

Катализаторы процесса каталитического крекинга
Приведенное выше рассмотрение механизма процесса каталитического крекинга основывалось на том, что химические превращения инициировались протоном. Таким образом, каталитический крекинг является процессом, который катализируется кислотами Бренстеда-Лаури.
Казалось бы, что в каталитическом крекинге в качестве катализаторов можно использовать обычные минеральные кислоты, например, серную, фосфорную и т.д. Подобного типа минеральные кислоты действительно способны привести к расщеплению высших углеводородов на низшие алканы и алкены. Однако в промышленной органической химии они не могут быть использованы. Связано это с тем, что жидкие кислоты (даже слабые карбоновые кислоты) при высоких температурах вызывают интенсивную коррозию металлической аппаратуры. Поэтому в каталитическом крекинге практическое применение нашли твердые кислоты, которые в минимальной степени способны взаимодействовать со стенками аппаратуры.
Использование твердых кислот для катализа предполагает, что углеводороды из газовой фазы первоначально должны адсорбироваться на поверхности твердой фазы. Затем, в ходе миграции адсорбированной молекулы углеводорода по поверхности твердой фазы, она должна встретиться с кислотным центром. На этом центре должны пройти процессы распада молекулы высшего углеводорода. В конце процесса образовавшиеся молекулы продуктов реакции с поверхности твердой фазы должны десорбироваться в газовую фазу.
Естественно, поскольку процессы каталитического крекинга протекают на поверхности твердого катализатора, то чем больше площадь этой поверхности, то тем выше должна быть активность катализатора. Поэтому катализаторы, используемые в каталитическом крекинге, должны обладать высокой пористостью. В качестве таких материалов наиболее подходящими оказались цеолиты.
Краткие сведения о цеолитах.
Цеолиты (буквальный перевод с греческого – кипящий камень) – это неорганические полимеры, получаемые поликонденсацией орто-кремниевой кислоты и гидроокиси алюминия. Эти полимеры относятся к классу алюмосиликатов. Алюмосиликаты составляют основу природных глин. Название цеолитов отражает тот факт, что они являются высокопористыми веществами.
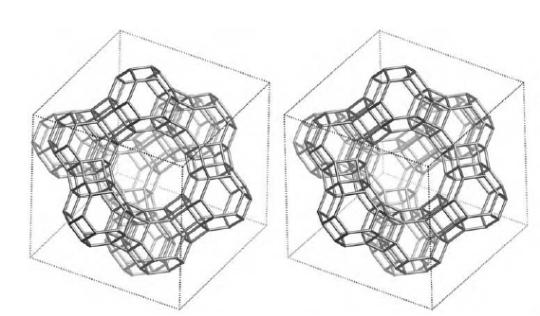
Рис. 1. Структура алюмосиликата типа “Цеолит Y”. Показаны виды спереди и сзади.

Рис. 2. Структура алюмосиликата типа “Морденит”.
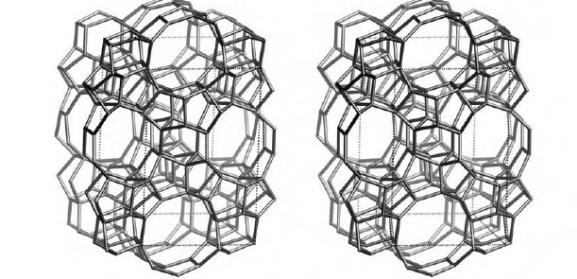
Рис.3. Структура алюмосиликата типа “ZSM-5”.
Структура алюмосиликатов значительно зависит от соотношения алюминия и кремния в этих материалах. В зависимости от этого соотношения образуются алюмосиликаты, обладающие разной кристаллической структурой. На рис. 1-3 в качестве примера приведены структуры некоторых алюмосиликатов.
Приведенные выше данные по структурам цеолитов указывают на то, что в них существуют поры. Благодаря этому цеолиты обладают высокой удельной поверхностью (отношение общей поверхности пористого тела к его объему или массе). У цеолитов удельная поверхность достигает величины более 1000 м2/г.
Исторически применение алюмосиликатов в процессах каталитического крекинга прошло ряд этапов. Первоначально в качестве катализаторов в этих процессах использовались природные алюмосиликаты – глины. Использование природных алюмосиликатов приводило к повышенной степени образования кокса в ходе крекинга. Оказалось, что к этому крайне нежелательному процессу приводят соединения тяжелых металлов (железа, никеля, кобальта, олова и т.д.), которые всегда в незначительных количествах присутствуют в природных алюмосиликатах. Поэтому встала задача получения алюмосиликатов, которые не содержали бы следов тяжелых металлов. Путь решения этой задачи потребовал организации получения синтетических алюмосиликатов. При получении синтетических алюмосиликатов используют очищенные до высокой степени гидроокись алюминия и ортокремневую кислоту. Тем самым исключается попадание в алюмосиликаты соединений тяжелых металлов. На первых этапах развития промышленности синтетических алюмосиликатов производились аморфные алюмосиликаты. Аморфные алюмосиликаты не обладали упорядоченной структурой. Их структура представляла беспорядочную трехмерную сеть из взаимосвязанных оксидов кремния и алюминия. Они обладали слабо развитой поверхностью. Поэтому аморфные алюмосиликаты обладали пониженной каталитической активностью. В дальнейшем было освоено производство высокопористых кристаллических алюмосиликатов (структуры некоторых кристаллических алюмосиликатов приведены выше), которые применяются в качестве катализаторов в настоящее время.
Причина появления каталитической активности у алюмосиликатов в процессах каталитического крекинга.
Приведенное выше рассмотрение механизма каталитического крекинга указывает на то, что необходимым условием для осуществления процессов является присутствие в системе кислот Бренстеда-Лаури. Поверхностный слой алюмосиликатов содержит гидроксильные группы. Поэтому можно было бы полагать, что эти гидроксильные группы и являются источником протонов. Однако оказалось, что сами алюмосиликаты не обладают никакой каталитической активностью в процессах каталитического крекинга. Алюмосиликаты начинают катализировать эти процессы только тогда, когда они предварительно проходят высокотемпературную обработку (8000С и выше). Если прокаленные алюмосиликаты выдержать во влажной атмосфере, то их каталитическая активность полностью исчезает, но она вновь появляется при их повторном прокаливании. Эти экспериментальные данные указывают на то, что не поверхностные гидроксильные группы в цеолитах являются ответственными за явление катализа в обсуждаемом процессе. При прокаливании алюмосиликатов в них протекают какие-то превращения, которые и являются ответственными за факт появления у них каталитической активности.
Выяснение причин появления каталитической активности у алюмосиликатов потребовало длительных исследований. Полное решение этой проблемы стало возможным только тогда, когда появились прецизионные методы определения содержания гидроксильных групп в них, и метода ядерного магнитного резонанса высокого разрешения в твердом теле. Последний метод позволял судить об электронном состоянии атомов в алюмосиликатах.
Родственными алюмосиликатам по химической природе являются различные модификации двуокиси кремния. Она также является неорганическим полимером. Поверхностный слой двуокиси кремния также содержит гидроксильные группы. Эти гидроксильные группы легко регистрируются методом ИК-спектроскопии. Оказалось, что при прокаливании различных модификаций двуокиси кремния происходит полная потеря гидроксильных групп. Прокаленная двуокись кремния не обладает никакой каталитической активностью в процессах каталитического крекинга. Наблюдаемое явление указывает на то, что при термической обработке происходит дегидратация поверхностного слоя двуокиси кремния:

В алюмосиликатах поверхностный слой также содержит гидроксильные группы при атомах кремния. Однако в этом случае гидроксильные группы пространственно разделены по сравнению с двуокисью кремния. Ответственными за это являются атомы алюминия, включенные в структуру полимерной цепи:
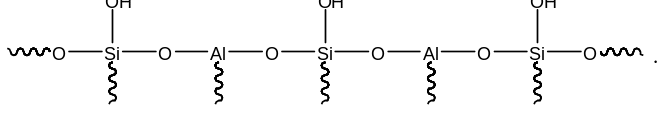
При термической обработке алюмосиликатов, как в случае двуокиси кремния происходит дегидратация за счет отщепления молекул воды при взаимодействии поверхностных гидроксильных групп друг с другом. Молекулы воды при этом удаляются в газовую фазу. В силу этого процесс становится необратимым. Однако пространственная разобщенность гидроксильных групп при дегидратации приводит к тому, что на поверхности остаются анионный кислородный центр и катионный центр – силициниевый ион:

В ходе указанного процесса исчезают два кислотных Бренстедовских центра и появляются один основной и один кислотный Льисовских центры. Эти изменения в структуре поверхностного слоя фиксируются современными методами.
Реакция каталитического крекинга инициируется возникшими при прокаливании алюмосиликатов силициниевыми ионами. Все процессы, происходящие при прокаливании алюмосиликатов, являются обратимыми. Поэтому, естественно, если прокаленный катализатор будет контактировать с парами воды, то все приведенные выше реакции будут протекать в обратном направлении. Это приведет к исчезновению катионных центров на поверхности алюмосиликатов. Соответственно, исчезает и их каталитическая активность.
Взаимодействие силициниевых ионов со связями С-С или С-Н приводит к возникновению карбкатионов:

Дальнейший распад карбкатионов приводит к возникновению протонов, метильных катионов, которые являются продолжателями катионной цепи превращений. Однако все эти превращения продолжают протекать на поверхности алюмосиликата. Поэтому, когда частицы катализатора покрываются пленкой кокса, препятствующей адсорбции молекул углеводородов сырья на поверхности алюмосиликатов, то активность катализатора падает. Отсюда следует, что для поддержания высокой активности катализатора пленку кокса на его поверхности необходимо удалять. Это делается путем выжига этой пленки при высоких температурах в атмосфере воздуха. В ходе этой операции возникшие на поверхности катализатора алкилсилановые и силановые фрагменты окисляются до силанольных (силанолами называют соединения, содержащие фрагмент Si-OH). Структура алюмосиликата при этом полностью восстанавливается.
Эволюция технологии каталитического крекинга.
Приведенные выше данные указывали на то, что чем больше время контакта углеводородного сырья с катализатором, тем более вероятным становиться протекание побочных реакций. При этом происходит безвозвратная потеря первичных продуктов каталитического крекинга и возрастает образование кокса. Вся эволюция технологии каталитического крекинга является демонстрацией того, что с уменьшением времени контакта сырья с катализатором возрастал выход полезных продуктов.
Первые установки каталитического крекинга (1930-1940 г.г.) имели реактора, в которых на полках находился неподвижный слой катализатора. Время контакта сырья с катализатором исчислялось часами. Эти установки давали только 18% полезных продуктов.
Далее было осознано, что коль реакция крекинга проходит на поверхности катализатора, то целесообразно его на полках перемешивать. Появились установки с перемешивающимся слоем катализатора (1940-1954 г.г.). Время контакта при этом было сокращено до 10-15 минут. Проведенные совершенствования технологии привели к увеличению выхода полезных продуктов до 28%.
Следующим этапом в развитии технологии каталитического крекинга явилось использование кипящего слоя катализатора (1954-1974 г.г.). Время контакта сырья с катализатором при этом было сокращено до 3-5 минут. Эти установки давали уже 42% полезных продуктов.
Дальнейшим совершенствованием технологии каталитического крекинга явилось использование лифт-реакторов. Эти установки появились в 1974 г., и они частично эксплуатируются до настоящего времени. Время контакта на этих установках составляет менее 3 секунд. Выход полезных продуктов составляет 53 %. В лифт-реакторах, которые представлют собой обычную обогреваемую трубу, сырье и катализатор подают снизу, а продукты вместе с катализатором выводятся сверху. Цеолиты используют в виде микросферических частиц, средний диаметр которых составляет 60 мкм. Частицы размером до 40 и свыше 105 мкм не используют, так как слишком малые частицы уносятся из реактора, а крупные не обладают прочностью, достаточной для избежания истирания в потоке. За счет протекающих одновременно с крекингом процессов полимеризации образуется кокс и катализатор теряет активность. После реактора катализатор отделяется от смеси углеводородов на циклонах. Далее катализатор поступает в регенератор, в котором при 650-6700С в атмосфере воздуха проводят выжигание кокса, а выделяющееся при этом тепло используют для нагрева сырья.
В новейших установках каталитического крекинга предусмотрена подача потоков катализатора и сырья во взаимно перпендикулярных направлениях. Эти установки получили название установок с ультракоротким временем контакта (УКВК). Оно доведено до миллисекунд. Эта технология позволяет конвертировать углеводородное сырье в полезные продукты с выходом 96%.