергічної природи.
Розрізняють гострий і хронічний гломерулонефрит.
Для гострого гломерулонефриту характерні бурхливий початок, олігурія, протеїнурія, азотемія, артеріальна гіпертензія, набряки, гематурія, порушення з боку центральної нервової системи.
Гострий гломерулонефрит виникає під час (або після) якої-небудь інфекції, частіше стрептококової природи. Вважають, що гемолітичний стрептокок групи А (типи 4,12) є специфічним "нефритогенним" штамом. Певну роль відіграють й інші інфекції, у тому числі вірусні, паразитарні. Встановлено етіологічну роль охолодження, дифузних уражень сполучної тканини (системний червоний вовчак, ревматоїдний артрит, вузликовий періартеріїт), опікової хвороби, попередньої вакцинації або використання з лікувальною метою гетерологічних сироваток.
Виділяють два основних патогенетичних варіанти гострого гломерулонефриту.
1. Ураження базальної мембрани клубочків нефронів антитілами проти її антигенів - нефротоксичний гломерулонефрит (має стрімкий перебіг, швидко прогресує). Носієм антигенних властивостей базальної мембрани є глікопротеїд.
2. Розвиток запального процесу в клубочках унаслідок фіксації на базальній мембрані та всередині її імунних комплексів - імунокомплексний гломерулонефрит.
У розвитку такого типу хвороби можуть брати участь антигени як екзогенного (інфекційної чи неінфекційної природи), так і ендогенного (білок тканин, ДНК) походження. Антитіла (Ig G, Ig М) прямо в крові вступають у взаємодію із зазначени-
ми антигенами, потім у вигляді імунних комплексів (антиген-антитіло-комплемент) надходять у клубочки, відкладаючись на їхній базальній мембрані. Реалізація ушкоджувальної дії імунних комплексів, як і нефротоксичних антитіл, відбувається через розвиток імунного запалення (див. розд. 10).
До імунокомплексних відносять гломерулонефрити, що розвиваються після стрептококової інфекції, при системному червоному вовчаку, сироватковій хворобі та ін. Більшість випадків гломерулонефриту (не менше ніж 80 %) є імунокомплексними.
32.30. Які існують експериментальні моделі гострого гломерулонефриту?
1. Введення тваринам нефротоксичної сироватки, тобто сироватки, що містить антитіла проти антигенів ниркової тканини.
Серед моделей цієї групи найчастіше використовують модель Ліндемана і модель Масугі.
Модель Ліндемана. Кролям внутрішньовенно вводять нефротоксичну сироватку, отриману від морської свинки, попередньо імунізованої суспензією нирки кроля. При цьому відбувається фіксація протиниркових антитіл на базальних мембранах клубочків з наступним зв'язуванням комплементу і розвитком ушкодження. Модель Масугі. Кролям внутрішньовенно вводять нефротоксичну сироватку качок, імунізованих тканиною нирок кролів. Особливість цієї моделі полягає в тому, що антитіла птахів фіксуються на базальній мембрані клубочків, але не можуть зв'язувати комплемент ссавців. Це призводить до утворення власних кролячих антитіл проти фіксованих качиних (для цього треба 6—8 діб). Останні в цьому випадку виступають антигенами. Кролячі антитіла, з одного боку, взаємодіють з фіксованими на базальній мембрані качиними, з другого - зв'язують комплемент, активація якого і призводить до ушкодження клубочків.
2. Охолодження нирки — заморожування хлороформом (Герцен). При цьому в крові дослідних тварин (кролів) з'являються специфічні протиниркові антитіла і зазнає уражень друга інтактна нирка. Зазначена модель доводить роль аутоімунних механізмів у розвитку так званого "окопного" нефриту (нефриту воєнного часу).
3. Введення в черевну порожнину кролів клітинної суспензії ниркової тканини і культури стрептококів (подружжя Ковелті).
4. Імунізація овець базальними мембранами клубочків нефронів нирки людини в повному ад'юванті Фрейнда (Стеблей).
5. Імунізація щурів суспензією гомологічної або аутологічної нирки з повним ад'ю-вантом Фрейнда (Хеймен).
6. Виведення чистих ліній тварин (наприклад, новозеландські миші лінії NZB/BL), у яких спонтанно виникають аутоімунні хвороби, у тому числі і гломерулонефрит.
32.31. Чим може виявлятися хронічний гломерулонефрит? Які його етіологія й патогенез?
Хронічний гломерулонефрит являє собою тривале прогресуюче дифузне двостороннє ураження нирок запальної природи, неоднорідне за походженням, клінічними проявами і патогенезом.
Залежно від клінічних проявів виділяють такі його форми.
1. Латентна форма виявляє себе ізольованим сечовим синдромом — помірною про-теїнурією, гематурією. У частини хворих спостерігають набряки і транзиторну артеріальну гіпертензію.
2. Гіпертонічна форма характеризується стійким підвищенням артеріального тиску, набряками, гематурією, протеїнурією, циліндрурією і лейкоцитурією.
3. Нефротична форма, яку супроводжують набряковий синдром, виражена протеї-нурія і циліндрурія, зміни крові (гіпопротеїнемія, гіперліпідемія).
4. Змішана, або нефротично-гіпертонічна форма, для якої характерні набряк і гіпертензія, але на відміну від нефротичної форми нема характерних змін крові.
Хронічний гломерулонефрит може бути наслідок гострого, але частіше розвивається первинно. Залежно від причин виділяють такі його форми:
1) інфекційного походження (постстрептококовий, при затяжному септичному ендокардиті, малярії, сифілісі, туберкульозі та інших інфекціях);
2) неінфекційні (сироватковий, вакцинний, медикаментозний, травматичний, при отруєннях, охолодженні, тромбозі ниркових вен);
3) зумовлені дифузними захворюваннями сполучної тканини (ревматоїдний артрит, системний червоний вовчак, геморагічний васкуліт та ін.);
4) особливі (після еклампсії, радіаційний, спадковий та ін.).
Загальновизнаною є імунологічна концепція розвитку хронічного гломерулонефриту. Поряд з нефротоксичними та імунокомплексними механізмами (див. запит. 32.29) певне значення в його патогенезі має гіперчутливість уповільненого типу.
32.32. Що таке пієлонефрит? Що може бути його причиною?
Пієлонефрит — це інфекційно-запальне захворювання слизової оболонки сечових шляхів і паренхіми нирок з переважним ураженням інтерстиціальної тканини.
Хвороба виникає у зв'язку із занесенням інфекції в нирки гематогенним шляхом або поширенням її у висхідному напрямку по сечових шляхах. Збудниками найчастіше є кишкова паличка, коки.
Виникненню захворювання сприяють умови, що викликають застій сечі (звуження, закупорка сечоводів, аденома простати), порушують трофіку сечових шляхів, знижують реактивність організму (цукровий діабет, хронічна інтоксикація та ін.).
Починається пієлонефрит як гостре захворювання, що у більшості випадків через латентну, бідну на симптоми фазу переходить у хронічну форму, яка завершується зморщенням і недостатністю нирок.
Для клініки пієлонефриту характерними є:
а) ознаки важкого інфекційного і запального процесу (інтоксикація, гарячка, зміни крові - лейкоцитоз, збільшення ШОЕ та ін.);
б) прояви, пов'язані з.розладами функції нирок- артеріальна гіпертензія, набряки, анемія; сечовий синдром. Останній, залежно від стадії хвороби, може виявлятися поліурією або олігурією, полакіурією (частим сечовипусканням), лейкоцитурією, гематурією, помірною протеїнурією, циліндрурією, гіпо- та ізостенурією).
Важливим є те, що при пієлонефриті, особливо на початкових стадіях хвороби, порушення функцій канальців є провідними, якщо порівнювати з розладами функцій клубочків (своєрідна функціональна дисоціація). Про це свідчать зменшення здат?5 ності нирок до концентрування сечі внаслідок порушення процесу реабсорбції, ранній і важкий канальцевий ацидоз, пов'язаний з порушенням ацидо- і амоніогенезу, синдром втрати солей, в основі якого — різке зниження канальцевої реабсорбції іонів натрію, калію, кальцію. Як наслідок зазначених розладів можуть розвиватися небезпечні для життя порушення водно-електролітного обміну і кислотно-основного стану організму.
Прогресування хвороби супроводжується наростанням описаних порушень і розвитком хронічної недостатності нирок.
32.33. Що таке сечокам 'яна хвороба ? Які її етіологія і патогенез ?
Сечокам 'яною називають хворобу, обумовлену утворенням каменів у паренхімі нирок і в мисково-сечовідному сегменті сечових шляхів. У вираженій формі сечокам'яна хвороба характеризується нападами ниркової кольки, причиною яких є гостра затримка сечі, викликана механічною закупоркою і спазмом миски і сечоводу; гематурією, що виникає внаслідок ушкодження сечових шляхів; гарячкою, лейкоцитозом. Ускладненням хвороби є приєднання інфекції і розвиток унаслідок цього калькульозного пієлонефриту, абсцесу нирки, інфікованого гідронефрозу і т. д. У цих випадках обов'язковим симптомом є піурія (наявність гною в сечі).
До складу каменів входять солі щавлевої і фосфорної кислот, сечова кислота, урати натрію і амонію, іноді цистин, ксантин. Частіше склад каменів має змішаний характер.
В етіології сечокам'яної хвороби мають значення такі фактори.
1. Якісний склад води і їжі, режим пиття і харчування. Від зазначених чинників залежать рН сечі, величина діурезу, концентрація в сечі солей, з яких утворюються камені, а також речовин, що забезпечують їхню розчинність і стабільний стан. Вважають, що споживання сильно мінералізованої, жорсткої питної води з великим вмістом солей кальцію, вживання їжі з надмірним вмістом солей, з яких утворюються камені, нестачею білків, вітамінів групи В, особливо піридоксину, сприяють утворенню каменів у нирках.
2. Інфекції сечових шляхів. Вони сприяють зміщенню реакції сечі в лужний бік унаслідок уролітичної дії деяких бактерій і осадження кристалів фосфатів; зниженню поверхневого натягу між сечею і ушкодженою слизовою оболонкою сечових органів; утворенню в ушкоджених місцях фібрину, клітинного детриту, згустків крові, які можуть служити кристалізаційними центрами; зниженню колоїдної стабільності сечі внаслідок обумовленого пієлонефритом порушення ниркової екскреції ряду речовин - сечовини, лимонної кислоти, кальцію, фосфатів та ін.
3. Жаркий і сухий клімат, робота в гарячих цехах. Такі умови викликають значну втрату організмом рідини при потінні і підвищення внаслідок цього густини сечі. Це сприяє частішому виникненню сечокам'яної хвороби в певних географічних районах і в осіб відповідних професій.
4. Ендокринні хвороби (наприклад, первинний гіперпаратиреоз).
5. Хвороби обміну речовин (цистиноз, оксалоз, гіперкальціурія, гіпергліцинемія та ін.).
6. Спадково обумовлені канальцеві синдроми, що характеризуються порушенням реабсорбції амінокислот (цистину, гліцину та ін.).
Наявність двох складових частин у структурі каменів — неорганічної й органічної — було покладено в основу відповідно кристалізаційної і колоїдної теорій їх утворення.
Відповідно до першої, утворення каменів цілком підпорядковане принципам кристалізації, а утворення органічної матриці (на її частку припадає 2—3 % сухої маси) є додатковим вторинним процесом.
На противагу цьому прихильники колоїдної теорії первинним при утворенні каменів вважають формування органічної матриці, а кристалізацію на ній сечових солей розглядають як процес вторинний.
Так чи інакше, для настання кристалізації потрібне зниження стабільності в сечі солей, з яких утворюються камені. Підвищення їхньої концентрації необхідне тільки на початкових етапах кристалізації. Подальше здійснення цього процесу може відбуватися і при нормальній концентрації солей. У цьому випадку провідну роль у патогенезі сечокам'яної хвороби відіграють фактори, що знижують розчинність солей. До їх числа відносять:
1) зниження в сечі вмісту солюбілізаторів — сечовини, креатиніну, гіпурової кислоти, ксантину, хлориду натрію, цитратів, магнію, які у нормі підтримують розчинений стан солей, з яких утворюються камені, і перешкоджають їхньому осадженню;
2) зниження в сечі вмісту речовин, що пригнічують кристалізацію,— інгібіторів кристалізації (неорганічний пірофосфат, іони цинку, марганцю, кобальту та ін.);
3) зниження в сечі рівня комплексоутворювачів, зокрема іонів магнію і цитратів, які у нормі зв'язують відповідно близько 30—40 % оксалатів і більш як 50 % іонізованого кальцію з утворенням розчинних комплексних сполук;
4) поява у сечі мукопротеїдів, піровиноградної кислоти, сульфаніламідів, продуктів колагену, еластину, що створюють умови для швидкої кристалізації солей у насиченому розчині.
33. Патологічна фізіологія ендокринної системи
33.1. Що таке істинні гормони? Які у них властивості?
Істинними гормонами називають продукти діяльності ендокринних залоз.
Ендокринні залози - це спеціалізовані популяції секреторних клітин, які утворюють і виділяють у кров (або інші циркулюючі рідини) свої специфічні сигнальні продукти — гормони або їхні найближчі біосинтетичні попередники.
Гормони можуть синтезуватися:
а) епітеліальними клітинами (власне залозистий епітелій);
б) нейроендокринними клітинами (клітини гіпоталамуса);
в) міоендокринними клітинами (м'язові волокна передсердь серця).
Секреція гормонів може здійснюватися:
а) ендокринними органами, що складаються із залозистих клітин одного типу (щитоподібна залоза);
б) ендокринними органами, що складаються із залозистих клітин різних типів (аде-ногіпофіз, кора надниркових залоз);
в) групами ендокринних клітин у неендокринних органах (підшлункова залоза).
Основні властивості гормонів:
1) утворюються спеціалізованими клітинами ендокринних залоз;
2) мають високу і специфічну біологічну активність. Гормони виявляють свій вплив у дуже низьких концентраціях - порядку 10"6 -10-11 моль/л. Специфічність дії гормонів пов'язана з існуванням "клітин-мішеней", які мають особливі рецептори до конкретного гормону;
3) секретуються в кров (або інші циркулюючі рідини);
4) мають дистантну дію, тобто чинять вплив на клітини, що знаходяться на великій відстані від місця утворення гормону.
33.2. Як класифікують гормони?
І. За анатомічним принципом розрізняють гормони гіпоталамуса, аденогіпофіза, нейрогіпофіза, кори і мозкової речовини надниркових залоз, щитоподібної і при-щитоподібних залоз і т. п.
II. За хімічною структурою виділяють:
а) стероїдні гормони (мінерало- і глюкокортикоїди, жіночі і чоловічі статеві гормони);
б) похідні амінокислот (тиреоїдні гормони, катехоламіни, мелатонін);
в) білково-пептидні гормони (рилізинг-гормони, вазопресин, окситоцин, гормони аденогіпофіза, інсулін, глюкагон, паратирин, кальцитонін).
III. За функціональними ефектами гормони можуть бути:
а) ефекторними (діють безпосередньо на органи-мішені);
б) тропними (регулюють синтез ефекторних гормонів);
в) рилізинг-гормонами (регулюють синтез і секрецію тропних гормонів).
IV. За значенням для організму виділяють:
а) гормони, що забезпечують фізичний, статевий і розумовий розвиток організму (соматотропний, гонадотропні, статеві гормони, нейропептиди);
б) адаптивні гормони, що забезпечують довгострокову адаптацію організму до змін зовнішнього середовища (тиреоїдні гормони, АКТГ, глюкокортикоїди);
в) гомеостатичні гормони, які беруть участь у підтриманні сталості внутрішнього середовища організму (альдостерон, вазопресин, паратирин, інсулін).
33.3. Що таке ендокринна функція? Назвіть її складові частини.
Ендокринною функцією називають участь ендокринних залоз у здійсненні гуморальної регуляції фізіологічних і біохімічних процесів в організмі. Ендокринна функція має такі складові частини:
1. Регуляція діяльності ендокринних залоз.
2. Власне функція ендокринних залоз —утворення і секреція гормонів.
3. Транспорт гормонів.
4. Взаємодія гормонів з периферичними клітинами — циторецепція.
5. Метаболізм гормонів і екскреція продуктів їх інактивації.
33.4. Які існують типи порушень ендокринних функцій?
I. Гіперфункція ендокринних залоз (ендокринна гіперфункція).
II. Гіпофункція ендокринних залоз (ендокринна гіпофункція).
III. Дисфункція ендокринних залоз (ендокринна дисфункція) — різноспрямовані зміни функції ендокринних залоз.
33.5. Які виділяють патогенетичні варіанти порушень ендокринних функцій?
Рис. 149. Патогенетичні варіанти порушень ендокринних функцій
I. Дисрегуляторні порушення - розлади регуляції ендокринних залоз.
II. Залозисті порушення — розлади біосинтезу гормонів та їх секреції.
III. Периферичні порушення — розлади транспорту, рецепції і метаболізму гормонів (рис. 149).
33.6. Як здійснюється регуляція ендокринних функцій? Чим можуть бути обумовлені їх дисрегуляторні порушення?
Регуляція діяльності ендокринних залоз може здійснюватися за допомогою чотирьох механізмів (рис. 150).
Рис. 150. Механізми регуляції ендокринних залоз
1. Нервова (імпульсно-медіаторна) регуляція. За допомогою прямих нервових впливів регулюється діяльність:
а) мозкового шару надниркових залоз;
б) нейроендокринних структур гіпоталамуса;
в) епіфіза.
2. Нейроендокринна (гіпоталамічна) регуляція. Здійснюється нейроендокринними клітинами гіпоталамуса, що трансформують нервові імпульси у специфічний ендокринний процес. Сутність цього процесу полягає в утворенні і секреції в систему портальних судин гіпофіза рилізинг-гормонів, що регулюють діяльність аде-ногіпофіза.
3. Ендокринна регуляція. Сутність її полягає в безпосередньому впливі одних гормонів на синтез і секрецію інших. Прикладом цього механізму регуляції є вплив тропних гормонів аденогіпофіза на діяльність кори надниркових залоз, щитоподібної залози, статевих залоз.
4. Неендокринна гуморальна регуляція. Здійснюється неспецифічними гуморальними факторами, зокрема метаболітами, іонами. Так, концентрація глюкози в крові прямо впливає на синтез і секрецію інсуліну і глюкагону; склад і рівень амінокислот — на утворення соматотропного гормону; вміст іонів калію — на виділення в кров альдостерону; концентрація кальцію - на секрецію паратирину і кальцитоніну. Розвиток дисрегуляторних порушень ендокринних функцій може бути пов'язаний з розладами всіх чотирьох механізмів регуляції. В одних випадках, при активацій зазначених механізмів розвивається ендокринна гіперфункція, в інших, при пригнд чені регуляторних впливів, - ендокринна гіпофункція.
33.7. Які принципи лежать в основі регуляції ендокринних функцій? Як вони можуть порушуватися?
І. Принцип прямих зв'язків. Лежить в основі діяльності таких ендокринних функціональних систем:
а) гіпоталамо-гіпофізарно-наднирникової;
б) гіпоталамо-гіпофізарно-тиреоїдної;
в) гіпоталамо-гіпофізарно-гонадальної.
Цей принцип реалізується через здійснення трьох послідовних етапів:
1) сприйняття гіпоталамусом вищих нервових регуляторних сигналів і секреція відповідних рилізинг-гормонів;
2) у відповідь на дію рилізинг-гормонів секреція аденогіпофізом відповідних троп-них гормонів;
3) у відповідь на дію тропних гормонів утворення і виділення периферичними залозами (корою надниркових залоз, щитоподібною залозою, статевими залозами) ефекторних гормонів (рис. 151).
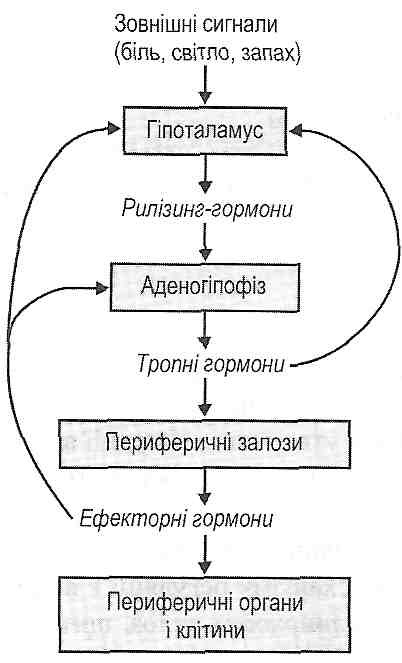
Рис. 151. Прямі і зворотні
зв 'язки в системі гіпоталамус-
аденогіпофіз-периферичні залози
Порушення прямих зв'язків у регуляції ендокринних функцій можуть бути обумовлені розладами синтезу і секреції:
а) рилізинг-гормонів;
б) тропних гормонів;
в) ефекторних гормонів.
II. Принцип зворотних зв'язків. Розрізняють зворотні негативні зв'язки в системах гіпоталамус-аденогіпофіз-периферичні залози й короткі парагі-пофізарні зворотні зв'язки.
Сутність негативних зворотних зв'язків у системах гіпоталамус-аденогіпофіз-периферич-ні залози полягає в тому, що утворювані гормони пригнічують діяльність структур, які здійснюють попередні етапи регуляції. Внаслідок цього посилення секреції ефекторного гормону через певні ланки призводить до зменшення його утворення і надходження в кров, і навпаки, зменшення вмісту гормону в крові викликає підвищення інтенсивності його утворення і секреції.
Порушення негативних зворотних зв'язків у системах гіпоталамус-аденогіпофіз-периферичні залози мають велике значення в розвитку ендокринної патології. Це положення можна проілюструвати схематично такими прикладами.
1. Система гіпоталамус-аденогіпофіз-щитоподібна залоза. Дефіцит йоду в продуктах харчування і питній воді → зменшення утворення тиреоїдних гормонів → стимуляція гіпоталамуса і аденогіпофіза → збільшення утворення тиреолі-берину і тиреотропного гормону → гіпертрофія щитоподібної залози → ендемічний зоб.
2. Система гіпоталамус—аденогіпофіз—кора надниркових залоз. Тривале застосування з лікувальною метою глюкокортикоїдних препаратів → пригнічення діяльності відповідних структур гіпоталамуса і аденогіпофіза → зменшення утворення кор-тиколіберину і АКТГ → атрофія пучкової зони кори надниркових залоз → зменшення утворення власних глюкокортикоїдів → при різкій відміні глюкокортикоїдних препаратів розвивається синдром гострої недостатності кори надниркових залоз (синдром відміни).
3. Система гіпоталамус-аденогіпофіз-статеві залози. Тривале застосування (наприклад, спортсменами-важкоатлетами) анаболічних стероїдів — похідних чоловічих статевих гормонів → пригнічення діяльності відповідних структур гіпоталамуса і аденогіпофіза → зменшення утворення гонадотропних гормонів → атрофія клітин сім'яників, що продукують чоловічі статеві гормони → розвиток імпотенції і безплідності.
Короткі парагіпофізарні зворотні зв'язки (рис. 152) здійснюються поза системою гіпоталамус-аденогіпофіз-периферичні залози. Вони відіграють провідну роль у регуляції діяльності прищитоподібних залоз, β-клітин острівців підшлункової залози. Ці зв'язки можна описати такими схемами.
Прищитоподібні залози', зменшення концентрації іонів кальцію в плазмі крові → збільшення продукції паратирину → збільшення вмісту іонів кальцію → пригнічення секреції паратирину.
β-клітини острівців підшлункової залози: збільшення концентрації глюкози в крові → активація утворення і секреції інсуліну → зменшення вмісту глюкози в крові → пригнічення секреції інсуліну.
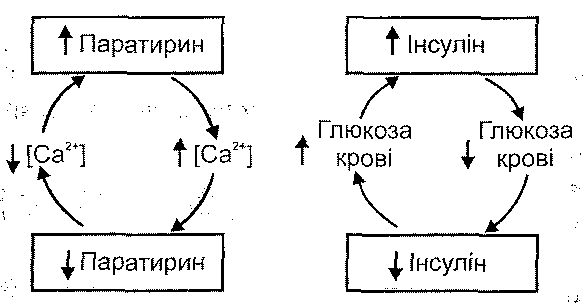
Рис. 152. Короткі парагіпофізарні зворотні зв'язки
33.8. Чим можуть бути обумовлені власне залозисті порушення ендокринних функцій?
I. Змінами кількості функціонально активних ендокринних клітин:
а) зменшенням їх кількості (видалення залози або її частин, ушкодження, некрози), що призводить до розвитку ендокринної гіпофункції;
б) збільшенням їх кількості (доброякісні і злоякісні пухлини залозистого епітелію), що супроводжується ознаками відповідної ендокринної гіперфункції.
II. Якісними змінами в ендокринних клітинах:
а) розладами біосинтезу гормонів;
б) порушеннями процесів їх секреції.
33.9. Назвіть основні причини порушень біосинтезу гормонів.
Причини порушення синтезу білково-пептидних гормонів:
1) порушення транскрипції;
2) порушення трансляції;
3) дефіцит необхідних амінокислот;
4) дефіцит АТФ;
5) порушення посттрансляційної модифікації і активації.
Причини розладів синтезу стероїдних гормонів:
1) порушення надходження в клітини, синтезу і депонування холестеролу - вихідної речовини для синтезу стероїдів;
2) набуті або спадково обумовлені дефекти ферментів, які беруть участь у реакціях біосинтезу стероїдних гормонів;
3) дефіцит кисню (гіпоксія), необхідного для реакцій гідроксилювання стероїдів;
4) дефіцит відновленого НАДФ (НАДФН) - основного джерела електронів і протонів у реакціях гідроксилювання стероїдів.
Причини порушення синтезу гормонів — похідних амінокислот:
1) дефіцит вихідних амінокислот (тирозину, триптофану);
2) дефіцит мікроелементів (йоду для утворення тиреоїдних гормонів);
3) набуті або спадково обумовлені дефекти ферментів синтезу цих гормонів;
4) дефіцит АТФ.
33.10. Як здійснюється секреція гормонів? Що може обумовлювати її порушення?
Існує три механізми секреції гормонів ендокринними клітинами:
1) вивільнення гормону із клітинних секреторних гранул (секреція білково-пептидних гормонів і катехоламінів);
2) вивільнення гормону з білковозв 'язаної форми (секреція тиреоїдних гормонів);
3) відносно вільна дифузія гормонів через клітинні мембрани (секреція стероїдних гормонів).
В основі розладів секреції гормонів можуть лежати такі механізми: а) порушення депонування гормонів. При цьому страждає утворення комплексів гормонів з речовинами — факторами депонування (білками-нейрофізинами для вазопреси-
ну і окситоцину, АТФ - для катехоламінів, цинком - для інсуліну). Як наслідок, гормони не здатні накопичуватися в необхідних кількостях у секреторних гранулах;
б) порушення передачі сигналів, що стимулюють секрецію. Вони часто пов'язані зі зменшенням утворення в клітинах або надходження вторинних посередників (цАМФ, іонів Са2+);
в) ураження контрактильних елементів (мікрофіламентів, мікротрубочок), що беруть участь у процесах екзо- і ендоцитозу. Ці процеси становлять основу секреції білково-пептидних гормонів (екзоцитоз) і тиреоїдних гормонів (ендоцитоз);
г) дефіцит АТФ, унаслідок чого порушується забезпечення енергозалежних процесів транспорту гормонів.
33.11. Які порушення можуть лежати в основі розвитку периферичних розладів ендокринних функцій?
1. Порушення транспорту гормонів в організмі.
2. Розлади метаболічної інактивації гормонів.
3. Порушення взаємодії гормонів з периферичними клітинами-мішенями - патологія циторецепції гормонів.
33.12. Як здійснюється транспорт гормонів в організмі? Які порушення ендокринних функцій можуть бути пов 'язані з його розладами?
Існує чотири форми транспорту гормонів в організмі.
1. Транспорт вільного гормону (розчиненого у воді). Саме в такій формі гормон виявляє свою біологічну активність, отже, від концентрації вільної форми гормону залежать його функціональні, структурні й біохімічні ефекти. У нормі вміст вільних гормонів у крові не перевищує 10 % від загальної їх кількості.
2. Комплекси гормонів зі специфічними транспортними білками плазми крові. Вміст цієї транспортної форми в крові становить 80 % і більше від сумарної концентрації даного гормону.
3. Неспецифічні комплекси гормонів з білками плазми крові (альбумінами, а-глобу-лінами).
4. Адсорбція гормонів на поверхні формених елементів крові (еритроцитів, лімфоцитів, моноцитів).
Порушення транспорту гормонів в організмі можуть виявлятися двома типами розладів ендокринної функції.
З одного боку, при збільшенні зв'язування гормону зменшується вміст його вільної форми, а отже, з'являються ознаки відповідної ендокринної гіпофункції. З другого боку, зменшення зв'язування гормону викликає збільшення в крові концентрації його вільної форми, що виявляється ознаками відповідної ендокринної гіперфункції.
33.13. Як здійснюється метаболічна інактивація гормонів? Які порушення ендокринної функції можуть бути пов'язані з розладами метаболізму гормонів ?
Руйнування білково-пептидних гормонів швидко відбувається в печінці під дією ферментів пептидаз.
Інактивація стероїдних гормонів здійснюється в печінці, кишках, нирках - практично у всіх органах і тканинах, за винятком тиміко-лімфоцитарної системи. У реакціях перетворення стероїдів беруть участь НАДФН-залежні ферменти. Інактивовані форми стероїдних гормонів, що утворилися в різних органах, надходять у печінку, де відбувається їх кон'югація із сірчаною і глюкуроновою кислотами з подальшим виведенням з організму у складі сечі і калу.
Інактивація катехоламінів може відбуватися трьома шляхами:
1) перетворення, обумовлені моноамінооксидазою (МАО-шлях);
2) вплив катехолоксиметилтрансферази (КОМТ-шлях);
3) хіноїдне окиснення з утворенням адренохрому.
Метаболічні перетворення тиреоїдних гормонів, що відбуваються переважно в печінці, охоплюють:
1) реакції дейодування;
2) окисне дезамінування і декарбоксилювання залишків аланіну;
3) кон'югацію із сірчаною і глюкуроновою кислотами.
У людини 65—95 % інактивованих метаболітів усіх гормонів виводиться з організму із сечею.
Порушення метаболічних перетворень гормонів можуть обумовлювати розвиток периферичних розладів ендокринної функції. Так, при уповільненні інактивації гормонів збільшується їх вміст у крові, що виявляється ознаками відповідної ендокринної гіперфункції. І навпаки, прискорене перетворення гормонів у неактивні форми супроводжується розвитком ендокринної гіпофункції.
33.14. Які можливі порушення взаємодії гормонів з периферичними клітинами?
Порушення взаємодії гормонів з периферичними клітинами-мішенями, як правило, обумовлене патологією клітинних рецепторів (цumopeuenmopiв). Можливі такі варіанти порушень.
I. Зменшення кількості рецепторів або їхньої спорідненості до гормону (десенсити-зація). При цьому, незважаючи на те що концентрація гормону в крові нормальна або навіть підвищена, розвиваються ознаки ендокринної гіпофункції.
II. Збільшення кількості рецепторів до гормону (сенситизація). Як правило, супроводжується розвитком елементів відповідної ендокринної гіперфункції.
33.15. Як відбувається реалізація біологічної дії гормонів на клітини?
Вплив гормонів на клітини-мішені здійснюється через їхню дію на специфічні
білки, що одержали назву циторецепшорів.
Розрізняють два принципово різних типи циторецепції гормонів.
І. Внутрішньоклітинний тип циторецепції. Лежить в основі механізму дії стероїдних і тирео'їдних гормонів. Пов'язаний з вільним проходженням гормону через/ плазматичну мембрану в клітину, у цитоплазмі якої відбувається взаємодія гормону із внутрішньоклітинними білками-рецепторами.
Ефекторною структурою клітини, на яку впливає утворений комплекс, є ядро, а основним біологічним ефектом — зміни інтенсивності процесів транскрипції, а отже, і синтезу клітинних білків.
II. Мембранний тип циторсцепції. Є основним механізмом дії білково-пептидних гормонів і катехоламінів. При цьому гормони не проникають усередину клітини, а зв'язуються з білками-рецепторами на поверхні плазматичної мембрани. Далі передача регуляторного сигналу з поверхні клітини до її ефекторних структур обумовлена появою в цитоплазмі так званих вторинних посередників, або ме-сенджерів. У результаті виникають швидкі біохімічні ефекти, пов'язані з активацією вже синтезованих ферментів чи інших білків. Серед відомих сьогодні вторинних посередників (месенджерів) такі сполуки:
1) циклічні нуклеотиди — цАМФ, цГМФ;
2) іониСа2+;
3) фосфоліпідні месенджери — діацилгліцерол (ДАГ) та інозитолтрифосфат (ІФ3).
33.16. Яке значення гіпоталамуса в регуляції ендокринних функцій?
Гіпоталамус є відділом центральної нервової системи, де відбувається інтегрування нервових і ендокринних механізмів регуляції. Це пов'язано з тим, що нейрони гіпоталамуса, об'єднані в окремі ядра, є особливими нейронами — нейроендокринними клітинами, здатними синтезувати і вивільняти гормони.
Гіпоталамус анатомічно і функціонально пов'язаний з адено- і нейрогіпофізом. Тому виділяють дві функціональні системи: гіпоталамо-аденогіпофізарну і гіпотала-мо-нейрогіпофізарну.
Діяльність гіпоталамо-аденогіпофізарної системи пов'язана з утворенням у гіпоталамусі гіпофізотропних гормонів - рилізипг-гормоиів. Залежно від функціональних ефектів (активація або пригнічення функції аденогіпофіза) їх поділяють на дві групи: ліберини і статини. До ліберинів (активаторів секреторної функції аденогіпофіза) відносять, зокрема, тиреоліберин, соматоліберин, кортиколіберин, гонадоліберин, пролактоліберин, меланоліберин. Статинами, що пригнічують функції аденогіпофіза, є соматостатин, пролактостатин, меланостатин.
В основі функціонування гіпоталамо-нейрогіпофізарної системи лежить утворення в супраоптичному і паравентрикулярних ядрах гіпоталамуса двох гормонів — вазопресину і окситоцину.
33.17. Які причини можуть викликати порушення функції гіпоталамо-аденогіпофізарної системи?
1. Патогенна дія факторів зовнішнього і внутрішнього середовища, які у нормі регулюють стан гіпоталамо-аденогіпофізарної системи (негативні емоції, біль, психічні порушення та ін.).
2. Ураження відділів центральної нервової системи, які чинять регуляторний вплив на гіпоталамус, - вищих центрів кори великих півкуль головного мозку, структур лімбічної системи, ретикулярної формації.
3. Ураження гіпоталамуса.
4. Ураження аденогіпофіза.
33.18. Які механізми можуть лежати в основі гіпер- і гіпофункції гіпоталамо-аденогіпофізарної системи?
I. Порушення центральної регуляції нейроендокринних зон гіпоталамуса. При збільшенні активуючих і зменшенні гальмівних впливів розвивається гіперфункція гіпоталамо-аденогіпофізарної системи. Зменшення активуючої дії і збільшення гальмівних впливів, навпаки, викликають гіпофункцію цієї системи.
II. Порушення утворення і виділення рилізинг-гормонів клітинами гіпоталамуса. При цьому гіперфункція гіпоталамо-аденогіпофізарної системи розвивається при збільшенні секреції ліберинів і зменшенні утворення статинів, а гіпофункція - при зменшенні виділення ліберинів і збільшенні секреції статинів.
Ш. Порушення утворення і секреції гормонів аденогіпофіза. Залежно від спрямованості цих порушень можуть розвиватися ендокринна гіпер- або гіпофункція.
33.19. Які гормони утворюються в аденогіпофізі? Які біологічне ефекти вони мають?
1. Соматотропний гормон (СТГ, соматотропін, гормон росту). Утворюється ацидофільними клітинами аденогіпофіза. Має ростову і метаболічну активність. Ростова активність пов'язана з дією СТГ на рецептори гепатоцитів, клітин ниркового епітелію та ін., унаслідок чого вивільняються тканинні фактори росту — со-матомедини. Соматомедини спричиняють такі ефекти:
а) збільшують поглинання сульфатів клітинами сполучної тканини та їх включення в хондроїтинсульфат. Це викликає посилений ріст хрящів;
б) збільшують кількість мітозів і стимулюють клітинний поділ;
в) є гуморальними факторами, що беруть участь у розвитку гіпертрофії різних органів.
Метаболічна активність. СТГ має цілий ряд відносно швидких метаболічних ефектів, не пов'язаних з утворенням соматомединів. До них відносять:
а) вплив на вуглеводний обмін. СТГ, стимулюючи а-клітини острівців підшлункової залози, викликає збільшення продукції глюкагону. При тривалій дії великих доз СТГ розвивається інсулінорезистентність (див. розд. 20). Усе це в кінцевому підсумку призводить до гіперглікемії;
б) вплив на жировий обмін. СТГ, активуючи ліполіз у жировій тканині, збільшує вміст вільних жирових кислот у крові, сприяє розвитку жирової інфільтрації печінки, викликає гіперпродукцію кетонових тіл;
в) вплив на білковий обмін. СТГ має анаболічну дію. Він збільшує транспорт амінокислот у клітини і активує біосинтез білків.
2. Адренокортикотропний гормон (АКТГ, кортикотропін). Утворюється в базо-фільних клітинах аденогіпофіза. Має гландотропну і негландотропну дію. Гландотропна дія. АКТГ, впливаючи на кору надниркових залоз (в основному на пучкову зону), активує синтез і секрецію кортикостероїдних гормонів, головним чином глюкокортикоїдів.
Негландотропні ефекти:
а) посилення пігментації шкіри (відтворює дію меланоцитстимулятивного гормону);
б) мобілізація жиру з жирових депо (відтворює ефекти Р-ліпотропіну).
3. Тиреотропний гормон (ТТГ). Утворюється в базофільних клітинах передньої частки гіпофіза. Діє на щитоподібну залозу, стимулюючи утворення і вивільнення тиреоїдних гормонів.
4. Фолікулостимулятивний гормон (ФСГ). Продукується базофільними клітинами аденогіпофіза. Викликає ріст фолікулів у яєчниках і утворення естрогенів.
5. Лютеїнізуючий гормон (ЛГ). Утворюється в тих же клітинах, що і ФСГ. Разом з ним складає групу гонадотропних гормонів. Стимулює утворення жовтого тіла в яєчниках і синтез прогестинів.
6. Пролактин. Гормон еозинофільних клітин аденогіпофіза. Стимулює ріст молочних залоз і секрецію молока.
7. Меланоцитстимулюючий гормон (МСГ). Утворюється залозистими клітинами проміжної (середньої) частки гіпофіза. Стимулює утворення пігменту (меланіну) у пігментних клітинах сполучної тканини.
33.20. Що таке гіпопітуїтаризм? Які існують його форми? Чим він виявляє себе?
Гіпопітуїтаризмом називають гіпофункцію аденогіпофіза.
Розрізняють пангіпопітуїтаризм і парціальний гіпопітуїтаризм.
Пангіпопітуїтаризм — це зменшення утворення всіх гормонів аденогіпофіза. В експерименті моделюють видаленням гіпофіза (гіпофізектомією). Відомі такі клінічні форми пангіпопітуїтаризму:
1) гіпофізарна кахексія Симондса;
2) післяпологовий некроз гіпофіза — синдром Шеєгана;
3) хромофобні аденоми гіпофіза, тобто пухлини, що ростуть із хромофобних клітин. При цьому пухлина здавлює і ушкоджує залозисті клітини аденогіпофіза.
Клінічні прояви пангіпопітуїтаризму пов'язані з дефіцитом гормонів аденогіпофіза і порушенням діяльності периферичних ендокринних залоз (щитоподібної залози, кори надниркових залоз, статевих залоз). Перші симптоми ураження аденогіпофіза з'являються при ушкодженні 70-75 % тканини залози, а для розвитку повної картини пангіпопітуїтаризму необхідне руйнування 90-95 % аденогіпофіза. Синдроми, що розвиваються при тотальному порушенні функцій аденогіпофіза, представлено в таблиці:
Дефіцит гормону аденогіпофіза |
Синдроми, що розвиваються |
стг |
Відставання у рості (у дітей), раннє старіння, схильність до гіпоглікемії |
ФСГ і ЛГ |
Вторинний гіпогонадизм |
ТТГ |
Вторинний гіпотиреоз |
АКТГ |
Вторинний гіпокортицизм |
Пролактин |
Порушення лактації після пологів |
МСГ |
Депігментація |
Парціальний гіпопітуїтаризм — це порушення утворення не всіх, а окремих гормонів аденогіпофіза. Описано такі варіанти парціального гіпопітуїтаризму:
1) гіпофізарний нанізм (карликовість) — дефіцит СТГ;
2) вторинний гіпогонадизм — дефіцит ФСГ і ЛГ;
3) вторинний гіпотиреоз — дефіцит ТТГ;
4) вторинний гіпокортщизм — дефіцит АКТГ
33.21. Що таке гіперпітуїтаризм? Чим він може бути обумовлений?
Гіперпітуїтаризм - це гіперфункція аденогіпофіза. Основною причиною його розвитку є доброякісні пухлини - аденоми ендокринних клітин. Розрізняють дві групи аденом.
I. Еозинофільні аденоми. Розвиваються з ацидофільних клітин аденогіпофіза, що утворюють СТГ. Клінічно гіперфункція СТГ виявляє себе гігантизмом (якщо аденома розвивається у дітей і молодих людей до закриття епіфізарних хрящів) або акромегалією (у дорослих).
Для гігантизму характерне пропорційне збільшення всіх складових частин тіла. Акромегалія виявляється посиленим ростом акральних (кінцевих) ділянок рук, ніг, підборіддя, носа, язика, печінки.
Крім того, розвиваються ознаки підвищеної метаболічної активності СТГ — гіперглікемія, інсулінорезистентність, аж до розвитку метагіпофізарного цукрово-го діабету, жирова інфільтрація печінки.
II. Базофільні аденоми. Ростуть із базофільних клітин аденогіпофіза, найчастіше з тих, що продукують АКТГ. При цьому розвивається хвороба Іценка-Кушинга. Вона характеризується:
а) вторинним гіперкортицизмом (див. запит. 33.40);
б) посиленою пігментацією шкіри (негландотропна дія АКТГ).
Досить рідко бувають пухлини, що продукують інші гормони аденогіпофіза: ТТГ, гонадотропні гормони, пролактин, МСГ.
33.22. Які гормони виділяються при активації гіпоталамо-нейрогіпофізарної системи? Які біологічні ефекти вони мають?
Основним структурним елементом гіпоталамо-нейрогіпофізарної системи є су-праоптичне і паравентрикулярні ядра гіпоталамуса, які у відповідь на надходження інформації від рецепторів, що контролюють показники гомеостазу (осмотичний тиск, об'єм циркулюючої крові, артеріальний тиск), і від вищих нервових центрів продукують вазопресин і окситоцин. Ці гормони по аксонах нейроендокринних клітин спускаються в нейрогіпофіз, звідки надходять у.кров.
Вазопресин (антидіуретичний гормон) виявляє такі впливи:
1) діючи на дистальні звивисті канальці і збірні трубки нирок, посилює реабсорбцію води (антидіуретичний гормон);
2) викликає скорочення гладких м'язів кровоносних судин;
3) посилює глікогеноліз і глюконеогенез у печінці;
4) сприяє консолідації слідів пам'яті і мобілізації інформації, що зберігається (гормон пам'яті);
5) є ендогенним анальгетиком (пригнічує біль).
Окситоцин виявляє такі функціональні ефекти:
1) стимулює виділення молока (лактацію), викликаючи скорочення міоепітеліальних клітин дрібних проток молочних залоз;
2) ініціює й посилює скорочення вагітної матки;
3) погіршує запам'ятовування і мобілізацію інформації (амнестичний гормон).
33.23. Які розлади в організмі виникають при порушенні функції гіпоталамо-нейрогіпофізарної системи ?
1. Синдром надмірної секреції вазопресину. Виникає при пухлинах різних тканин, що утворюють вазопресин (ектопічна продукція), а також при розладах регуляції ендокринної функції гіпоталамуса. Основним його проявом є гіперволемія, що призводить до розвитку стійкої артеріальної гіпертензії.
2. Нецукровий діабет. Розрізняють два його патогенетичних варіанти: центральний (нейрогенний), при якому утворюється мало вазопресину, і нефрогений, в основі якого - нечутливість епітеліальних клітин дистальних відділів нефронів і збірних трубок до дії вазопресину (відсутність або мало У2-рецепторів).
У патогенезі основних проявів нецукрового діабету провідну роль відіграє зменшення факультативноїреабсорбції"води в нирках. Це призводить до поліурії^(добовий діурез зростає до 25 л — сечовиснаження) і зневоднення. Останнє зумовлює спрагу (полідипсію). У декомпенсованому стані зменшується об'єм циркулюючої крові (гіповолемія) і падає артеріальний тиск, розвивається гіпоксія.
3. Зменшення продукції окситоцину. Виявляє себе порушеннями лактації, слабкістю пологової діяльності.
Синдромів, обумовлених збільшенням секреції окситоцину, не описано.
33.24. Які гормони утворюються в корі надниркових залоз?
Кора надниркових залоз гістологічно складається з трьох зон, кожна з яких синтезує свою групу гормонів.
У клубочковій (зовнішній) зоні утворюються мінеролокортикоїди - гормони, що регулюють в основному водно-сольовий обмін. Основним представником цієї групи є альдостерон, інтенсивність секреції якого становить 0,125 мг/добу.
Пучкова (середня) зона є джерелом глюкокортикоїдів — гормонів, що впливають на обмін речовин в організмі. У людини основний глюкокортикоїд — кортизол. Менше значення мають кортизон і кортикостерон. Інтенсивність секреції глюкокортикоїдів становить 20-25 мг/добу.
Клітини сітчастої (внутрішньої) зони синтезують чоловічі статеві гормони -андрогени.
33.25. Як регулюється утворення і секреція мінералокортиШїдів? Назвіть основні біологічні ефекти альдостерону.
В організмі існує три механізми активації! утворення і секреції альдостерону.
1. Ангіотензииний механізм. Пов'язаний з активацією ренін-ангіотензинної системи і утворенням ангіотензинів II і III. Ці пептиди чинять тропну дію на клубочко-
ву зону кори надниркових залоз, викликаючи надходження альдостерону в кров (докладно див. розд. 23).
2. АКТТ-оїіосередкований механізм. Виявляє себе пермісивною дією АКТГ. Інакше кажучи, коли АКТГ нема, то зменшується або зовсім зникає тропна дія ангіо-тензинів II і III на клубочкову зону.
3. Пряма стимуляція секреції альдостерону високими концентраціями іонів калію плазми крові.
Секрецію альдостерону пригнічують передсердний натрійуретичний гормон і дофамін.
Виділяють дві групи біологічних ефектів альдостерону.
I. Ниркові ефекти. Альдостерон, діючи на дистальні звивисті канальці нефронів, збільшує:
а) реабсорбцію іонів натрію;
б) секрецію іонів калію;
в) секрецію іонів водню (ацидогенез).
II. Позаниркові ефекти. Пов'язані із впливом альдостерону на слинні і потові залози, дистальні відділи товстої кишки і спрямовані на затримку натрію в організмі та виведення калію.
33.26. Як здійснюється регуляція утворення і секреції глюкокортикоїдів? Які біологічні ефекти мають ці гормони?
Утворення і секреція глюкокортикоїдів, на відміну від альдостерону, повністю залежать від АКТГ.
Виділяють три ефекти АКТГ при його дії на пучкову зону надниркових залоз:
1) гострий ефект (протягом кількох хвилин). Виявляється зв'язуванням холестеро-лу з цитохромом Р450 і посиленням трансляції наявної інформаційної РНК. Це викликає істотне збільшення інтенсивності утворення стероїдів;
2) підгострий ефект (через десятки годин). Відбувається посилення процесів транскрипції генів, що кодують структуру ферментів стероїдоге-незу. Як наслідок, кількість таких ферментів зростає і, природно, збільшується синтез кортикостероїдів; 3) хронічний ефект (через кілька діб). Виявляється гіпертрофією і гіперплазією пучкової зони кори надниркових залоз.
Прояви біологічної дії глюкокортикоїдів поділяють на дві групи: низькодозові і високодозові.
Низькодозовими називають ефекти фізіологічних концентрацій глюкокортикоїдів (так званих замісних доз). Вони визначаються за типом порушень, що їх усувають замісні дози глюкокортикоїдів після видалення надниркових залоз (адреналектомії).
Високодозовими позначають ефекти великих доз глюкокортикоїдів. Високі концентрації глюкокортикоїдів у крові можуть створюватися при:
а) дії на організм сильних ушкоджувальних агентів (стрес);
б) патологічній гіперфункції кори надниркових залоз (хвороба і синдром Іценка-Кушинга);
в) введенні глюкокортикоїдів та їх фармакологічних аналогів з лікувальною метою.
Вивченню біологічних ефектів глюкокортикоїдів було присвячено роботи одного з учнів О.О. Богомольця - В. П. Комісаренка.
33.27. Назвіть основні низькодозові ефекти глюкокортикоїдів.
ї. Вплив на вуглеводний обмін — посилення глюконеогенезу, зменшення чутливості тканин до інсуліну.
2. Вплив на білковий обмін - активація протеолізу в периферичних тканинах зі збільшенням надходження амінокислот у кров (аміноацидемія), пригнічення біосинтезу білків у тих же тканинах.
3. Пермісивна дія. Глюкокортикоїди необхідні для прояву дії деяких інших гормонів, зокрема, катехоламінів і глюкагону.
4. Тонізуючий вплив на центральну нервову систему і скелетні м'язи.
5. Вплив на процеси росту і диференціювання органів і тканин в ембріогенезі.
33.28. Назвіть основні високодозові ефекти глюкокортикоїдів.
1. Антианаболічна дія - пригнічення біосинтезу білків у різних органах і тканинах. Клінічно виявляється слабкістю м'язів (особливо рук і плечового пояса), пригніченням росту дітей, уповільненням загоєння ран.
2. Гіперглікемічна дія. Обумовлена збільшенням інтенсивності глюконеогенезу в печінці, підвищенням секреції глюкагону, зменшенням використання глюкози периферичними клітинами, зокрема м'язовими, лімфоцитами. Як відповідна реакція розвивається вторинний гіперінсулінізм з наступним виснаженням Β-клітин острівців підшлункової залози і появою вторинної інсулінової недостатності. Як наслідок, розвивається метасшероїднш цукровий діабет.
3. Ліпотропна дія. Глюкокортикоїди стимулюють утворення і дію на жирову тканину гормонів, що мають ліполітичні властивості (адреналін, глюкагон). У результаті збільшується вміст вільних жирових кислот у крові (гіперліпацидемія), відбувається відкладення жиру на тулубі й обличчі. Останнє набуває місяцеподібної форми.
4. Гіпсртензивна дія. Підвищення артеріального тиску почасти пов'язане з тим, що глюкокортикоїди у великих дозах мають деякі властивості мінералокортикоїдів, тобто, збільшуючи реабсорбцію іонів натрію, сприяють підвищенню об'єму циркулюючої крові. Крім того, кортизол підвищує пресорну дію катехоламінів і пригнічує утворення депресорних факторів у нирках, зокрема простагландинів.
5. Ліпокортинові ефекти (див. запит. 33.29).
6. Антимітотична і цитолітична дія. Глюкокортикоїди, пригнічуючи клітинний поділ, порушують процеси фізіологічної і репаративної регенерації, пригнічують ріст пухлин, зменшують утворення соматомединів у печінці. Крім того, вони викликають руйнування (цитоліз) деяких типів клітин, зокрема лімфоцитів. З цією дією пов'язана інволюція вилочкової залози, еозинопенія, протилейкозні ефекти.
7. Імунодепресивна дія. Обумовлена пригніченням проліферації лімфоцитів (глю-кокортикоїди пригнічують алергічні реакції уповільненого типу) і зменшенням утворення антитіл (прояв антианаболічної дії).
8. Ульцерогенна дія. Глюкокортикоїди сприяють розвитку виразок у шлунку і дванадцятипалій кишці. Це обумовлено:
а) зменшенням утворення простагландинів, у результаті чого збільшується шлункова секреція;
б) зменшенням утворення слизу (антианаболічна дія);
в) пригніченням фізіологічної регенерації епітелію травного каналу (антимі-тотична дія).
9. Остеопатична дія. Виявляється розвитком остеопорозу, частими переломами хребців і довгих кісток. У розвитку цих ефектів має значення пригнічення синтезу білкових елементів органічної матриці кісткової тканини.
10. Психотропна дія. Механізми впливу глюкокортикоїдів на психічні процеси людини ще не з'ясовано. Крайнім проявом цього ефекту є розвиток психозів.
33.29. Що такеліпокортинові ефекти глюкокортикоїдів? Щодо них відносять?
Jlinoкopmuнoвuмu називають високодозові ефекти глюкокортикоїдів, пов'язані з утворенням у клітинах білка ліпокортину. Глюкокортикоїди індукують транскрипцію гена, що кодує структуру цього білка (рис. 153).
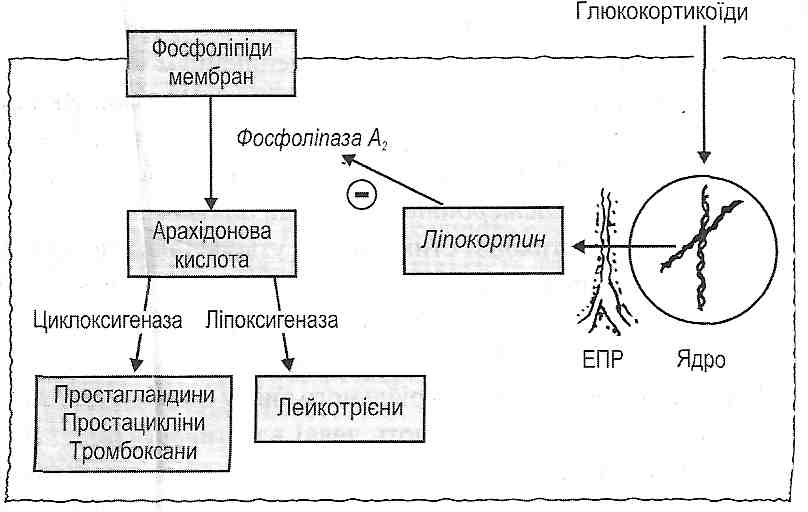
Рис. 153. Ліпокортиновий механізм дії глюкокортикоїдів
Ліпокортин є ендогенним внутрішньоклітинним інгібітором фосфоліпазиАг У разі пригнічення цього ферменту припиняється вивільнення арахідонової кислоти з фосфо-ліпідів мембран, унаслідок чого не утворюються дві важливі групи біологічно активних речовин: продукти циклоксигеназного (простагландини, простациклін, тромбоксани) і ліпоксигеназного (лейкотрієни) шляхів перетворення арахідонової кислоти.
Основні ліпокортинові ефекти:
1. Протизапальна дія. З одного боку, вона пов'язана зі зменшенням утворення лізо-фосфоліпідів, які є сильними детергентами. Унаслідок нього зменшується ушкодження мембран (стабілізація мембран), а отже, і явища альтерації. З другого боку, зменшується утворення медіаторів запалення (простагландинів, лейкотрієнів), у результаті чого менш вираженими стають судинні реакції у вогнищі запалення, підвищення проникності судин, хемотаксис лейкоцитів.
2. Протиалергічна дія. Обумовлена зменшенням утворення медіаторів шіергічних реакцій негайного типу (І типу за Кумбсом і Джеллом), зокрема повільно реагуючої субстанції анафілаксії — речовини, що є лейкотрієном.
3. Жарознижувальна дія. Глюкокортикоїди зменшують утворення простагландинів у центрі терморегуляції в гіпоталамусі, внаслідок чого гарячка не розвивається або, якщо вона вже виникла, зменшується (див. розд. 15).
4. Шлункова гіпєрсекреція. Пов'язана зі зменшенням гальмівної дії простагландинів на секрецію соляної кислоти парієтальними клітинами залоз шлунку.
5. Антиагрегантна дія. Обумовлена зменшенням утворення тромбоксанів — сполук, що викликають агрегацію тромбоцитів.
6. Гіпертензивна дія. Пов'язана зі зменшенням утворення в нирках простагландинів - природних депресорних факторів.
7. Тератогенна дія - поява "вовчої пащі", "заячої губи". У розвитку цих дефектів має значення порушення виходу гідролітичних ферментів з клітин епітелію слизової оболонки піднебіння у довкружну тканину. Це пояснюють тим, що за відсутності активної фосфоліпази А2 мембрана лізосом залишається непроникною для своїх ферментів, вони не можуть вийти за межі клітин, зруйнувати епітелій країв двох половин твердого піднебіння і створити тим самим умови для їх зрощення.
33.30. Які існують порушення функції кори надниркових залоз?
Найчастіше бувають такі порушення:
1) гіпофункція кори надниркових залоз — недостатність цієї залози;
2) гіперфункція клубочкової зони — гіперальдостеронізм;
3) гіперфункція пучкової зони — синдром Іценка-Кушинга;
4) дисфункція кори надниркових залоз - адреногенітальний синдром.
33.31. Як класифікують недостатність кори надниркових залоз? Що може бути причиною гіпофункції цієї ендокринної залози?
I. За етіологією розрізняють первинну і вторинну недостатність кори надниркових залоз. Первинна виникає як результат власне ураження надниркових залоз, вторинна пов'язана або з ураженнями гіпоталамуса (дефіцит кортиколіберину), або з гіпофункцією аденогіпофіза (дефіцит АКТГ).
II. Залежно від характеру порушень виділяють тотальну (порушено утворення всіх гормонів) і парціальну (як правило, спадково обумовлені дефекти синтезу окремих гормонів) недостатність кори надниркових залоз.
III. За клінічним перебігом недостатність кори надниркових залоз може бути гострою і хронічною.
Прикладами гострої недостатності є:
а) стан після адреналектомії (видалення надниркових залоз);
б) крововиливи в надниркові залози, що виникають при сепсисі, особливо при менінгококовій інфекції (синдром Уотерхауза—Фредріксена);
в) синдром відміни глюкокортикоїдних препаратів.
Хронічна недостатність кори надниркових залоз відома під назвою хвороби Ад-дісона (бронзовоїхвороби). Найчастіші її причини:
а) туберкульозне руйнування надниркових залоз;
б) аутоімунне їх ушкодження.
33.32. Назвіть основні прояви недостатності кори надниркових залоз.
I. Прояви, пов'язані з випадінням мшералокортикоХдних функцій кори надниркових залоз:
1) зневоднення (дегідратація). Розвивається внаслідок втрати іонів натрію (зменшується реабсорбція) з наступною втратою води (поліурія);
2) артеріальна гіпотензія. Обумовлена зменшенням об'єму циркулюючої крові (результат зневоднення);
3) гемоконцентрація (згущення крові). Пов'язана із втратою рідини. Призводить до розладів мікроциркуляції й гіпоксії;
4) зменшення ниркового кровообігу (обумовлено падінням артеріального тиску) з порушенням клубочкової фільтрації і розвитком інтоксикації (азотемії);
5) гіперкаліємія. Обумовлена зменшенням канальцевої секреції іонів К+ і виходом їх з ушкоджених клітин. Викликає порушення функції збудливих тканин (див. розд. 23);
6) дистальний канальцевий ацидоз. Пов'язаний з порушенням ацидогенезу в дистальних звивистих канальцях нефронів;
7) шлунково-кишкові порушення (нуцота, блювота, проноси). У їхньому розвитку певне значення мають втрата натрію (осмотична діарея) та інтоксикація. Зазначені порушення мінералокортикоїдної функції кори надниркових залоз без відповідної корекції призводять до смерті.
II. Прояви, обумовлені порушенням глюкокортикоїдної функції кори надниркових залоз. Пов'язані з випадінням низькодозових ефектів кортизолу, а отже, усуваються введенням замісних доз глюкокортикоїдів. До таких проявів відносять:
1) гіпоглікемію, що виникає при голодуванні (непереносність голодування);
2) артеріальну гіпотензію (зменшення пермісивної дії стосовно катехоламінів);
3) зменшення реакції о/сирової тканини на звичайні ліполітичні стимули;
4) зменшення опірності організму до дії різних патогенних факторів — порушення механізмів неспецифічної резистентності (стресу);
5) зменшення здатності виводити воду при водному навантаженні (водне отруєння);
6) м 'язова слабкість і швидка стомлюваність;
7) емоційні розлади (депресія);
8) затримка росту і розвитку дітей;
9) сенсорні порушення — втрата здатності розрізняти окремі відтінки смакових, нюхових, слухових відчуттів;
10) дистрес-синдром у новонароджених (гіаліновий мембраноз). Обумовлений порушенням утворення сурфактантів у легенях, унаслідок чого легені не розправляються при народженні дитини.
ПІ. Прояви, пов'язані з впливом інших гормонів. До них, зокрема, відносять деякі ознаки, обумовлені негландотропною дією АКТГ. Зменшення концентрації глюкокортикоїдів у крові спричиняється до активації певних нейроендокринних структур гіпоталамуса і базофільних клітин аденогіпофіза. Це, у свою чергу, викликає гіперпродукцію АКТГ, який, діючи на пігментні клітини сполучної тканини, стимулює утворення меланіну і обумовлює пігментацію шкіри (звідси назва "бронзова хвороба").
33.33. Що таке пперальдостєронізм? Які існують Його види? Чим він виявляється?
Пперальдостєронізм — це патологічний стан, що виникає в результаті гіперфункції клубочкової зони кори надниркових залоз, яка продукує мінералокортикоїди.
Розрізняють первинний і вторинний гіперальдостєронізм.
Первинний пперальдостєронізм (синдром Конна) виникає в результаті аденоми клубочкової зони, що утворює великі кількості альдостерону. Основні прояви цього захворювання:
1) артеріальна гіпертензія (низькоренінова). Пов'язана зі збільшенням вмісту натрію у крові і стінках кровоносних судин, унаслідок чого підвищується чутливість їхніх гладких м'язів до дії пресорних факторів, зокрема катехоламінів;
2) гіпокаліємія (результат посиленої секреції іонів К+у канальцях нирок). Вона веде до порушень діяльності збудливих органів і тканин (порушення роботи серця, міастенія, парези);
3) негазовий алкалоз. Пов'язаний з посиленням ацидогенезу в дистальних звивистих канальцях нефронів;
4) поліурія. Виникає як наслідок втрати чутливості епітелію ниркових канальців до дії вазопресину (антидіуретичного гормону). Цим, зокрема, пояснюють той факт, що при первинному гіперальдостеронізмі об'єм циркулюючої крові не зростає і набряки не розвиваються.
Вторинний гіперальдостеронізм є наслідком активації ренін-ангіотензинної системи (див. розд. 23), у процесі якої утворюються ангіотензини П і III, що діють на кору надниркових залоз. Цей стан виявляється:
а) артеріальною гіпертензією (високореніновою);
б) набряками (гіперволемічними);
в) гіпокаліємією;
г) негазовим алкалозом.
33.34. Які існують клінічні форми гіперфункції пучкової зони кори надниркових залоз? Чим вони виявляються?
Існує дві клінічні форми гіперфункції пучкової зони кори надниркових залоз. 1. Хвороба Іценка-Кушинга - базофільна аденома передньої частки гіпофіза.
2. Синдром Іценка-Кушинга:
а) пухлинний - аденома пучкової зони кори надниркових залоз;
б) ектопічна продукція АКТГ деякими злоякісними пухлинами (наприклад, рак легень);
в) ятрогенний — введення глюкокортикоїдів в організм з лікувальною метою. Гіперфункція пучкової зони кори надниркових залоз виявляє себе утворенням і
секрецією великих кількостей глюкокортикоїдів, високодозові ефекти яких і визначають клінічну картину (див. запит. 33.28). Для цього стану характерні:
1) артеріальна гіпертензія;
2) гіперглікемія — метастероїдшй іскровий діабет;
3) ожиріння;
4) інфекційні захворювання з мінімальними ознаками запалення або без них;
5) шлункова гіперсекреція і утворення виразок у шлунку і дванадцятипалій кишці;
6) остеопороз;
7) м'язова слабкість;
8) уповільнене загоєння ран.
33.35. Що таке адреногенітальний синдром? Які існують його варіанти?
Адреногенітальний синдром є відображенням дисфункції кори надниркових залоз. Він виникає в результаті спадково обумовленої блокади синтезу кортизолу і
посиленого утворення андрогенів із загальних проміжних продуктів (рис. 154).
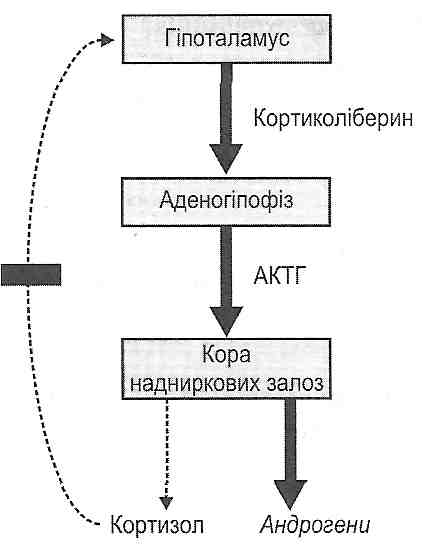
Рис. 154. Схема розвитку адреногенітального синдрому
Залежно від рівня блокади синтезу кортизолу розрізняють три варіанти адреногенітального синдрому.
I. Порушення ранніх етапів синтезу— дефіцит глюкокортикоїдів, мінералокортикоїдів і гіперпродукція андрогенів. Прояви: ознаки недостатності глюко- і мінералокортикоїд-ної функцій кори надниркових залоз, ознаки раннього статевого дозрівання в осіб чоловічої статі, вірилізація у жінок (поява чоловічих статевих ознак).
II. Порушення проміжних етапів- дефіцит глюкокортикоїдів, надлишок андрогенів, утворення мінералокортикоїдів не порушено (класичний адреногенітальний синдром). Прояви ті ж самі, що і в першому випадку, тільки без ознак недостатності мінералокор-тикоїдної функції.
III. Порушення на кінцевих етапах синтезу кортизолу — дефіцит глюкокортикоїдів, гіперпродукція андрогенів і мінералокортикоїдів. До проявів класичного адреногенітального синдрому додаються ознаки гіперальдостеронізму.
33.36. Які гормони утворюються мозковою речовиною надниркових залоз? Які механізми їхньої дії та біологічні ефекти?
У мозковій речовині надниркових залоз утворюються два гормони: адреналін і норадреналін (катехоламіни). У людини 70-90 % становить адреналін і 10-30 % - но-радреналін.
Джерелами катехоламінів є хромафіннІ клітини, що являють собою видозмінені клітини симпатичних гангліїв. Регуляція їх секреторної активності здійснюється нервовими механізмами — прегангліонарними симпатичними нейронами. Вивільнення катехоламінів у кров відбувається при активації симпатичної нервової системи. Функціональний синергізм цієї частини вегетативної нервової системи і мозкової речовини надниркових залоз дає підстави розглядати їх як одне ціле. Звідси термін -симпатоадреналова система.
Катехоламіни є гормонами з мембранним типом циторецепції. їхній вплив на периферичні клітини здійснюється через:
1) а1-адренорецептори. Вони є в кровоносних судинах, матці, гладких м'язах кишок, м'язах зіниці ока. Виникнення біологічних ефектів пов'язано з активацією фос-фоліпази С, утворенням інозитолтрифосфату і збільшенням надходження в цитоплазму іонів кальцію;
2) а2-адренорецептори. їх виявляють у тромбоцитах, пресинаптичних терміналях симпатичних і парасимпатичних нервів. Взаємодія катехоламінів з ос, адреноре-цепторами викликає зменшення активності аденілатциклази і вмісту цАМФ у клітинах, з чим, власне, і пов'язані біологічні ефекти, що виникають;
3) р1-адренорецептори. Є в серці, гладких м'язах травного каналу, жировій тканині, печінці. Реалізація дії катехоламінів відбувається через активацію аденілатциклази і збільшення вмісту цАМФ;
4) р2-адренорецептори. Виявлені в кровоносних судинах, бронхах, матці. їх взаємодія з катехоламінами супроводжується активацією аденілатциклази і утворенням цАМФ.
Дія катехоламінів на зазначені рецептори викликає дві групи змін.
І. Функціональні ефекти. Пов'язані із впливом адреналіну і норадреналіну на м'язові органи і тканини. Головними з них є:
1) кардіотонічна дія (дія на серце). Катехоламіни, зумовлюючи позитивні іно-, хроно-, батмо- і дромотропні ефекти, збільшують силу і частоту серцевих скорочень, підвищують збудливість і провідність у серці;
2) пресорна дія (дія на кровоносні судини). Впливаючи на aj-адренорецепто-ри, катехоламіни викликають звуження судин, а діючи на Р2-адренорецеп-тори, - їхнє розширення. Оскільки загальна кількість aj-адренорецепто-рів у судинах значно перевищує число р2-адренорецепторів, то загальним ефектом є збільшення периферичного судинного опору і підвищення артеріального тиску;
3) бронхорозширювальна дія. Обумовлена розслабленням гладких м'язів бронхів у зв'язку з активацією Р2-адренорецепторів.
II. Метаболічні ефекти. Обумовлені впливом на Р,-адренорецептори. Серед них:
1) гіперглікемічна дія. Пов'язана з активацією фосфоролітичного розщеплення глікогену в печінці;
2) ліполітична дія. Обумовлена активацією гормончутливої тригліцеридліпа-зи. Виявляється збільшенням вмісту в крові вільних жирових кислот (гіпер-ліпацидемією);
3) теплоутворювальна дія. Пов'язана з активацією окиснення в мітохондріях бурої жирової тканини, яке тут відбувається без фосфорування, тобто без утворення АТФ. У результаті цього істотно збільшується теплоутворення (нескорочувальний термогенез).
33.37. Які існують порушення функції мозкової речовини надниркових залоз?
I. Гіпофункціональні стани. Бувають рідко, мабуть, у зв'язку з тим, що функції мозкової речовини надниркових залоз можуть брати на себе хромафінні клітини, розташовані за межами цих ендокринних органів. Описано спадкове ауто-сомно-рецесивне захворювання — сімейна дизавтономія (синдром Райлі—Даї). Сутність генетичного дефекту полягає в порушенні структури або в повній відсутності дофамін-р-гідроксилази - ферменту, що перетворює дофамін у норадреналін.
II. Гіперфункціональні стани. Виникають, зокрема, при пухлині хромафінних клітин— феохромоцитомі. Виявляються артеріальною гіпертензією, тахікардією, гіперглікемією, гіперліпацидемією, гіпертермією.
33.38. Що таке стрес і загальний адаптаційний синдром?
Стрес - це стан напруження неспецифічних адаптаційних механізмів, що виникає у разі дії на організм надмірних за силою або патогенних факторів (рис. 155).
Клінічно стрес виявляє себе комплексом структурних, функціональних і біохімічних змін, що отримали назву загального адаптаційного синдрому.
Термін "стрес" уперше ввів у вжиток Г. Сельє у 1936 р.
33.39. Які морфологічні ознаки характерні для загального адаптаційного синдрому?
Дія на організм численних патогенних факторів, незалежно від їхніх властивостей і походження, дає стандартну відповідь — так звану морфологічну тріаду:
1) гіпертрофію кори надниркових залоз;
2) інволюцію тиміко-лімфоцитарної системи (атрофію вилочкової залози і лімфатичних вузлів);
3) утворення виразок і ерозій у шлунку і в кишках.
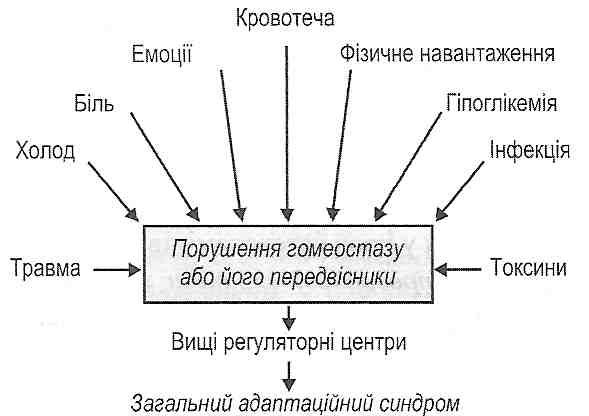
Рис. 155. Фактори, що викликають стрес
33.40. Які стадії виділяють у розвитку стресу?
I. Стадія тривоги:
а) підстадія шоку. Характеризується короткочасним зменшенням резистентності до патогенного фактора;
б) підстадія контршоку. Опірність організму спочатку поновлюється, а потім підвищується.
II. Стадія резистентності. Характеризується стійким і тривалим збільшенням опірності організму як до фактора, що викликав стрес, так і до інших патогенних агентів.
III. Стадія виснаження. Настає при дуже інтенсивній або тривалій дії патогенного фактора, а також в умовах функціональної слабкості адаптаційних механізмів. Супроводжується зменшенням резистентності організму до патогенних впливів (рис. 156). '
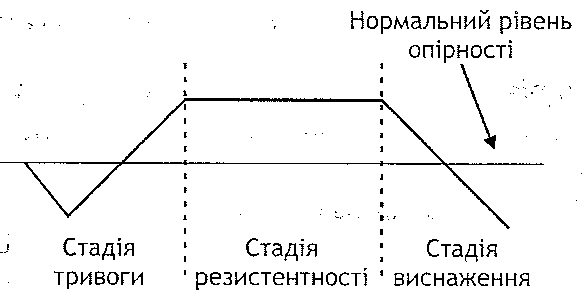
Рис. 156. Стадії загального адаптаційного синдрому
33.41. Які механізми беруть участь у реалізації стресу?
Численні ініціатори стресу (cmpecopu) - травма, холод, біль, емоції, кровотеча, фізичне навантаження, гіпоглікемія, інфекції та ін. — через порушення гомеостазу або передвісників такого порушення викликають збудження вищих нервових регупя-
торних центрів і пов'язане з цим вивільнення великої кількості гормонів. При цьому велике значення мають такі процеси.
I. Активація системи гіпоталамус-аденогіпофіз. Як наслідок відбувається виділення АКТГ, СТГ, ТТГ, які відповідно стимулюють секрецію глюкокортикоїдів, соматомединів, тиреоїдних гормонів.
II. Активація вегетативної нервової системи (симпатичної і парасимпатичної) супроводжується надходженням у кров катехоламінів, інсуліну, глюкагону.
III. Активація альдостерон-вазопресинової системи веде до збільшення вмісту в крові ангіотензинів, альдостерону, вазопресину (АДГ) (рис. 157).
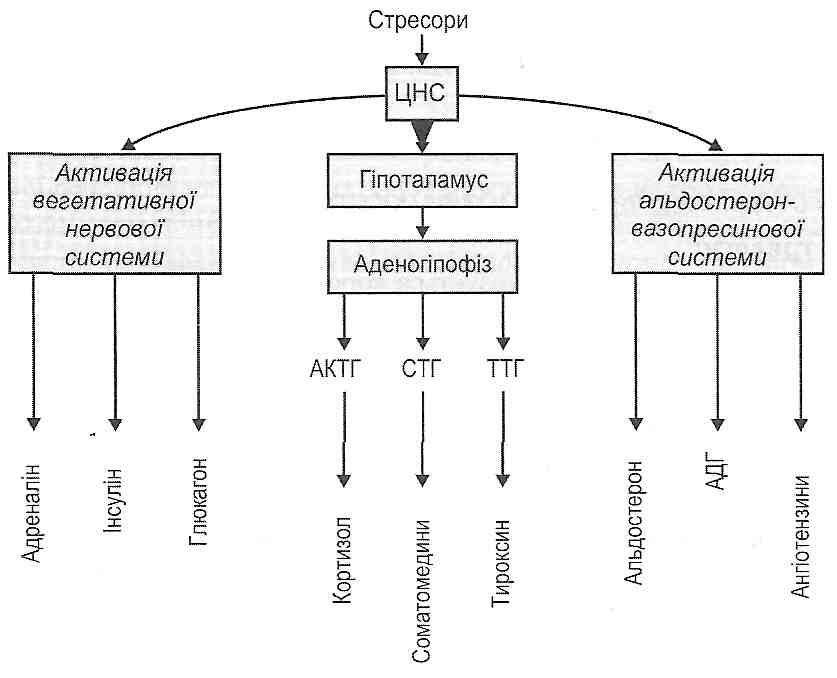
Рис. 157. У реалізації стресу беруть участь різні гормони
33.42. Яка участь різних гормонів у розвитку стресу?
Гормони, що виділяються під час стресу, визначають розвиток трьох послідовних фаз цієї реакції.
І. Гостра фаза. Гормони, що вивільнюються в цю фазу, забезпечують захист від падіння артеріального тиску і об 'ему циркулюючої крові. Це досягається збільшенням загального периферичного опору і збереженням води в організмі. Зазначені реакції пов'язані з посиленим надходженням кров катехоламінів, глюкокортикоїдів, ангіотензину II, альдостерону, вазопресину. її. Підгостра фаза. Характеризується мобілізацією ресурсів для енергетичного й пластичного забезпечення систем, що здійснюють адаптацію. Цьому слугує перерозподіл зазначених ресурсів між активно функціонуючими органами (серце, головний мозок) і структурами, що перебувають у відносному спокої (скелетні
м'язи, травний канал, лімфоїдна і жирова тканини). Метаболічні зміни в цю фазу обумовлені збільшенням секреції катехоламінів, глюкокортикоїдів, глюкагону і зменшенням виділення інсуліну. Наведені вище контрінсулярні гормони, посилюючи глікогеноліз, глюконеогенез, ліполіз і протеоліз, викликають збільшення вмісту в крові глюкози, амінокислот, вільних жирових кислот. III. Фаза довгострокової адаптації. Характеризується структурними змінами (гіпертрофією, гіперплазією) органів і тканин, що забезпечують адаптацію і перебувають у стані гіперфункції. У її реалізації беруть участь інсулін, СТГ, соматомедини, тиреоїдні гормони, фактори росту - істинні та тканинні гормони, що активують анаболічні процеси і формують так званий структурний слід адаптації.
33.43. Що таке "хвороби адаптації"? Які захворювання до них відносять ?
Хвороби адаптації- це захворювання, у розвитку яких провідна роль належить надмірному стресу і так званим стресорним механізмам ушкодження.
При великій інтенсивності і тривалості стрес із механізму адаптації може перетворитися у механізм патогенезу.
До хвороб адаптації відносять:
а) психосоматичні захворювання (ішемічну хворобу серця, гіпертонічну хворобу, виразкову хворобу шлунку і дванадцятипалої кишки);
б) хвороби обміну речовин (цукровий діабет);
в) алергічні і запальні захворювання (бронхіальну астму, ревматизм).
33.44. Назвіть гормони щитоподібної залози. Як здійснюється регуляція їх утворення і секреції?
У щитоподібній залозі утворюються тиреоїдні гормони - тироксин (Т4) і три-йодтиронін (Т3). Крім того, С-клітинами (парафолікулярними) синтезується кольци-тонін, який бере участь у регуляції фосфорно-кальцієвого обміну (див. розд. 24).
Вважають, що тироксин є прогормоном трийодтироніну. На думку про це наводять такі факти:
а) Т3 в 5 разів активніший, ніж Т4;
б) ефекти Т4 розвиваються після більш тривалого латентного періоду, якщо порівнювати зі змінами, що їх викликає Т ;
в) Т4 може перетворюватися в Т3 у периферичних тканинах завдяки процесам дейо-дування.
Регуляція утворення і секреції тиреоїдних гормонів здійснюється системою гі-поталамус-аденогіпофІз за схемою: гіпоталамус → тиреоліберин → аденогіпофіз → тиреотропний гормон (ТТГ) → щитоподібна залоза. ТТГ, діючи на щитоподібну залозу, викликає такі ефекти:
а) посилює захоплення і введення йоду в органічні сполуки;
б) посилює протеоліз депонованого тиреоглобуліну;
в) посилює секрецію Т3 і Т4;
г) у разі тривалої дії викликає гіпертрофію й гіперплазію щитоподібної залози.
33.45. Які механізми діїй біологічні ефекти тиреоїдних гормонів?
Тиреоїдні гормони є гормонами з вігутрішньоклітинним типом циторецепції. Установлено три внутрішньоклітинні мішені для їхньої дії: плазматична мембрана, мітохондрії, ядро.
На плазматичній мембрані чутливих до тиреоїдних гормонів клітин виявлено високоафінні ділянки зв'язування трийодтироніну (Т ). Результатом взаємодії Т3 з такими ділянками є стимуляція транспорту амінокислот. Відповідь виникає дуже швидко і не вимагає синтезу відповідних інформаційної РНК і білка.
У мітохондргях Т3 зв'язується з ферментом внутрішньої мембрани — транслока-зою аденінових нуклеотидів - і активує його. Наслідком цього є посилення транспорту АДФ із цитоплазми в мітохондрії. В результаті концентрація АДФ у мітохондріях зростає, що викликає збільшення інтенсивності біологічного окиснення (принцип акцепторного контролю).
Ядро є основною внутрішньоклітинною мішенню для Т3. Це визначає довгострокові ефекти тиреоїдних гормонів. При зв'язуванні Т3 (у меншій мірі Т4) з ядерними рецепторами відбувається індукція транскрипції і синтезу цілого ряду функціонально важливих білків. Серед них:
а) Na-K-АТФ-аза плазматичних мембран;
б) ферменти ліпогенезу (зокрема, НАДФ-малатдегідрогеназа);
в) ферменти мітохондрій (а-гліцерофосфатдегідрогеназа);
г) білкові компоненти р-адренорецепторів.
Усі біологічні ефекти, обумовлені дією тиреоїдних гормонів на клітини, можна поділити на три групи.
I. Анаболічна дія — вплив на ріст і диференціювання тканин. Є низькодозовим ефектом. Відсутність цього ефекту або його зменшення виявляється при гіпотиреозі.
II. Метаболічні ефекти - збільшення інтенсивності катаболічних процесів (окиснення, ліполізу). Будучи високодозовими, вони виявляються в умовах гіпертиреозу.
III. Сенсибілізуючі ефекти - збільшення чутливості клітин до дії інших гормонів, зокрема естрогенів і катехоламінів. Щодо останніх сенсибілізуючий вплив тиреоїдних гормонів пов'язаний зі збільшенням кількості р-адренорецепторів на поверхні клітин.
33.46. Назвіть основні причини гіпотиреозу.
В основі розвитку гіпофункції щитоподібної залози - гіпотиреозу - можуть лежати такі причини.
I. Центральні порушення: зменшення утворення і секреціїтиреоліберину і тире-отропного гормону (ТТГ) у зв'язку з розладами діяльності гіпоталамуса і адено-гіпофіза (вторинний гіпотиреоз).
II. Залозисті порушення, що призводять до розвитку первинного гіпотиреозу:
а) руйнування тканини залози, наприклад, радіоактивним йодом;
б) дефіцит йоду в питній воді та їжі — ендемічний зоб;
в) аутоімунне ушкодження клітин залози — аутоімунний тиреоїдит Хашимото;
г) уроджені порушення - гіпо- і аплазія щитоподібної залози, ензимопатії.
III. Периферичні порушення:
а) нечутливість периферичних клітин до дії тиреоїдних гормонів;
б) підвищене зв'язування тиреоїдних гормонів білками плазми крові;
в) посилений їх метаболізм у печінці.
33.47. Який патогенез основних проявів гіпотиреозу?
У розвитку проявів гіпотиреозу мають значення такі механізми.
I. Порушення росту і диференціювання тканий. При цьому важливу роль віді-
грають випадіння низькодозових (анаболічних) ефектів тиреоїдних гормонів і
зменшення секреції СТГ.
Оскільки тиреоїдні гормони потрібні для нормального процесу енхондральної
осифікації на межі діафіза й епіфіза, то в умовах гіпотиреозу порушується ріст
кісток у довжину. При цьому періостальний ріст кісток зберігається, у зв'язку з
чим вони стають товстими. Розвивається комплекс змін скелета — гіпотиреоїдна
карликовість.
Поряд з цим затримується і розумовий розвиток — поступово виникає кретинізм.
II. Зменшення теплоутворювальної дії тиреоїдних гормонів, що виявляється:
а) зменшенням основного обміну (падіння на 20-40 %). Воно обумовлено зниженням інтенсивності біологічного окиснення в мітохондріях і функціональної активності збудливих тканин;
б) зменшенням теплопродукції, у зв'язку з чим падає температура тіла;
в) поганою адаптацією до холоду при збереженні адаптації до високої температури;
г) гіпофагією - малим споживанням енергетичних ресурсів.
ІІІ. Зменшення функціональної активності збудливих тканий. Пов'язане з падінням активності Na-K-АТФ-аз і змінами процесів активного транспорту іонів. З другого боку, має значення зменшення чутливості тканин до катехоламінів, що обумовлено зменшенням кількості р-адренорецепторів на клітинах. Функціональні зміни збудливих органів і тканин виявляються:
а) порушеннями діяльності центральної нервової системи — уповільненням розумової діяльності, млявістю, загальмованістю, сонливістю і т. п.;
б) зменшенням функціональної активності скелетних м'язів — слабкістю, зменшенням тонусу, швидкою стомлюваністю;
в) порушеннями діяльності серцево-судинної системи - брадикардією, зменшенням хвилинного об'єму серця, падінням артеріального тиску;
г) зменшенням скорочувальної функції гладких м'язів кишок - закрепами;
ґ) порушеннями процесів всмоктування і екскреції. Зменшення всмоктування глюкози в кишках призводить до гіпоглікемії, а порушення екскреції холес-теролу в складі жовчі — до гіперхолестеролемії і атеросклерозу.
IV. Порушення з нез'ясованими механізмами розвитку. До них відносять слизовий набряк —мікседему. Характеризується збільшенням у тканинах кількості глі-козаміногліканів, що зв'язують воду; потовщенням шкіри, одутлим обличчям. Існує гіпотеза, згідно з якою мікседема є наслідком дії на сполучну тканину тире-отропного гормону (ТТГ), кількість якого при власне залозистій і периферичній формах гіпотиреозу істотно зростає.
33.48, Назвіть основні причини гіпертиреозу.
В основі розвитку гіпертиреозу можуть бути такі причини.
I. Центральні порушення - збільшення секреції тиреоліберину і тиреотропного гормону (ТТГ) при гіперфункції гіпоталамуса і аденогіпофіза (вторинний гіпертиреоз).
II. Власне залозисті порушення (первинний гіпертиреоз). Найпоширенішими клінічними формами первинного гіпертиреозу є дифузний токсичний зоб (хвороба Гревса, базедова хвороба) та аденоми щитоподібної залози.
Вважають, що дифузний токсичний зоб є аутоімунним захворюванням, у виникненні якого мають значення ТТГ-міметичні антитіла, тобто антитіла, що імітують дію ТТГ при взаємодії з поверхневими антигенами клітин щитоподібної залози (V тип алергічних реакцій за Кумбсом і Джеллом). На роль таких антитіл претендують LATS (long acting thyroid stimulator) i TSI (thyroid stimulating immunoglobuline), здатні взаємодіяти із ТТГ-рецепторами. Оскільки ці антитіла не мають місць зв'язування комплементу, то клітини щитоподібної залози не ушкоджуються, а активно функціонують.
III. Периферичні порушення:
а) збільшення чутливості клітин до дії Т3 і Т4;
б) зменшення зв'язування тиреоїдних гормонів транспортними білками;
в) уповільнення метаболізму тиреоїдних гормонів у печінці при її недостатності.
33.49. Який патогенез основних проявів гіпертиреозу?
У патогенезі проявів гіпертиреозу мають значення такі механізми. І. Антианаболічні ефекти. Є високодозовими ефектами тиреоїдних гормонів. До них відносять:
а) затримку росту;
б) атрофію м'язів і слабкість;
в) схуднення;
г) негативний азотистий баланс.
II. Посилення теплоутворювальної дії тиреоїдних гормонів. Воно виявляється:
а) збільшенням основного обміну;
б) збільшенням теплоутворення й підвищенням температури тіла;
в) збереженою адаптацією до холоду і поганою — до високої температури;
г) гіперфагією - підвищеним споживанням енергетичних ресурсів.
III. Збільшення функціональної активності збудливих тканин. Пов'язане з підвищенням активності Na-K-насосів клітинних мембран і збільшенням чутливості клітин до катехоламінів. Цим, зокрема, обумовлені такі прояви гіпертиреозу:
а) порушення діяльності центральної нервової системи — прискорення психічних процесів, занепокоєння, збудження, безсоння;
б) постійна спонтанна скорочувальна активність волокон скелетних м'язів -фібрилярні посмикування, тремор. З цим пов'язана м'язова слабкість, стомлюваність;
в) зміни діяльності серцево-судинної системи - тахікардія, збільшення хвилинного об'єму серця, артеріального тиску;
г) підвищення скорочувальної активності гладких м'язів кишок — проноси;
ґ) збільшення інтенсивності процесів усмоктування і екскреції. З цим, зокрема, пов'язана гіперглікемія і гіпохолестеролемія.
IV. Катехоламінові ефекти. Обумовлені підвищенням чутливості клітин до дії катехоламінів у зв'язку зі збільшенням на клітинній поверхні кількості )3-адреноре-цепторів.
У клініці гіпертиреозу велике значення мають функціональні ефекти катехоламінів, зокрема, їхній вплив на серцево-судинну систему (див. запит. 33.36), і метаболічні зміни. З останніми пов'язані такі порушення:
а) посилення глікогенолізу в печінці → гіперглікемія → гіперфункція β-клітин острівців підшлункової залози з наступним їх виснаженням → тиреогдний цукровий діабет',
б) посилення ліполізу в жировій тканині → гіперліпацидемія → збільшення кетогенезу в печінці → метаболічний ацидоз;
в) активація не пов'язаного з фосфоруванням окиснення в бурій жировій тка^ нині → збільшення теплопродукції —> підвищення температури тіла і основного обміну.
V. Порушення з нез'ясованими механізмами розвитку- орбітопатія і двосто-
ронній екзофтальм (витрішкуватість). В основі їхнього розвитку — набряк і лім-фоїдна інфільтрація м'язів очного яблука і ретробульбарної тканини. Ці зміни не залежать від концентрації тиреоїдних гормонів у плазмі крові і від рівня ТТГ-мі-метичних антитіл (LATS, TSI). Вважали, що в умовах гіпертиреозу виділяється особливий екзофтальмічний фактор, однак його дотепер не виявлено.
33.50. Що таке зоб? Які його види виділяють? Який патогенез ендемічного зоба?
Зобом називають видиме збільшення щитоподібної залози. Виділяють три види зоба.
1. Дифузний токсичний зоб- гіпертиреоїдний (хвороба Гревса, базедова хвороба). Характеризується ознаками гіперфункції щитоподібної залози.
2. Спорадичний зоб — еутиреоїдний. Збільшення маси щитоподібної залози не супроводжується вираженими змінами її функціональної активності.
3. Ендемічний зоб - гіпотиреоїдний. Виявляє себе клінікою гіпофункції щитоподібної залози.
Причиною ендемічного зоба є недостатній вміст йоду в питній воді і продуктах харчування, що пов'язано з особливостями ґрунту і підземних вод у певних регіонах і місцевостях земної кулі. Дефіцит йоду призводить до порушення утворення тире-оїдних гормонів, вміст яких у крові зменшується. Це викликає посилення продукції тиреоліберину і тиреотропного гормону (ТТГ). Останній, впливаючи на тканину щитоподібної залози, стимулює процеси гіпертрофії і гіперплазії — розвивається зоб. Оскільки збільшення залози не усуває дефіцит тиреоїдних гормонів (причину — недостатність йоду - не ліквідовано), то підвищена продукція ТТГ зберігається і триває його дія на тканину залози: зоб прогресує.
33.51. Як здійснюється регуляція утворення і секреції паратирину прищитоподібними залозами? Назвіть основні біологічні ефекти
паратгормону.
Утворення і секреція паратирину (паратгормону) регулюється вмістом іонів кальцію в плазмі крові. Секреція цього гормону зростає при зменшенні концентрації іонів Са2+ у плазмі, і навпаки, зменшується при збільшенні вмісту цих іонів. Крім того, вивільнення паратирину в кров пригнічує 24,25-(ОН)2 вітамін D, що утворюється в нирках (див. розд. 24).
Біологічні ефекти паратирину (докладно див. розд. 24):
1. Дія на кісткову тканину - активація функції остеокластів.
2. Дія на нирки — пригнічення реабсорбції фосфату.
3. Активація перетворення в нирках вітаміну D у його гормональну форму— 1,25-(ОН)2 вітамін D.
Наслідком усіх зазначених ефектів є збільшення концентрації іонів кальцію в плазмі крові.
33.52. Які причини і чим виявляє себе гіпопаратиреоз?
Причини гіпопаратиреозу -гіпофункції прищитоподібних залоз:
1) випадкове ушкодження або видалення прищитоподібних залоз при операціях на щитоподібній залозі;
2) ушкодження прищитоподібних залоз при лікуванні радіоактивним йодом хвороб щитоподібної залози;
3) аутоімунні ушкодження прищитоподібних залоз;
4) уроджене недорозвинення прищитоподібних залоз;
5) нечутливість клітин-мішеней до дії паратирину — псевдогіпопаратиреоз.
Основним проявом гіпопаратиреозу є гіпокальціємія. Вона обумовлює розвиток паратиреопривної тетанії, що виявляється різким підвищенням нервово-м'язової збудливості, множинними фібрилярними скороченнями м'язів усього тіла. Потім
виникають напади клонічних судом, що переходять у тонічні. Судомні скорочення можуть поширюватися і на внутрішні органи (пілороспазм, ларингоспазм). Під час одного з таких нападів настає смерть.
При хронічному гіпопаратиреозі у тварин розвивається клінічна картина парати-реопривної кахексії. Вона характеризується схудненням, анорексією, підвищеною нервово-м'язовою збудливістю, диспепсією й різноманітними трофічними порушеннями.
33.53. Які причини і чим виявляє себе гіперпаратиреоз?
Причини гіперпаратиреозу — гіперфункції прищитоподібних залоз:
1) пухлина — аденома прищитоподібної залози;
2) гіперфункція прищитоподібних залоз, обумовлена зменшенням чутливості їхніх ендокринних клітин до іонів кальцію, — порушення регуляції за принципом негативного зворотного зв'язку.
Гіперпаратиреоз виявляється двома групами пов'язаних між собою змін.
І. Порушення кісткової тканини- генералізована фіброзна остеодистрофія. Обумовлена підвищенням активності остеокластів і пригніченням функції остеобластів. Виявляється болем у кістках і суглобах, розм'якшенням кісток, різкою деформацією скелета. Розвивається демінералізація кісткової тканини (остеомаляція), що обумовлює підвищення вмісту іонів кальцію в плазмі крові — гіпер-кальціємію.
II. Гіперкальціємія. З нею пов'язані:
а) кальцифікація м'яких тканин (нирок, судин, легень). У важких випадках розвивається ниркова недостатність;
б) утворення кальцієвих каменів у нирках;
в) порушення збудливості нервової системи і м'язів - м'язова слабкість, депресія, порушення пам'яті;
г) артеріальна гіпертензія;
ґ) посилення шлункової секреції.
33.54. Як здійснюється регуляція утворення і секреції чоловічих статевих гормонів? Назвіть їх основні біологічні ефекти.
Ендокринні клітини сім'яників (інтерстиціальні клітини, або клітини Лейдига) синтезують і секретують чоловічі статеві гормони - андрогени, серед яких основним є тестостерон.
Регуляція утворення андрогенів здійснюється за участю системи гіпоталамус-аденогіпофіз за схемою: гіпоталамус → гонадоліберин → аденогіпофіз → лютеї-нізуючий гормон → інтерстиціальні клітини сім'яників → тестостерон → клітини Сертолі сім'яників → інгібін → гальмування функціональної активності відповідних утворень гіпоталамуса і аденогіпофіза → зменшення утворення гонадоліберину і лютеїнізуючого гормону → зменшення синтезу і секреції андрогенів → активація гіпоталамуса і т. д.
Основні біологічні ефекти тестостерону: 1. Ембріональне диференціювання. Андрогени забезпечують диференціювання статевих залоз і статевих органів у період ембріонального розвитку. Утворюючись
під впливом хоріонічного гонадотропіну (у період між 6 і 16 тижнями), тестостерон активує формування чоловічих статевих органів з вольфової протоки, урогеніталь-ного синуса і горбка, одночасно пригнічуючи розвиток жіночих статевих органів. Крім того, андрогени забезпечують диференціювання структур центральної нервової системи "за чоловічим типом", визначаючи надалі чоловічий тип статевої поведінки.
2. Статеве дозрівання і розвиток вторинних статевих ознак в особин чоловічої статі. Оскільки андрогени стимулюють діяльність сальних залоз, то в пубертатному періоді шкіра стає жирною, часто відбувається інфікування сальних залоз - з'являються вугрі.
3. Регуляція сперматогенезу. Тестостерон активує діяльність міоепітеліальних клітин сім'яників (клітин Сертолі) та епітеліальних клітин проток, які у свою чергу здійснюють вплив на інтенсивність поділу статевих клітин та їхнє дозрівання. Така дія тестостерону не пов'язана з надходженням його в кров, а є місцевою.
4. Регуляція статевої поведінки. Андрогени мають стосунок до формування статевого потягу (лібідо) і здатності до копуляції. Це пов'язано з тим, що тестостерон впливає на преоптичну зону гіпоталамуса (центр статевої поведінки) і полегшує спинномозкові рефлекси, важливі для копуляції. Тимчасова недостатність андрогенів у період статевого диференціювання і розвитку мозку призводить до формування в подальшому гомосексуальної орієнтації.
5. Анаболічна дія. Тестостерон активує процеси білкового синтезу. Саме з цим пов'язаний вплив андрогенів на ріст скелета і м'язів (посилення росту кісток у довжину, збільшення м'язової маси).
33.55. Які причини й основні прояви чоловічого гіпогонадизму?
Чоловічий гіпогонадизм — це гормональна недостатність чоловічих статевих за-
лоз. Його причинами можуть бути:
ї. Центральні (дисрегуляторні) порушення-зменшення утворення гонадолі-берину в гіпоталамусі, лютеїнізуючого гормону в аденогіпофізі. Це може бути пов'язано з ураженням зазначених структур, а також з гіперфункцією епіфіза, у разі якої збільшення утворення мелатоніну супроводжується пригніченням синтезу гонадотропних гормонів.
II. Власне залозисті порушення:
а) кастрація — видалення сім'яників;
б) фіброз яєчок після деяких вірусних інфекційних захворювань, ускладнених орхітом (наприклад, після епідемічного паротиту);
в) порушення розвитку яєчок.
Після втрати яєчок розвивається комплекс порушень за назвою євнухізм. Гіпофункцію яєчок при їх збереженні позначають терміном "євнухоїдизм".
III. Периферичні порушення:
а) зменшення чутливості клїтин-мтшенеи до дії андрогенів;
б) збільшене зв'язування тестостерону з білками плазми крові;
в) посилене руйнування андрогенів у печінці.
Прояви чоловічого гіпогонадизму залежать від того, у який період онтогенезу він розвивається.
Гіпогонадизм до періоду статевого дозрівання може виявлятися:
• синдромом тестикулярної фемінізації (спадково обумовленим чоловічим псевдо-гермафродитизмом). Цей синдром обумовлений порушеннями диференціювання статевої системи за чоловічим типом в період ембріогенезу;
• слабко вираженими вторинними статевими ознаками;
• пригніченням сперматогенезу;
• відсутністю статевого потягу;
• порушенням окостеніння епіфізарних хрящів при збереженому впливі СТГ — розвивається євнухоїдний, або гіпогонадний, гігантизм;
° слабким розвитком скелетної мускулатури (відсутність анаболічних ефектів андрогенів). Гіпогонадизм після завершення статевого дозрівання характерний, зокрема, для процесу старіння. Він виявляється:
• деяким зменшенням вираженості вторинних статевих ознак;
• порушеннями сперматогенезу;
• розвитком імпотенції;
• анаболічними порушеннями — зменшенням маси скелетних м'язів і працездатності.
33.56. Які причини і основні прояви чоловічого гіпергонадизму?
Чоловічий гіпергонадизм може розвиватися в дитячому віці до періоду статевого дозрівання і у дорослих.
У дітей гіпергонадизм найчастіше пов'язаний з пухлинами і запальними процесами в гіпоталамусі, а також гіпофункцією епіфіза. Усі ці причини викликають збільшення продукції гонадотропних гормонів і, як наслідок, андрогенів. Клінічно такий гіпергонадизм виявляється передчасним статевим дозріванням.
Причиною гіперфункції статевих залоз у дорослих є добро- і злоякісні пухлини, що ростуть з інтерстиціальних клітин. При цьому виражених клінічних ознак, пов'язаних з гіперандрогенемією, немає. У випадку злоякісних новоутворень характерні відсутність кахексії, збереження м'язової маси і сили м'язів.
33.57. Які гормони відносять до жіночих статевих? Як здійснюється регуляція їх утворення і секреції?
Місцем синтезу жіночих статевих гормонів є яєчники. Тут утворюється три групи гормонів.
I. Естрогени: естрадіол, естрон, естріол. Продукуються епітеліальними клітинами фолікулів.
II. Прогестіши, серед яких основним є прогестерон. Утворюються клітинами жовтого тіла.
III. Мінорні гормони: інгібін (пригнічує утворення ФСГ в аденогіпофізі) ірелаксин (розм'якшує лобкове зрощення, тазові зв'язки, шийку матки, розслаблює гладкі м'язи матки).
У регуляції утворення і секреції жіночих статевих гормонів вирішальне значення мають система гіпоталамус-аденогіпофіз-яєчники і механізми позитивного та негативного зворотного зв'язку.
У передовуляційний період під впливом ФСГ посилюється утворення і секреція естрогенів, які за принципом позитивного зворотного зв'язку стимулюють утворення гонадоліберину в гіпоталамусі і лютеїнізуючого гормону (ЛГ) в аденогіпофізі (забезпечують "пік ЛГ"). Останній викликає підвищення секреторної активності клітин, що продукують прогестини.
У післяовуляційний період естрогени і прогестини за принципом негативного зворотного зв'язку пригнічують утворення гонадоліберину і гонадотропних гормонів аденогіпофіза, що веде в підсумку до зменшення секреції і самих жіночих статевих гормонів.
33.58. Назвіть основні біологічні ефекти жіночих статевих гормонів.
I. Статеве дозрівання і розвиток вторинних статевих ознак в особин жіночої статі. Ці ефекти обумовлені естрогенами, секреція яких починає зростати в період статевого дозрівання.
II. Забезпечення! циклічних змін в організмі жінки — підготовка статевих органів до запліднення та імплантації яйцеклітини. Це здійснюється завдяки участі жіночих статевих гормонів разом з ФСГ і ЛГ у гормональному контролі овуляції й менструального циклу.
III. Забезпечення вагітності, пологів і лактації. Так, естрогени викликають швидкий ріст м'язів матки, стимулюють ріст системи проток молочних залоз. Прогестини блокують скорочувальну активність вагітної матки, стимулюють ріст залозистого епітелію молочних залоз. Зі зменшенням продукції прогестинів плацентою пов'язують настання пологів.
IV. Екстра генітальні ефекти. Естрогени впливають на:
а) кісткову тканину, забезпечуючи умови для своєчасного окостеніння епіфі-зарних хрящів;
б) печінку, активуючи синтез цілого ряду білків (транспортних білків, факторів зсідання, ангіотензиногену, ліпопротеїдів високої густини);
в) кровоносні судини, регулюючи процеси ангіогенезу, судинний тонус.
З дією прогестинів пов'язують збільшення базальної температури тіла в післяовуляційний період.
33.59. Які причини і основні прояви жіночого гіпогонадизму?
Виділяють такі групи причин жіночого гіпогонадизму.
I. Центральні (дисрегуляторнї) порушення. Можуть бути обумовлені психогенними факторами, ураженнями гіпоталамуса (дефіцит гонадоліберину), гіпофункцією аденогіпофіза (дефіцит ФСГ і ЛГ), гіперфункцією епіфіза (зменшення утворення гонадотропних гормонів при збільшенні синтезу мелатоніну).
II. Власне залозисті порушення:
а) спадково обумовлена аплазія яєчників;
б) дегенерація яєчників (запальна, кістозна);
в) аутоімунне ушкодження жіночих статевих залоз;
г) хірургічне видалення яєчників. III. Периферичні розлади:
а) зменшення чутливості клітин-мішеней до дії жіночих статевих гормонів;
б) підвищене зв'язування їх білками плазми крові;
в) посилене руйнування жіночих статевих гормонів у печінці.
Клінічні прояви гіпофункції статевих залоз залежать від часу настання гіпо-гонадизму.
Якщо він розвивається до настання статевого дозрівання, то формується комплекс порушень під назвою "оваріальний євнухоїдизм": слабкий розвиток вторинних статевих ознак, первинна аменорея, високий зріст.
Розвиток гіпогонадизму в дітородному віці супроводжується порушеннями циклічних процесів в організмі жінки (розлади менструального циклу), безплідністю, передчасним клімаксом.
Після менопаузи розвиваються клімактеричні зміни - нестабільність судинного тонусу, остеопороз та ін.
33.60. Які причини і основні прояви жіночого гіпергонадизму?
В основі жіночого гіпергонадизму, при якому збільшується секреція жіночих статевих гормонів, можуть лежати центральні (дисрегуляторні) порушення і власне залозисті причини.
Центральні порушення, обумовлені збільшенням секреції гонадоліберину і гонадотропних гормонів, можуть виявлятися розвитком синдрому передчасного статевого дозрівання і синдрому уявної вагітності.
Для першого характерні рання поява вторинних статевих ознак, ранній початок менструацій і ймовірність завагітніти (у віці 7—8 років). Другий синдром виявляється аменореєю й усіма зовнішніми ознаками вагітності. При цьому має місце підвищена продукція пролактину клітинами аденогіпофіза.
До власне залозистих причин жіночого гіпергонадизму відносять кістозні процеси і пухлини яєчників, що продукують гормони. При цьому розвивається гіпертрофія ендометрію, порушується менструальний цикл, поновлюються маткові кровотечі в менопаузі.
33.61. У чому сутність ендокринної функції епіфіза ? Чим можуть виявлятися її порушення?
Пінеалоцити (ендокринні клітини епіфіза) продукують гормон мелатонін — похідну сполуку амінокислоти триптофану. Рівень його секреції залежить від освітленості сітківки ока, інформація від якої через складні провідникові шляхи, у тому числі гіпоталамус і спинний мозок, надходить в епіфіз. У світлий час доби секреція мелатоніну гальмується, у темряві, навпаки, зростає. Так, у людини в нічний час (з 23 до 7 год.) вивільняється 70 % загальної добової кількості мелатоніну. З секрецією мелатоніну пов'язують функцію епіфіза, що її позначають як "біологічний годинник".
Серед відомих нині біологічних ефектів мелатоніну — пригнічення утворення і секреції гонадотропних гормонів аденогіпофіза, зменшення продукції ТТГ, АКТГ і СТГ.
Патологію епіфіза мало вивчено. Відомо, що при збільшенні продукції мелатоні-ну відбувається затримка статевого розвитку, а при зменшенні його секреції, навпаки — передчасне статеве дозрівання.
33.62. Якими змінами в організмі можуть виявлятися порушення ендокринної функції підшлункової залози?
Ендокринну функцію виконують клітини острівців підшлункової залози, що продукують інсулін (бета-клітини) і глюкагон (альфа-клітини).
Зменшення синтезу й секреції інсуліну призводить до розвитку цукрового діабету (див. розд. 20).
Збільшення утворення інсуліну — гіперінсулінізм — буває у разі виникнення пухлин, що розвиваються з β-клітин панкреатичних острівців і продукують інсулін; при спонтанній ідіопатичній гіпоглікемії у дітей; на початкових стадіях цукрового діабету; при ожирінні, демпінг-синдромі (стан після резекції шлунку), деяких ендокринних захворюваннях (акромегалія, тиреотоксикоз, хвороба Іценка-Кушинга).
Основні прояви гіперінсулінізму зумовлені гіпоглікемічною, анаболічною і міто-генною дією інсуліну (див. розд. 20). Під впливом надлишку інсуліну розвивається синдром гіпоглікемії (аж до гіпоглікемічної коми), ожиріння, склеротичні ураження кровоносних судин.
Другий гормон підшлункової залози - глюкагон має гіперглікемічну і ліполітич-ну дію, стимулює секрецію інсуліну.
Збільшення секреції глюкагону буває у разі розвитку пухлини з а-клітин острівців підшлункової залози (глюкагонома) і виявляє себе розвитком вторинного цукрового діабету (див. розд. 20).
Описано синдроми недостатності глюкагону. Для них характерна спонтанна гіпоглікемія, яку спостерігають у дітей.
34= Патологічна фізіологія нервової системи
34.1. Як класифікують порушення діяльності нервової системи?
І. За анатомічним принципом виділяють:
1) порушення периферичної нервової системи;
2) порушення центральної нервової системи, у тому числі розлади функції спинного мозку, довгастого, середнього і т. д.
її. За походженням виділяють спадково обумовлені і набуті порушення нервової системи. Набуті можуть бути первинними і вторинними.
Первинні розлади виникають унаслідок прямої дії на нервову систему патогенних факторів: фізичних (травма, радіація, термічні впливи), хімічних (токсини, отрути), біологічних (віруси, бактерії), соціальних (слово). Вторинні розлади обумовлені насамперед порушеннями гомеостазу (гіпоксія, гіпоглікемія, ацидоз і т. п.), імунними факторами (аутоалергічні реакції), розладами мозкового кровообігу.
III. Клітинний принцип передбачає такі види порушень функції нейронів:
1) порушення електрофізіологічних процесів;
2) розлади нейрохімічних (медіаторних) процесів;
3) порушення аксоплазматичного транспорту.
IV. Залежно від виду порушених функцій розрізняють такі розлади діяльності нервової системи:
1) порушення сенсорних функцій (чутливості);
2) порушення ефекторних функцій: рухової, вегетативної, трофічної;
3) порушення інтегративних функцій.
34.2. Які виділяють види порушень соматовісцеральної чутливості?
Поняття соматовісцеральної чутливості охоплює чутливість шкіри (тактильну, температурну, больову), глибоку чутливість (пропріорецепцію) і больову чутливість усього тіла (ноцицепцію).
Розрізняють такі види порушень соматовісцеральної чутливості:
1) гіперестезія — підвищення чутливості;
2) гіпестезія - зменшення чутливості;
3) анестезія — відсутність чутливості.
34.3. Які провідникові шляхи забезпечують надходження інформації про різні види чутливості від периферичних рецепторів у центральну нервову систему?
1. Лемнісковий шлях (рис. 159). Проводить усі види глибокої (пропріоцептивної) чутливості, а також тактильну чутливість від спеціалізованих механорецепторів шкіри (складна тактильна чутливість). Складається з трьох нейронів.

Рис. 159. Схема лемніскового
шляху
Рис. 160. Схема антеро-латерального шляху
Перші нейрони (І) - псевдоуніполярні клітини спинномозкових гангліїв. Аксони цих клітин входять через задні корінці в спинний мозок і піднімаються догори в складі задніх канатиків, утворюючи пучки Голля і Бурдаха.
Тіла других нейронів (II) містяться в довгастому мозку (n. gracilis і n. cuneatus). їхні аксони переходять на протилежний бік, перехрещуючись з аксонами контр-латеральних нейронів, і утворюють медіальну петлю (lemniscus medialis). Тіла третіх нейронів (III) містяться в таламусі. їхні аксони йдуть у кору головного мозку, у відповідні сенсорні зони.
2. Антеро-латеральний шлях (неоспіноталамічний тракт) (рис. 160). Проводить температурну, найпростіші види тактильної (від механочутливих вільних нервових закінчень) і шкірну больову (ранній біль) чутливість.
Перші нейрони (І) є клітинами спинномозкових гангліїв. Тіла других (II) нейронів містяться в задніх рогах спинного мозку. їхні аксони посегментно переходять на протилежний бік спинного мозку, перехрещуються і у складі бічних канатиків піднімаються в таламус, де містяться тіла третіх нейронів (III), що посилають свої відростки в сенсорні зони кори головного мозку.
3. Екстралемнісковий шлях (рис. 161). Проводить больову чутливість (пізній біль, глибокий і вісцеральний біль). На відміну від двох попередніх, є багатонейронним і філогенетично більш давнім.
Тіла перших нейронів (І) містяться в спинномозкових гангліях, а других (II) - у задніх рогах спинного мозку. Аксони останніх частково переходять на другий бік, а частково з цього ж боку йдуть нагору в складі бічних канатиків, утворюючи два тракти: спіноретикулярний і палеоспіноталамічний.
У ретикулярній формації стовбура мозку, куди піднімаються аксони других нейронів, відбувається багаторазове перемикання на інші нервові клітини. Від ре-
Рис. 161. Схема екстралемніскового шляху
тикулярної формації інформація надходить в утвори лімбічної системи (емоційні компоненти реакцій), центри гіпоталамуса (вегетативні компоненти реакцій), таламус, кору головного мозку та ін.
34.4. Які механізми можуть лежати в основі порушень соматовісцеральної чутливості?
1. Порушення рецепції. При збільшенні порогу збудження рецепторів виникає гіпестезія, при зменшенні - гіперестезія.
2. Ушкодження периферичних нервів. При цьому випадають усі види чутливості в зоні іннервації даного нерва. Як правило, зона випадання чутливості менша зони іннервації, що пояснюють деяким перекриванням зон іннервації різних нервів.
3. Ушкодження задніх корінців спинного мозку. Характеризується випаданням усіх видів чутливості в зоні відповідних сегментів.
4. Ушкодження спинного мозку. При перетинанні половини спинного мозку (лівої або правої) розвивається
синдром Броун-Секара (див. запит. 34.5). Повний перетин спинного мозку веде до зникнення усіх видів чутливості по обидва боки нижче рівня перетинання.
5. Порушення функції підкіркових структур, що беруть участь у здійсненні сенсорних функцій. Найбільше значення має ураження ядер таламуса.
6. Ураження сенсорних зон кори головного мозку. Порушення нейронів постцен-тральної звивини спричиняються до розладів складної тактильної і пропріоцеп-тивної чутливості на протилежному боці тіла. Ушкодження тім'яної частки викликає розвиток комплексу порушень під назвою "аморфосинтез".
У людини (що не є шульгою) аморфосинтез виявляють після видалення кори правої півкулі великого мозку. При цьому зникає уява про просторове розташування частин тіла на протилежному боці. Людина не може надягти одяг або привести його в порядок на лівій половині, не може поголити ліву половину обличчя або зачесати волосся на лівому боці.
Якщо ж уражено тім'яну частку з лівого боку, то аморфосинтез доповнюється агнозією - нездатністю розпізнавати частини тіла, предмети, їхнє зображення і розташування в просторі.
34.5. Коли виникає синдром Броун-Секара? Чим він виявляється?
Синдром Броун-Секара розвивається після перетинання половини спинного мозку (лівої або правої). Характеризується дисоціацією розладів чутливості. Так, нижче рівня перетину з того ж боку випадають пропріоцептивна і складні види тактильної чутливості (ушкоджується лемнісковий шлях до його перехрещення), а з протилежного боку — температурна, проста тактильна й частково больова чутливість (ушкоджується антеро-латеральний шлях після перехрещення).
34.6. Що таке біль? Чим він відрізняється від інших видів чутливості?
Біль — це неприємне сенсорне й емоційне відчуття, пов'язане з загрозою або самим ушкодженням тканин.
Особливості болю як виду чутливості:
1. Біль дає мало інформації про навколишній світ, зате інформує про небезпеку, що може виникнути або вже виникла внаслідок дії ушкоджувальних факторів, - захисна функція болю.
2. На відміну від інших видів чутливості до болю не розвивається адаптація. У зв'язку з цим біль може бути причиною страждань хворого.
3. Біль супроводжується складними емоційними, вегетативними й руховими реакціями.
4. Біль може бути патогенетичним механізмом розвитку генералізованих патологічних процесів, зокрема шоку.
34.7. Як класифікують біль?
I. За клінічною характеристикою (суб'єктивним відчуттям) біль може бути гострий і тупий, локалізований і дифузний, мати характер пощипування, поколювання, жару і т. п.
II. Залежно від тривалості больових відчуттів біль може бути гострим і хронічним. Гострий біль швидко проходить після припинення дії больових стимулів, хронічний є тривалим, він завдає страждань хворому.
III. За значенням для організму біль може бути фізіологічним і патологічним. Фізіологічний біль має захисне значення. Він сигналізує про ушкодження або його можливість, ініціює певні поведінкові реакції, спрямовані на усунення ушкодження, обмежує функції ураженого органа. Патологічний біль не несе сигнальної функції, він стає механізмом порушення життєдіяльності, у тому числі і мозку, призводить до розладів функцій різних органів і систем.
IV. За механізмами розвитку розрізняють соматичний і вісцеральний біль. Соматичний біль поділяють на поверхневий і глибокий (рис. 162).
Рис. 162. Класифікація болю за механізмами розвитку
34.8. Що таке соматичний поверхневий біль? На які види його поділяють?
Соматичний поверхневий біль — це біль, що виникає в шкірі. Розрізняють два його види: ранній і пізній біль.
Якщо нанести сильну механічну травму, то відразу ж виникає гострий, різкий, добре локалізований біль, що швидко минає після завершення дії патогенного фактора — це так званий ранній біль.
Через певний час (0,5—1 с) виникає пізній біль. Це тупий, ниючий, дифузний біль. Він триває ще якийсь час після припинення дії патогенного фактора.
34.9. Дайте порівняльну характеристику раннього і пізнього болю.
Ранній біль |
Пізній біль |
Виникає відразу |
Виникає через 0,5—1 с |
Гострий, різкий |
Тупий, ниючий |
Чітко локалізований |
Дифузний |
Швидко зникає після закінчення дії патогенного фактора |
Зникає поступово |
Низький поріг больового відчуття |
Високий поріг |
Ступінь болю зростає зі збільшенням інтенсивності ушкодження (градуальність больового відчуття) |
Відсутність градуальності - закон «все або нічого» |
Проводиться мієлінізованими волокнами Ад зі швидкістю 10—25 м/с |
Проводиться немієлінізованими волокнами С зі швидкістю 0,5-1 м/с |
Проводиться антеро-латеральною системою |
Екстралемнісковою системою |
Формується в сенсорних центрах кори великого мозку |
У таламічних сенсорних центрах |
Вегетативним компонентом є збудження симпатичної нервової системи |
Збудження парасимпатичної нервової системи |
Є засобом попередження про ушкодження |
Є засобом нагадування про .ушкодження |
Має захисне значення — вмикає захисні рефлекси і реакції |
Відіграє патогенну роль - викликає афективні (емоційні) реакції, загальне нездужання, хворобливий стан |
Є епікритичним (еволюційно більш молодим) |
Є протопатичним (еволюційно більш давнім) |
34.10. Що таке соматичний глибокий біль? Наведіть приклади.
Соматичний глибокий біль — це біль, що виникає в глибоких тканинах. До нього відносять головний, зубний біль, біль у м'язах і суглобах.
Має багато спільного з пізнім соматичним поверхневим болем. Він часто тупий, не має чіткої локалізації, супроводжується афективними (загальне нездужання, хворобливий стан) і вегетативними (нудота, потовиділення, зменшення артеріального тиску) реакціями. Докладно див. характеристику пізнього болю (запит. 34.9).
34.11. Що таке вісцеральний біль? Чим він характеризується?
Вісцеральний біль - це біль, що виникає у внутрішніх органах.
Його джерелами можуть бути парієтальна очеревина і корінь брижі, парієтальна плевра і перикард, м'язові органи (серце, артерії, порожнисті органи). Розвиток болю в гладком'язових порожнистих органах може бути пов'язаний як зі швидким і різким їх розтягненням, так і з сильним скороченням (спазмом) гладких м'язів, унаслідок чого порушується кровопостачання і виникає ішемія. Напади такого болю одержали назву кольок.
Для вісцерального болю характерні:
1) афективні реакції (пригнічений емоційний стан, загальне нездужання, стан хвороби);
2) вегетативні реакції'— нудота, потовиділення, падіння артеріального тиску;
3) рефлекторне скорочення скелетних м'язів (напруження м'язів черевної стінки, вимушена поза та ін.).
34.12. Які бувають види болю залежно від його локалізації?
1. Місцевий біль - локалізується в місці дії подразника; там, де розвивається патологічний процес.
2. Проекційний біль - місце, на яке діє больовий подразник, не збігається з тим, де біль відчувається. Наприклад, при ушкодженні міжхребцевих дисків здавлюються спинномозкові нерви. При цьому болить та ділянка, що отримує іннервацію від защемленого нерва, тобто має місце проекція болю на ділянки, іннервацію яких здійснює ушкоджений нерв.
3. Відбитий (іррадійований, рефлекторний) біль. Больове відчуття, що виникає внаслідок впливу на внутрішні органи, часто локалізується не в даному органі (або не тільки в ньому), а у віддалених поверхневих ділянках шкіри.
Біль завжди відбивається на ділянки периферії, які отримують іннервацію від того ж сегмента спинного мозку, що і уражений внутрішній орган. Якщо йдеться про поверхню шкіри, то біль відбивається на певному дерматомі. Оскільки органи одержують іннервацію більш ніж від одного спинномозкового сегмента, то біль відбивається на кількох дерматомах. Разом вони утворюють зону Геда для даного органа.
34.13. Назвіть можливі причини болю.
Біль викликають больові стимули - фактори, що обумовлюють сильне подразнення або ушкодження тканини. До них відносять:
1) механічні больові стимули;
2) термічні больові стимули (температура вище 45 °С);
3) хімічні больові стимули - ацетилхолін, серотонін, гістамін, кініни, простагландини Е субстанція Р, іони Н+ (при рН < 6), іони К+ (при концентрації понад 20 ммоль/л);
4) ушкодження нервових провідників.
34.14. Які існують теорії механізмів болю?
I. Теорія інтенсивності. її прихильники вважають, що в організмі немає спеціальних больових рецепторів. Біль виникає в тому випадку, коли низькопорогові механо- і терморецептори стимулюються з інтенсивністю, що перевищує певний рівень. Якщо фактор діє з низькою або середньою інтенсивністю, то виникає тактильне або температурне відчуття, якщо ж інтенсивність висока - то відчуття болю.
II. Теорія розподілу імпульсів. її сутність полягає в тому, що больовий стимул викликає особливий хід нервових імпульсів, що відрізняється від поширення розрядів, які виникають при дії неушкоджувальних факторів. На цьому будується так звана "ворітна теорія" болю (Мелзак, Волл), що надає велике значення у формуванні больових відчуттів желатинозній субстанції спинного мозку (substantia gelatmosa, SG; рис. 164).
Нейрони SG здійснюють пресинаптичне гальмування, блокуючи проходження імпульсів у нейрони задніх рогів спинного мозку по товстих і тонких нервових волокнах. Якщо нейрони SG збуджуються, відбувається пресинаптичне гальмування - "ворота" закриті. Якщо нейрони SG самі загальмовані, то пресинаптичне гальмування знімається -"ворота" відкриті. Інтенсивна стимуляція товстих мієлінізованих нервових волокон викликає збудження нейронів SG - "ворота" закриваються, і проведення імпульсів у спинний мозок зменшується.
При інтенсивному збудженні тонких немієлінізованих волокон, що проводять біль, відбувається гальмування нейронів SG — знімається пресинаптичне гальмування і полегшується надходження імпульсів у задні роги спинного мозку.
III. Теорія специфічності. Передбачає існування специфічних больових рецепторів - ноцщепторів. Вони відповідають тільки на інтенсивні стимули і таким чином беруть участь у формуванні больових відчуттів.
Рис. 164. Схема, що ілюструє "ворітний"механізм
болю (за Мелзаком і Воллом):
А — товсті мієлінізовані; С — тонкі немієлінізовані
волокна; SG - желатинозна субстанція; Т
- нейрони задніх рогів спинного мозку
Виділяють такі види ноцицепторів:
а) механочутливі ноцицептори (містяться у шкірі, скелетних м'язах);
б) термочутливі ноцицептори - збуджуються при температурі вище 45 °С (рецептори гарячого);
в) полімодальні ноцицептори — збуджуються як механічними, так і температурними больовими стимулами;
г) хемочутливі ноцицептори — збуджуються хімічними больовими стимулами;
д) вісцеральні ноцицептори — збуджуються розтягненням стінки гладком'язо-вих органів або спастичним скороченням.
34.15. Що таке хронічний біль? Назвіть його клінічні форми.
Хронічним називають сильний, тривалий біль, що виснажує, завдає страждань хворому. Виділяють такі форми хронічного болю.
1. Невралгія— больовий синдром, пов'язаний з порушеннями функції периферичного нерва при вірусних інфекціях, авітамінозах, порушеннях кровообігу, розладах обміну речовин (цукровий діабет). Особливо важкою є невралгія трійчастого нерва, яка виявляється нападами настільки сильного болю, що хворі не в змозі приймати їжу і розмовляти. Виникнення такого болю провокує дія дуже слабких подразників, наприклад, дотик до кута рота.
2. Каузалгія - сильний пекучий біль, що виникає при ушкодженні великих соматичних нервів (неповне перетинання нерва). У хворого виникає відчуття, начебто на шкіру ллють окріп або прикладають руку до розпечених предметів, або тримають її у вогні. Навіть легкий дотик до шкіри, що її іннервує ушкоджений нерв, викликає нестерпний біль. Крім того, біль можуть провокувати несподівані зорові та слухові подразники.
3. Фантомний біль. Виникає після ампутації кінцівок — "болить" кінцівка, якої вже немає. При цьому біль дуже сильний і часто нестерпний.
4. Таламічний біль — важкий спонтанний біль у всій половині тіла з гіперпатією (суб'єктивним враженням підвищеної чутливості). Розвивається при ураженнях ядер таламуса.
34.16. Які механізми можуть лежати в основі формування хронічного болю?