

Лекция 9. Хроматографические методы анализа
Хроматографические методы в системах экологического мониторинга также получили довольно широкое практическое применение. Наиболее часто используются методы газовой и жидкостной хроматографии, причем первые распространены больше. Кроме указанных, выделяют еще методы тонкослойной хроматографии и другие.
Газохроматографический метод основан на разделении соединений между двумя несмешивающимися фазами, одна из которых неподвижна (жидкость или твердое тело), а другая – подвижна (инертный газ-носитель). Рассматриваемый метод позволяет определять ничтожно малые количества веществ, в т.ч. не обладающих специфической реакционной способностью, анализируя смеси, состоящие из десятков и сотен компонентов с близкими свойствами.
Существует два основных вида газовой хроматографии (ГХ): газоадсорбционная и газожидкостная (ГЖХ). В газоадсорбционной хроматографии неподвижной фазой служит твердый сорбент, а в газожидкостной – жидкость, наносимая тонким слоем на инертный твердый носитель.
Метод газовой хроматографии позволяет в одном анализе определить качественный и количественный состав сложной смеси, содержащей до 100-200 летучих компонентов Высокочувствительные селективные детекторы позволяют определять микропримеси с концентрацией 10-4 % (мкг/мл) и иногда менее.
Принципиальная схема газового хроматографа показана на рис. 1.
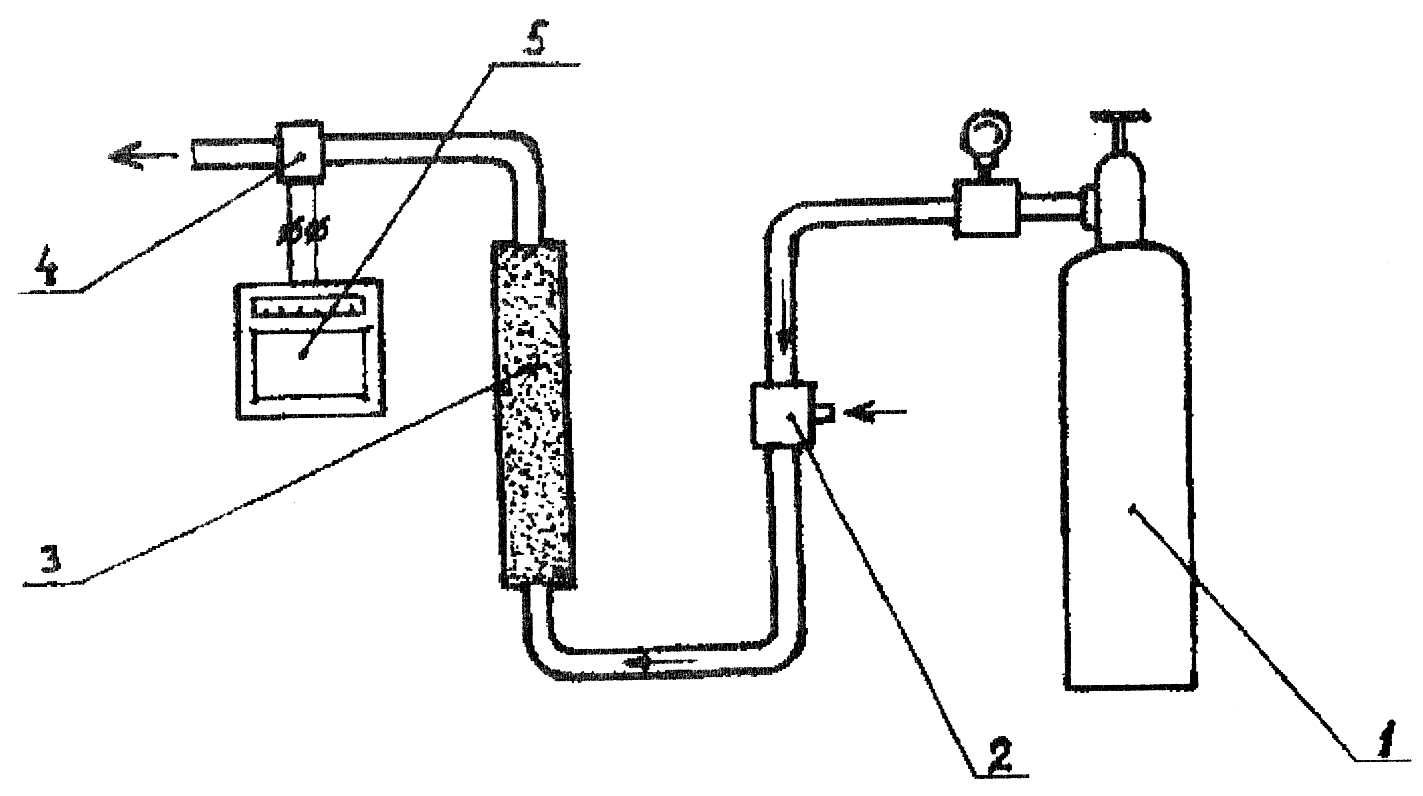
1 – баллон с инертным газом; 2 – устройство для ввода пробы; 3 – колонка; 4 – детектор; 5 – самопишущий прибор
Рис. 1. Принципиальная схема газового хроматографа
Основной частью хроматографа является колонка, в которой происходит разделение анализируемой смеси на составляющие компоненты. Разделение основано на распределении молекул различных компонентов между подвижной газовой фазой и неподвижной фазой сорбента. Хорошо сорбируемые компоненты продвигаются вдоль слоя сорбента медленнее, чем плохо сорбируемые. Разделенные компоненты выходят вместе с газом-носителем и фиксируются детектором. Сигнал детектора записывается на ленте регистрирующего (самопищущего) прибора. На хроматограмме каждому компоненту соответствует свой пик. Время от момента ввода пробы до выхода максимума пика называют временем удерживания. Для идентификации компонентов определяют индекс относительного удерживания компонента и сравнивают его с индексами в справочных таблицах. Так проводится идентификация веществ.
Газовая хроматография (как и другие виды хроматографии) относится к гибридным методам, в которых объединены способ разделения (на хроматографической колонке) и способ определения разделенных компонентов (с помощью детектора). Благодаря разделению метод селективен, а благодаря свойствам детектора – чувствителен.
Графическую временную зависимость сигнала детектора, реагирующего на изменения концентрации разделяемых на хроматографической колонке веществ, называют хроматограммой. При достаточно полном разделении хроматограмма представляет собой совокупность пиков, каждый из которых соответствует одному из компонентов анализируемой смеси. Положение пиков на хроматограмме служит для идентификации компонентов пробы, а площадь под пиками или высота пиков характеризуют концентрацию анализируемого вещества в пробе.
Наиболее важными параметрами пика являются (рис. 2): h – высота пика; r – ширина основания; 0,5 – ширина пика на половине высоты; h' – приведенная высота пика.
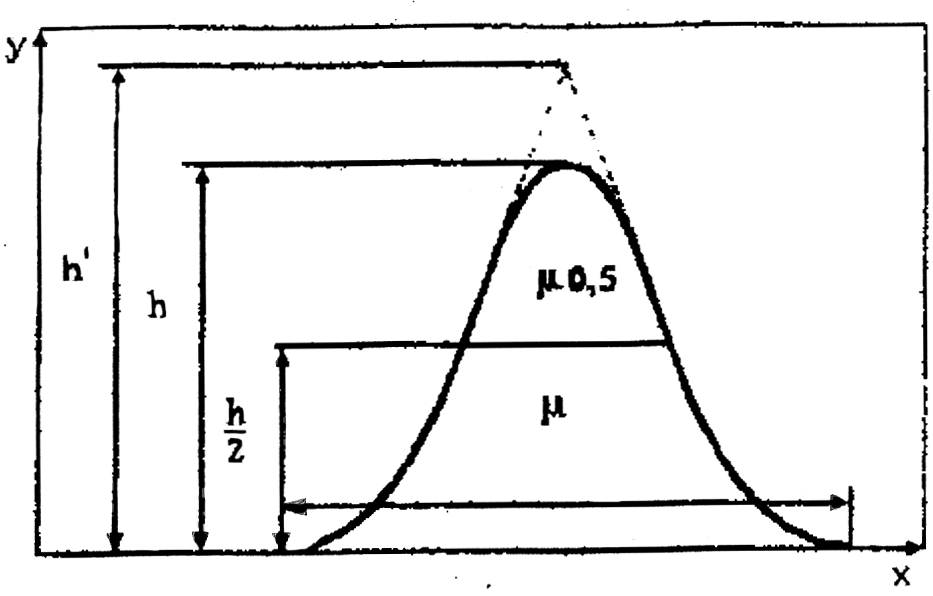
Рис. 2. Симметричный хроматографический пик
Если ширину пика целесообразно измерять в единицах времени , с или объема газа V, см3, то используются соотношения:
![]()
где – ширина пика, мм; В – скорость диаграммной ленты прибора, мм/с; Va – расход газа-носителя на выходе из колонки, см3/мин.
Площади хроматографических пиков автоматически измеряются при помощи интеграторов, входящих в состав современных хроматографов. Наряду с этим используют другие способы, в частности:
а) графически находят высоту (h) и ширину пика на середине высоты (); если форма пика близка к кривой нормального распределения, то площадь пика Q может быть вычислена из соотношения: Q = 1,065h;
б) строят треугольник, проводя касательные к обеим сторонам пика и соединив их третьей линией, параллельной нулевой линии (площадь полученного треугольника приблизительно равна:
0,96Q : Q = 0,516rh').
В основе количественного анализа в хроматографии лежит зависимость между площадью или высотой хроматографического пика от содержания определяемого компонента. Количественный анализ по высоте пиков менее надежен, так как эта величина зависит от условий эксперимента. Наиболее часто используют следующие приемы количественного хроматографического анализа.
Метод абсолютной калибровки. В этом методе концентрацию
i-го компонента в пробе определяют как
![]()
где Q – площадь пика на хроматограмме; Vпр – объем пробы при фиксированной температуре; kq – калибровочный коэффициент, определяемый на основе калибровочных смесей известного состава, причем
![]()
где Ск – концентрация компонента в калибровочной смеси, % (об.); VK –объем смеси; QK – площадь пика компонента на хроматограмме.
Если определяющим параметром является высота, то вместо площади пика QK подставляют его высоту.
Метод внутренней нормализации. Концентрацию i-го компонента определяют по формуле:
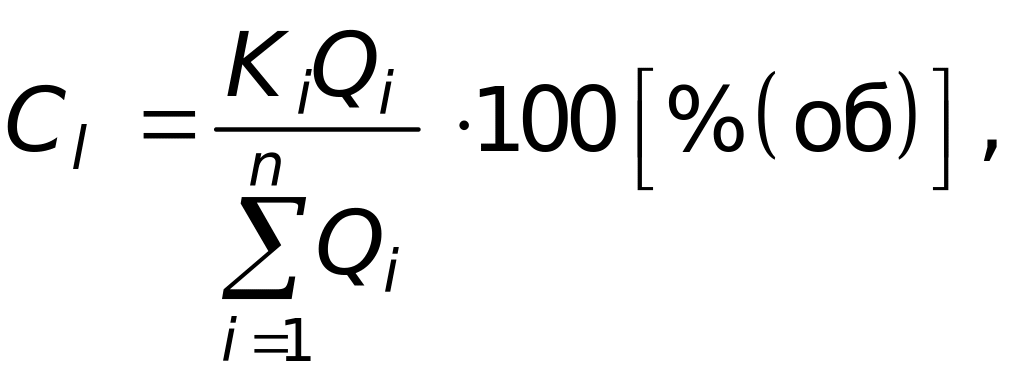
где n – число компонентов в смеси; Ki и Qi – поправочные коэффициенты и площади пиков компонентов смеси.
Если анализируемая смесь состоит из веществ близкой природы, и чувствительность хроматографических детекторов к этим веществам практически одинакова, то Сi определяют без учета поправочных коэффициентов:

Метод внутреннего стандарта. Его используют в тех случаях, когда на хроматограмме отсутствуют или полностью не регистрируются пики некоторых компонентов смеси. Концентрацию i-го компонента рассчитывают по уравнению:
Ki Qi
Сi = ------ . 100 . r
Kcт Qст
где r – отношение массы стандарта к массе анализируемой пробы; Qi и QСТ– площади пиков i-го компонента и добавленного к пробе стандарта; Ki и КСТ – поправочные коэффициенты, учитывающие чувствительность детектора к соответствующим веществам.
Детектор – важнейший элемент газового (да и любого) хроматографа, в котором изменения состава проходящей через него газовой смеси преобразуются в изменение выходного сигнала. Различают потоковые и концентрационные детекторы.
Сигнал потокового детектора пропорционален мгновенному значению массовой скорости поступающей на него определяемого вещества. Сигнал концентрационного детектора пропорционален мгновенному значению концентрации определяемого вещества в объеме камеры детектора. Из большого числа газохроматографических детекторов наиболее часто на практике используют концентрационный детектор по теплопроводности (катарометр) и потоковый – пламенно-ионизационный детектор (ПИД).
Детекторы также разделяют на неселективные (катарометр, ПИД, электрокондуктометрический и др.) и селективные (ТИД, ЭЗД, ПФД и др.). первые используются на практике чаще.
Чувствительность детектора по теплопроводности Sc определяется по уравнению:
![]()
где Q – площадь регистрируемого пика, см2; Va – объем газа-носителя, см3/мин; B1 – чувствительность самописца регистратора, см/мВ; B2 – скорость движения диаграммной ленты, см/мин; gпр – масса введенной пробы, мг.
Обычно чувствительность детекторов по теплопроводности составляет порядка 103 мВсм3/мг; минимальная определяемая концентрация вещества порядка 10–3 % (об.).
Чувствительность пламенно-ионизационного детектора (ПИД) Sj определяют по уравнению:
![]()
где Q – площадь регистрируемого пика, см2; B1 – чувствительность регистратора, см/А; B2 – скорость движения диаграммной ленты, см/мин; g – масса анализируемого компонента, мг; 60 – коэффициент.
Обычно чувствительность детектора ионизации в пламени составляет порядка 4·10–6 Кл/мг по пропану; минимально определяемый поток вещества порядка 210–8 мг/с.
Существуют и другие детекторы: электронно-захватный (ЭЗД), пламенно-фотометрический (ПФД), фотоионизационный (ФИД) и другие. У каждого из детекторов есть своя группа веществ, к которым он специфичен, для них он и применяется.
На рис. 3 приведен общий вид серийно выпускаемого в России аппаратно-программного комплекса на базе газожидкостного хроматографа «Кристалл-2000», оснащённого комплектом детекторов.
Подробности метода ГХ и ГЖХ описаны в учебной и специальной литературе [24], [25], [26], [27], [28].

ЗАО Специальное конструкторское бюро «ХРОМАТЭК» разработало с использованием аппаратно-программного комплекса «Хроматэк-кристалл 2000 М» большое количество методик определения в воде пестицидов, нефтепродуктов, фенолов, ароматических соединений, ацетона, метанола, предельных углеводородов и галогенсодержащих соединений.
Жидкостная хроматография представляет собой разделение во времени компонентов смеси в соответствии с различием их физико-химических свойств и последующем детектировании в потоке подвижной жидкой фазы. Жидкостная хроматография (ЖХ) основана на распределении веществ между двумя несмешивающимися фазами: неподвижной и подвижной жидкой фазой, которая проходит через слой неподвижной фазы. Различают «твердожидкостную» и «жидко-жидкостную» хроматографию.
Твердожидкостная хроматография является адсорбционной. В качестве адсорбента используют силикагель, уголь, оксид алюминия и другие сорбенты. Жидко-жидкостная хроматография является распределительной. Жидкость, используемая в качестве неподвижной фазы, наносится на инертный носитель и через нее пропускают поток жидкости подвижной фазы. Жидкости должны быть несмешивающимися. Этот метод применяется, как правило, при высоких давлениях для разделения нелетучих соединений. Такой метод называют «хроматографией высокого давления». В качестве неподвижной фазы используются специальные смеси, например, полиэтиленгликоль ПЭГ-600 или ПЭГ-4000. В качестве подвижной фазы используют неполярные растворители или их смеси, например, гексан с диэтиловым эфиром или гексан с этанолом.
В данном методе используются колонки с внутреннем диаметром в несколько миллиметров и объемы пробы порядка десятков микролитров, поэтому используемые объемы растворов чрезвычайно малы, и для хорошего разделения компонентов смесей нужно использовать детекторы с довольно малым внутренним объемом.
В ЖХ используют как универсальные, так и специфические детекторы. Последние фиксируют вещества по их конкретным параметрам. Обычно применяют фотометрические детекторы с переменной длиной волны, флуоресцентные и электрохимические детекторы.
В проточных анализаторах непрерывного действия пробы засасывают и инжектируют в поток жидкости, которая транспортирует их к детектору. Во время переноса веществ их часто специально химически модифицируют различными способами. Видом жидкостной хроматографии является ионная хроматография (ИХ), с помощью которой селективно анализируют различные катионы и анионы.
Непрерывное титрование в проточных жидкостных системах сочетает простоту оценки концентрации определяемого вещества, характерную для прямых методов, с высокой точностью титрометрических. Все эти методы основаны на достижении эквивалентных соотношений определяемого вещества и титранта во время непрерывного смешения реагента с раствором пробы и различаются выбором переменных параметров, а также применяемой аппаратурой. В таких системах широко используют электрохимический и, в частности, потенциометрический детектор. Важно, что в этом случае допускаются не слишком строгие требования к скорости отклика детектора и (за отдельными исключениями), к воспроизводимости его результата.
На рис. 4 приведен общий вид жидкостного хроматографа
«Милихром-6».
"Милихром-6" Эффективная жидкостная система: - автоматическое устройство ввода пробы на 30 образцов;
- высокоточное дозирование в диапазоне от 1 до 99 мкл;
- два микрошприцевых насоса, объёмом по 2500 мкл;
- максимальное давление 8+1,0 МПа;
- расход – 2-999 мкл/мм;
- нестабильность расхода, не более – 0,8%;
- кювета из химически инертного материала с регулируемым клапаном противодавления исключает образование воздуха в магистралях;
- гидродинамический смеситель обеспечивает воспроизводимость смешиваемых жидкостей при скорости потока до 200 мкл/мин;
- коррозийная стойкость материалов позволяет работать с концентрированными кислотами;
- изократический и градиентный (любой формы) режим работы;
- микроколонки с эффективностью не ниже 5000-6000 т.т. объёмом 250-300 мкл с расходом элюента 1300-1600 мкл на один анализ;
- термостат колонки с диапазоном устанавливаемых температур 35÷850С, с погрешностью термостатирования 10С.
Малогабаритный электронный блок из импортных компонентов высокой надёжности.
Система управления прибором и обработки хроматографической информации на базе современного компьютера и удобной для Пользователя программой UniChrom, функционирующей в среде Windows.
Превосходство хроматографа Милихром-6 заложено в уникальной совокупности его функциональных возможностей и позволяет экономично проводить анализ любой сложности одновременно для нескольких соединений с высокой точностью и воспроизводимостью, используя ранее сформированные библиотеки спектров и спектральных отношений веществ.

Рис. 4. Хроматограф жидкостный микроколоночный
Технические характеристики
Спектрофотометрический детектор: Диапазон длин волн, нм ………………………………………….......…...190-360
Дискретность смены длины волны, нм………………………………...……2
Точность установки длины волны, нм…..…………………………...………1
Уровень флуктуационных шумов нулевого сигнала, е.о.п …...................1х10-4
Дрейф нулевого сигнала е.о.п........................................................... .....…5х10-5
СКО высоты пиков, % ..................................................................................<1
Режимы детекции: одноволновой, многоволновой, спектр
Воспроизводимость установки длинны волны, нм …………....…..........…0,01
Чувствительность по контрольным веществам, г/мл …….......……........1х10-8
Устройство ввода пробы автоматическое: ·Количество проб…………………….....…………………………..……………30
Микрошприцевый насос: ·Объем шприца, мкл…………….………………………………….………..…2500
Диапазон расхода, мкл/мин….…………………………………......…..…2-999
Максимальное рабочее давление, МПа……….……………..……...........8+0,5
Нестабильность расхода, %………………………………...………...……….0,8
Термостат колонки: ·Диапазон температур, 0С…………………………....………..……………….35-85
Погрешность термостатирования, 0С………………......……....………………1
Размеры (без компьютера), мм…………….....….……...............…320х360х550
Вес (без компьютера), кг.....................................................................................26
Питание (без компьютера), Вт……………………………….….……………200
Питание осуществляется от электрической сети 220В
Программное обеспечение: Лицензионные “ UNICHROM ”, “ Windows ” XP. Все блоки и узлы запатентованы.
Более подробно об особенностях метода ЖХ можно прочитать в учебной и специальной литературе [29], [30], [31], [32].
Тонкослойная хроматография – это хроматография на стеклянной или металлической пластинке, на которую нанесен слой сорбента. На стартовую линию наносят пробы веществ «свидетелей» и исследуемой смеси. Край пластинки погружают в растворитель. По мере продвижения жидкости по пластинке происходит разделение смеси. Пластинку сушат и проявляют, а идентификацию веществ производят по положению пятен разделенных компонентов и веществ свидетелей, как показано на рис.5.

1, 2 – вещества-«свидетели»; 3 – анализируемая смесь
Рис. 5. Хроматографирование на пластинке в током слое
Для углубленного изучения тонкослойной хроматографии можно воспользоваться специальной литературой [33], [34]. Существует еще ряд публикаций на русском языке, посвященных хроматографии [35], [36].