Контрольные вопросы для проверки знаний по теме : «Алкилирование непредельных углеводородов предельными углеводородами»
1. Проведите анализ термодинамики реакции алкилирования изобутана изобутиленом. При каких температурах необходимо проводить данный процесс?
2. Приведите уравнения реакций алкилирования изобутилена пропаном. Какие продукты будут получаться в указанном процессе?
3. Приведите уравнения реакций алкилирования изобутана пропиленом. Какие продукты образуются в указанном процессе?
4. В какой ряд активности расположатся кислоты Бренстеда-Лаури -фторсульфоновая кислота F-SO2-OH, серная кислота, трифторметансульфокислота CF3-SO2-OH – в реакции алкилирования изобутана изобутиленом? Чем обусловлено возникновение определенного ряда активности кислот?
5. В качестве катализатора в реакции алкилирования изобутана изобутиленом используется нанесенная на двуокись кремния серная кислота. Активность катализатора возрастает при введении в каталитическую систему кислот Льюиса, например трехфтористого бора, хлористого алюминия и т.д. Какова причина увеличения каталитических свойств серной кислоты при введении в нее кислот Льюиса?
6. Основным продуктом в реакции алкилирования изобутилена изобутаном должен быть 2,2,4-триметилпентан:
СН3-С(СН3)2-СН2-СН(СН3)-СН3 .
Однако в ходе реакции в существенных количествах образуется также 2,3,4-триметилпентан:
СН3-СН(СН3)- СН(СН3)- СН(СН3)-СН3 .
Укажите пути образования этого изомера октана в ходе реакции.
7. При алкилировании метанола непредельными соединениями в присутствии кислот:
СН3ОН + СН2=СН-R → CH3O-CH(CH3)R ,
олефены располагаются в следующий ряд активности:
CH2=CH2 < CH2=CH-CH3 < CH2=C(CH3)2 .
Чем обусловлен указанный ряд активности алкенов?
8. При взаимодействии метанола с изобутиленом в кислой среде:
CH3OH + CH2=C(CH3)2 → CH3O-C(CH3)3 ,
изменение свободной энергии Гиббса ( ΔG) при 298К составляет минус 6,53 ккал/моль, а при 400К – минус 0,65 ккал/моль. Рассчитайте константы равновесия реакций при указанных температурах. При каких температурах целесообразно проводить данный процесс?
Алкилирование ароматических соединений непредельными соединениями
Получение алкилпроизводных ароматических соединений является широко используемым процессом в нефтехимической отрасли РТ. Алкилированнием бензола этиленом (ОАО «Нижнекамскнефтехим») получают этилбензол:
C6H6 + CH2=CH2 → C6H5CH2CH3.
Этилбензол далее дегидрируется и превращается в стирол (С6Н5СН=СН2). Стирол является одним из важнейших мономеров в химии полимеров. Стирол используется для получения полистирола. Большое количество стирола потребляется в производстве сополимеров на его основе – АБС-пластика (термопластичный тройной сополимер акрилонитрила, бутадиена и стирола), бутадиен-стирольных, бутадиен-изопрен-стирольных каучуков. Сополимеры стирола с малеиновым ангидридом находят применение в лакокрасочной промышленности. Высокими конструкционными свойствами обладает сополимер стирола, метилакрилата и акрилонитрила. На основе стирола получают бутадиен-стирольные, изопрен-стирольные термоэластопласты, которые называют материалами XXI века. В 2000 г. в России было произведено 90,5 тыс. тонн сополимеров на основе стирола. При этом из-за рубежа (импорт) дополнительно было завезено 78,2 тыс. тонн этих сополимеров. Эти данные указывают на то, что отечественная промышленность пока не в состоянии обеспечить потребности внутреннего рынка в этих полимерах.
На ОАО «Казаньоргсинтез» алкилированием бензола пропиленом получают изопропилбензол (кумол):
С6Н6 + СН3-СН=СН2 → С6Н5СН(СН3)2 .
Окислением изопропилбензола кислородом получают гидроперекись изопропилбензола, которая при разложении в присутствии кислот образует фенол и ацетон. Фенол является мономером при получении фенолформальдегидных смол, он является исходным продуктом при получении таких важнейших мономеров, как капролактам, адипиновая кислота, дифенилолпропан. При алкилировании фенола образуются его алкилпроизводные. Они используются при производстве поверхностно-активных веществ, антиоксидантов, фенол-формальдегидных смол. На основе фенола получают пластификаторы. Аминирование фенола аммиаком является экологически чистым способом получения анилина.
Алкилированием бензола высшими α-олефинами (СН2=СН-СnH2n-1, n = 12-14) получают высшие алкилбензолы (их называют линейными алкилбензолами, ЛАБ). Сульфирование высших алкилбензолов (ОАО «Нефис», г.Казань) серным ангидридом приводит к получению сульфокислот высших алкилбензолов. Их натриевые соли (алкилбензолсульфонаты) являются основой синтетических анионных моющих веществ.
Приведенные данные указывают на то, что алкилароматические соединения являются базовыми соединениями, на использовании которых основан большой круг химических производств. Поэтому их производство является одним из важнейших направлений промышленной органической химии.
Механизм алкилирования бензола этиленом. Все реакции алкилирования ароматических углеводородов непредельными соединениями протекают сходным образом. Эти превращения протекают в присутствии сильных кислот Бренстеда. В настоящем разделе рассматривается механизм алкилирования бензола этиленом.
Роль кислоты Бренстеда в обсуждаемых реакциях заключается в том, что протон этих соединений, взаимодействуя с непредельными соединениями, приводит к получению алкильных катионов. Протонирование этилена приводит к образованию этильного катиона:
СН2=СН2 + Н+ → СН3-СН2+ .
Образовавшийся этильный катион далее вступает с бензолом в реакцию электрофильного ароматического замещения. В ходе этого процесса происходит замещение атома водорода в бензоле на этильную группу:
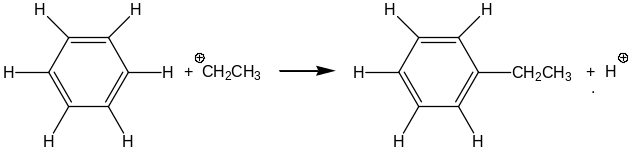
Процесс замещения называют электрофильным по той причине, что в этом превращении происходит атака молекулы бензола положительно заряженной частицей.
По предложению английского химика Кристофера Ингольда (1893-1970) все реагенты разделяются на электрофильные и нуклеофильные. Электрофильными (буквальный перевод – любящие электроны. Электроны имеют отрицательный заряд.) называют такие соединения, которые имеют на реакционном центре дефицит электронов, т.е. активный центр несет либо полный, либо частичный положительный электрический заряд. Нуклеофильными (буквальный перевод – любящие ядро. Ядро атомов несет положительный заряд.) называют такие соединения, реакционный центр которых несет полный или частичный отрицательный электрический заряд. С позиций электростатики взаимодействие противоположно электрически заряженных тел сопровождается выделением энергии. Такого типа взаимодействия благоприятны для осуществления химических превращений. К.Ингольд предложил рассматривать все химические реакции как взаимодействия электрофилов с нуклеофилами. Этот принцип лег в основу классификации органических реакций (например, нуклеофильное замещение у насыщенного атома углерода, электрофильное присоединение к кратным связям, нуклеофильное присоединение к карбонильной группе и т.д.). Эта классификация реакций используется до настоящего времени.
Процесс взаимодействия этильного катиона с бензолом включает ряд стадий. Первоначально в ходе этого процесса образуется π-комплекс:

Образование этих комплексов сопровождается незначительным выигрышем в энергии (около 5 ккал/моль). В π-комплексе этильный катион не связан с каким-либо определенным атомом углерода бензольного ядра. Молекула бензола и катионная частица удерживаются друг около друга за счет электростатических сил взаимодействия между отрицательно заряженной π-системой бензола и катионным центром электрофила, который несет положительный заряд. Есть еще один фактор, который способствует понижению энергии при образовании π-комплекса. Это межорбитальное межмолекулярное донорно-акцепторное взаимодействие между реагирующими частицами (между высшей занятой молекулярной орбиталью бензола и низшей свободной молекулярной орбиталью этильного катиона).
Образовавшийся π-комплекс на последующей стадии превращается в σ-комплекс:

Этот промежуточный продукт образован за счет взаимодействия этильного катиона с одним из атомов углерода бензола. При этом атакуемый атом углерода из sp2-гибридного состояния переходит в sp3-гибридного состояние. Для образования химической связи между атомом углерода бензольного ядра и атомом углерода этильного катиона нужна пара электронов. Эта пара электронов на атакуемом атоме углерода бензольного кольца возникает в результате локализации электронов, которые первоначально находились на одной из занятых молекулярных орбиталей бензола. Локализация пары электронов на одном из атомов углерода бензола приводит к тому, что все другие атомы углерода бензольного ядра приобретают положительный заряд. Образование σ-комплекса приводит к разрушению устойчивой сопряженной системы бензола. Поэтому этот процесс сопровождается повышением энергии системы.
Целевой продукт – этилбензол – образуется в результате отщепления протона от σ-комплекса:

При этом сопряженная система ароматического ядра полностью восстанавливается. Этот процесс протекает с выделением энергии.
Дальнейшее развитие процесса идет по пути повторения рассмотренных стадий.
Побочные процессы в реакциях алкилирования бензола этиленом.
Полиалкилирование бензола. Образующийся в ходе алкилирования бензола этиленом этилбензол не является инертным по отношению к этильным катионам. Этильная группа в ароматическом ядре проявляет электронодонорный характер. Она увеличивает электронную плотность на атомах углерода ароматического ядра, в первую очередь, на пара- и орто-атомах углерода. Коль ароматическое ядро этилбензола по сравнению с бензолом более обогащено электронами, то, естественно, частица с положительным зарядом, этильный катион, будет более легко реагировать с этилбензолом, чем с бензолом. Это взаимодействие приводит к образованию пара- и орто-диэтилбензола:

В молекулах диэтилбензолов электронная плотность на ароматическом ядре возрастает уже под действием двух электронодонорных групп. Это приводит к тому, что диэтилбензолы более легко реагируют с этильными катионами, образуя 1,2,4-триэтилбензол:

Процесс алкилирования может повторяться снова и снова, пока в ходе этого процесса не образуется гексаэтилбензол.
Процесс полиалкилирования бензола является нежелательным процессом. Он связан с бесполезной потерей дефицитного бензола. Поэтому технология производства этилбензола предусматривает решения, позволяющие уменьшить вклад этих побочных направлений. Логика одного из подходов заключается в том, что в реакционной системе всегда бензол должен находиться по сравнению с образующимся бензолом в избытке. Тогда возникающие в ходе протонирования этилена этильные катионы преимущественно будут сталкиваться с молекулами бензола, а не этилбензола. Это обстоятельство приводит к тому, что при избыточном содержании бензола в реакционной смеси вклад направлений, приводящих к образованию полиэтилбензолов, уменьшается, хотя полностью избавиться от этих нежелательных процессов не удается. В промышленных установках при получении этилбензола соотношение бензол : этилен составляет 2 ÷ 3 : 1, но в некоторых технологических установках оно достигает 8 ÷ 16 : 1 (в молях).
Другим направлением, позволяющим уменьшить затраты бензола на побочные процессы, является использование в процессе алкилирования реакции трансалкилирования.
Реакция трансалкилирования. Реакции алкилирования ароматических соединений являются обратимыми. В ходе процесса алкилирования бензола этиленом протекают взаимодействия не только приводящие к получению полиалкилпроизводных, но и обратные процессы. Молекулы полиалкилбенолов могут протонироваться. Образующийся σ-комплекс может либо отщепить протон, либо этильный катион. Образующийся этильный катион далее может прореагировать с молекулой бензола, например:

Краткая запись этих процессов в виде химического уравнения выглядит следующим образом:

В ходе этого процесса от молекулы полиалкилбензола алкильная группа переносится на молекулу бензола. Процессы подобного типа получили название реакций трансалкилирования или переалкилирования.
В технологии получения этилбензола образующиеся в ходе процесса ди- и триэтилбензолы выделяют, и их вновь подают в реактор алкилирования. В реакторе алкилирования полиэтилбензолы расходуются в процессах трансалкилирования. Тем самым происходит сокращение непроизводительного расхода бензола в процессе получения этилбензола.
Образование высших линейных алкилбензолов. Другим побочным направлением в процессе получения этилбензола является образование высших линейных алкилбензолов. Это направление связано с тем, что в условиях алкилирования частично идет катионная олигомеризация этилена, которая приводит к получению бутильных, гексильных, октильных и т.п. катионов:
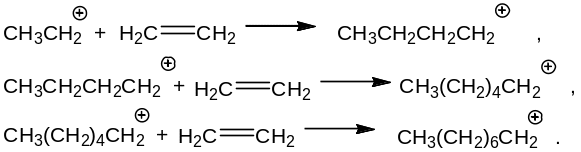
Каждый из образующихся катионов способен алкилировать бензол, что приводит к появлению в реакционной смеси н-бутил-, н-гексил-, н-октилбензола:

Высшие линейные алкилбензолы имеют более высокие температуры кипения, и они отделяются от этилбензола в процессе ректификации.
Побочные процессы, связанные с протонированием боковой цепи этилбензола. В качестве побочного продукта в реакции алкилирования бензола этиленом образуется толуол. Образование толуола связано с тем, что в ходе реакции протекает побочный процесс – протонирование простой углерод-углеродной связи в этильном фрагменте этилбензола:

Промежуточный карбониевый ион может распадаться не только по приведенной выше схеме, но и по альтернативному пути, приводящему к бензильному катиону и метану. Взаимодействие бензильного катиона с бензолом приводит к получению дифенилметана:

Протонированию могут подвергаться и углерод-водородные связи этильного фрагмента этилбензола. Это направление взаимодействия приводит к получению изомерных дифенилэтанов:

Таким образом, процесс получения этилбензола является сложным. В условиях реакции образуется не только целевой продукт, но и ряд побочных соединений, включая такие неожиданные продукты, как водород, метан.
Термодинамика процессов алкилирования бензола этиленом.
В таблицах 10-12 приведены изменения свободной энергии Гиббса и константы равновесия при разных температурах для процессов обсуждаемых процессов алкилирования.
Таблица 10. Изменения свободной энергии Гиббса (ΔG, ккал/моль) и константы равновесия (Кр) для алкилирования бензола этиленом: С6Н6 + С2Н4 → С6Н5С2Н5 в газовой фазе
T, K |
ΔG |
Kр |
298 |
-16,06 |
6,2·1011 |
400 |
-12,96 |
1,2·107 |
500 |
-9,94 |
2,3·104 |
Таблица 11. Изменения свободной энергии Гиббса (ΔG, ккал/моль) и константы равновесия (Кр) для алкилирования этилбензола этиленом: С2Н5С6Н5 + С2Н4 → п-С2Н5С6Н4С2Н5 в газовой фазе
T, K |
ΔG |
Kр |
298 |
-14,54 |
4,7·1010 |
400 |
-11,00 |
1,0·106 |
500 |
-7,56 |
2,0·103 |
Таблица 12. Изменения свободной энергии Гиббса (ΔG, ккал/моль) и константы равновесия (Кр) для трансалкилирования бензола п-диэтилбензолом: С6Н6 + п-С2Н5С6Н4С2Н5 → 2 С2Н5С6Н5 в газовой фазе
T, K |
ΔG |
Kр |
298 |
-1,52 |
13,06 |
400 |
-1,96 |
11,80 |
500 |
-2,38 |
11,00 |
Из приведенных в таблицах 10,11 данных видно, что константы равновесия, как образования этилбензола, так и п-диэтилбензола, увеличиваются с уменьшением температуры. Таким образом, изменяя температуру проведения реакции алкилирования невозможно добиться преимущественного образования одного лишь этилбензола. Указанные константы равновесия сохраняются большими в широком интервале температур, что обеспечивает практически необратимое образование указанных соединений в обсуждаемых условиях.
Равновесие реакции трансалкилирования сдвинуто в сторону образования этилбензола и константы равновесия процесса трансалкилирования мало чувствительны к температуре.
Казалось бы, опираясь на полученные данные, реакцию алкилирования бензола этиленом необходимо проводить при пониженных температурах. Однако реально этот процесс проводится при повышенных температурах, которые могут достигать 5000С. Это обусловлен желанием полнее использовать потенциал реакции трансалкилирования при получении этилбензола. Равновесие этой реакции медленно устанавливается при низких температурах, и быстро – при повышенных.
Реакции алкилирования протекают с выделением тепла. Энтальпия реакции алкилирования бензола этиленом равна минус 25,2 ккал/моль, этилбензола этиленом – минус 24,94 ккал/моль. Процесс трансалкилирования практически термонейтрален. Энтальпия реакции трансалкилирования бензола п-диэтилбензолом составляет минус 0,26 ккал/моль.
Катализаторы реакции алкилирования. В процессе алкилирования долгое время в качестве катализатора использовался хлористый алюминий. Он продолжает использоваться до настоящего времени на существующих производствах алкилирования ароматических соединений.
Основы использования хлористого алюминия как катализатора в различных органических реакциях заложил известный российский химик Густавсон Г.Г.
Гавриил Гавриилович Густавсон (1842-1908) закончил в 1865 г. физико-математический факультет Петербургского университета. С момента окончания университета по 1875 г. состоял лаборантом при кафедре технической химии этого университета. В эти годы Густавсон Г.Г. являлся ближайшим помощником выдающегося русского химика-органика Бутлерова А.М. В 1873 г. Густавсон защитил магистерскую диссертацию и в 1875 г. возглавил кафедру органической химии в Петровской сельскохозяйственной академии. В стенах этой академии им была выполнена докторская диссертация на тему: «Органические соединения в их отношении к галоидным солям алюминия» (1884 г.). Эта работа, изложенная на 84 страницах, была оценена очень высоко. С нее началось широкое использование хлористого алюминия как катализатора в органической химии. Наряду с французскими химиками Фриделем и Крафтсом, Густавсон является полноправным разработчиком методов получения алкилароматических соединений.
Густавсоном было открыто превращение α,ω-дигалоидпроизводных под действием цинка в циклические углеводороды. Так им был получен из 1,3-дибромпропана циклопропан. Впоследствии это превращение получило название реакции Густавсона.
Густавсон Г.Г. является одним из организаторов исследований в области агрохимии. Его учебное пособие «Двадцать лекций агрономической химии» (1889 г.) имело большой успех.
Из приведенных выше данных видно, что алкилирование ароматических соединений непредельными углеводородами возможно в присутствии кислот Бренстеда-Лаури. Хлористый алюминий не относится к этому классу соединений. Поэтому он не должен катализировать рассматриваемые процессы. Это действительно наблюдается экспериментально. Химически чистый хлористый алюминий не проявляет никакой активности в реакциях алкилирования. Каталитическая активность хлористого алюминия начинает проявляться тогда, когда к нему дополнительно в реакционную систему вводят соединения с подвижным атомом водорода, например, воду, спирты, хлористый водород. Промоторами могут быть галоидные алкилы.
Такие соединения, как вода, спирты энергично реагируют с хлористым алюминием с выделением хлористого водорода:
AlCl3 + ROH → Al(OR)Cl2 + HCl ,
AlCl3 + H2O → Al(OH)Cl2 + HCl .
Эти процессы приводят к появлению в реакционной системе кислоты Бренстеда-Лаури – хлористого водорода. Это соединение не является инертным по отношению к хлористому алюминию. На атоме хлора в хлористом водороде на внешней электронной оболочке локализованы три неподеленные пары электронов. Поэтому этот атом способен проявлять свойства основания Льюиса. Атом же алюминия в хлористом алюминии имеет вакантную атомную орбиталь, и этот атом проявляет свойства кислоты Льюиса. При взаимодействии хлористого алюминия с хлористым водородом в системе генерируется новой соединение – алюминийхлористоводородная кислота:
HCl + AlCl3 → H+[AlCl4]− .
В тетрахлоралюминат-анионе отрицательный заряд делокализован на пяти атомах. Это обстоятельство приводит к тому, что кулоновское взаимодействие протона с комплексным анионом становиться малым. Поэтому алюминийхлористоводородная кислота является по сравнению с хлористым водородом намного более сильной кислотой. Величина функции кислотности Ho, которая характеризует протонирующую способность соединения, для алюминийхлористоводородной кислоты равна минус 15, в то время, как концентрированная серная кислота характеризуется величиной Но, равной минус 12. Алюминийхлористоводородная кислота является по функции кислотности в 1000 раз более сильной кислотой, чем концентрированная серная кислота.
При наличии в системе избыточного хлористого алюминия тетрахлоралюминат-анион образует комплексы со следующими молекулами хлористогоалюминия, образуя полиядерные анионные комплексы:


В этих комплексных ионах делокализация отрицательного заряда увеличивается. Это приводит к тому, что протонирующая способность соответствующих этим анионам кислот возрастает.
Таким образом, при алкилировании ароматических соединений непредельными соединениями при катализе реакции хлористым алюминием на первой стадии принимают меры для генерации алюминийхлористоводородная кислота.
Алюминийхлористоводородная кислота является кислотой Бренстеда-Лаури. С позиций рассмотренных выше механизмов превращений она, казалось бы, должна инициировать все указанные взаимодействия. Однако алюминийхлористоводородная кислота не растворима в углеводородах. При введении этой кислоты в реакционную систему в ходе процесса наблюдается индукционный период, после которого реакция начинает нормально протекать. Наличие индукционного периода в химических реакциях свидетельствует о том, что в реакционной системе возникают какие-то промежуточные продукты, которые являются непременными участниками химического взаимодействия. Наблюдая за состоянием реакционной смеси в реакциях алкилирования ароматических соединений непредельными соединениями при катализе хлористым алюминием, Густавсон Г.Г. обратил внимание на то, что в ходе реакции на поверхности хлористого алюминия первоначально медленно образуется темно-коричневая маслянистая жидкость, которая частично растворима в углеводородах. Индукционный период в реакции алкилирования превращается тогда, когда на поверхности катализатора появляется эта темная жидкость. Исследование процессов, протекающих на поверхности алюминийхлористоводородной кислоты, показало, что первоначально на этой поверхности медленно идет полиалкилирование бензола (например, при алкилировании бензола этиленом образуется 1,2,4-триэтилбензол). Основность образующихся полиалкилбензолов столь высока, что далее они образуют устойчивые катионные σ-комплексы с алюминийхлористоводородной кислотой.:
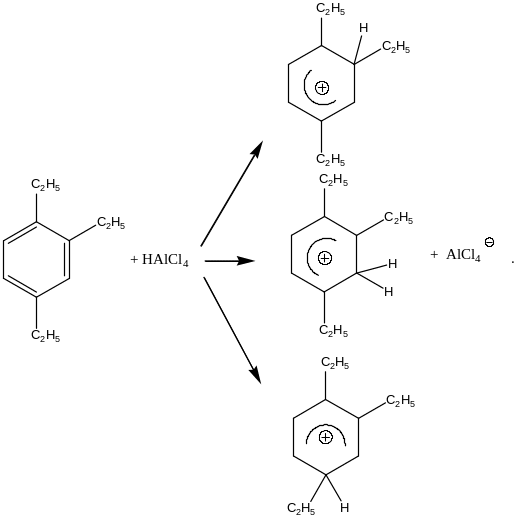
В ходе взаимодействия образуются σ-комплексы всех возможных структур. На приведенной выше схеме показаны только часть из образующихся σ-комплексов. Эти σ-комплексы окрашены. Они существуют в виде тесной ионной пары и обладают частичной растворимостью в углеводородах. Истинными катализаторами в реакциях алкилирования бензола этиленом являются не хлористый алюминий, не алюминийхлористоводородная кислота, а катионные σ-комплексы полиалкилбензолов с алюминийхлористоводородной кислотой. Появление отмеченных σ-комплексов в углеводородной фазе связано с переходом от гетерогенного катализа к гомогенному катализу. Протонирование молекул этилена в этом случае в углеводородной фазе ведется катионными σ-комплексами. При их взаимодействии с молекулами непредельных соединений генерируются алкильные катионы, например:

Катионные σ-комплексы, образующиеся в системе: непредельное соединение - алюминийхлористоводородная кислота – бензол, как знак признания заслуг Густавсона Г.Г. в развитии представлений о протекании реакций алкилирования ароматических соединений непредельными соединениями, в настоящее время называют катализаторами (комплексом) Густавсона.
Катализаторы Густавсона образуются также при взаимодействии молекул галоидалкилов с полиалкилбензолами в присутствии хлористого алюминия. В этом случае первоначально в ходе реакции молекул галоидалкилов с хлористым алюминием генерируются карбениевые ионы, имеющие в качестве противоиона тетрахлоралюминат-анион, например:
СН3СН2Cl + AlCl3 → CH3CH2+ AlCl4− .
При взаимодействии этой ионной пары с молекулами полиалкилбензолов образуются катионные σ-комплексы:

Наличие индукционного периода в химических реакциях является неблагоприятным фактором. Такие процессы трудно управляются. Поэтому для исключения индукционного периода в реакциях алкилирования бензола непредельными соединениями в присутствии хлористого алюминия катализатор Густавсона получают в отдельном реакторе, и уже полученный катализатор подают в реактор алкилирования.
Первоначально процессы алкилирования ароматических соединений алкенами с использованием хлористого алюминия как катализатора являлись периодическими. В дальнейшем был совершен переход на непрерывные схемы производства. Большим недостатком этих технологий является то, что в них появляется отдельная стадия – отделение органических компонент реакционной среды от хлористого алюминия. Эта процедура включает водно-щелочную обработку. При этом возвратный бензол обводняется, что влечет за собой появление узла по сушке бензола. Все это ведет к усложнению технологической схемы.
Современные процессы алкилирования являются непрерывными, и они основаны на использовании нанесенных катализаторов. При этом в технологии полностью исчезает стадия отделения катализаторов от органических компонент реакционной среды.
Среди таких катализаторов используются фосфорная кислота на различных формах цеолитах (процесс UOP). При использовании этого катализатора процесс ведут при 3000С и давлении 40-65 атм. Фирма Mobil-Badger разработала процесс получения этилбензола на цеолите ZSM-5, который проводится при 435-4500С и давлении 14-28 атм. Фосфорная кислота на двуокиси кремния, активированная трехфтористым бором уже много лет используется фирмой UOP для производства этилбензола. Процесс ведут при 2900С и давлении 60-65 атм.