

§ 5. Общие сведения о хроматографии газов
Физико-химическая сущность любого хроматографического метода анализа газовых смесей состоит в селективной сорбции компонентов смеси твердыми или жидкими поглотителями с последующей их раздельной десорбцией при помощи инертного к данному сорбенту газа-носителя.
Анализ проводится в трубках (колонках), наполненных сорбентом. Анализируемый газ вводится в колонку через дозаторы в потоке газа-носителя. Так как газ-носитель инертен к веществу сорбента, то он, не задерживаясь, выходит из колонки. Что же касается компонентов исследуемой газовой смеси, то они, обладая различным сродством к сорбенту, распределяются по длине колонки на отдельные зоны в порядке уменьшения своих сорбционных свойств. Обозначим компоненты газовой смеси А, Б, В и газ-носитель П. Тогда графически можно изобразить формирование зон сорбции, как это показано на рис. 8.
Процесс сорбции обратим. При промывке колонки газом-носителем резко снижается концентрация компонентов газовой смеси под зонами сорбции и происходит десорбция. Очевидно, что десорбция компонентов газа будет осуществляться в порядке, обратном их сорб-ционной активности. Иначе говоря, отдельные компоненты газа будут двигаться по колонке с различными скоростями и их время удерживания в колонке будет тоже различным. Таким образом, во время движения газа-носителя через колонку происходит распределение компонентов анализируемой смеси между подвижной и неподвижной фазами.
Представленная на рис. 8 несколько упрощенная схема достаточно наглядно показывает, что на выходе из колонки вполне возможно разделение, а следовательно, и фиксация отдельных компонентов газовой смеси. Практика показывает, что это достигается даже при хроматографировании смесей, содержащих компоненты, весьма близкие по химическим и физическим свойствам.
Хроматографический метод разделения веществ впервые предложенный еще в 19013 г. русским ботаником М. С. Цветом, в настоящее время получил исключительно широкое распространение при анализе самых различных сложных смесей веществ, находящихся в жидком и газообразном состоянии-. В качестве примера можно указать, что в практику исследовательских и заводских нефтеперерабатывающих и нефтехимических
 Схема
зон сорбции.
Схема
зон сорбции.
Рис. 8.
предприятий внедрены хроматографические методы анализа для определения содержания;
углеводородов С2—С5 в сырой нефти;
предельных и непредельных углеводородов, а также неуглеводородных компонентов (Н2, О2, N2, CO, СO2, H2S) в сухом газе при любых соотношениях;
предельных и непредельных углеводородных газов во фракции С4 (при контроле производства бутадиена из бутана);
различных углеводородов в прямогонных бензинах и во фракциях вторичного происхождения;
сероводорода в газах;
микропримесей сераорганических соединений (сероуглерода, метил- и этилмеркаптанов, тиофена, некоторых сульфидов) в сырье для каталитических процессов при суммарной концентрации до 100 мг/м3;
целевых синтетических жирных кислот во фракциях C5 – C6, С7—С9, С9—С10, C10—С13;
примесей в нефтяном бензоле;
основного вещества в нефтяном ксилоле и многих других компонентов нефтепродуктов, кипящих до 350 °С.
Существует несколько разновидностей хроматографического анализа, но все они основаны на том же принципе распределения компонентов анализируемой смеси между двумя нёсмешивающимися фазами: неподвижной и подвижной. Неподвижной фазой является твердый или жидкий сорбент, а подвижная фаза (газ или жидкость) пропускается через колонку с сорбентом, участвуя в переносе разделяемых компонентов. Если подвижная фаза — газ, то хроматография называется газовой, если подвижная фаза — жидкость, хроматография называется жидкостной.
При анализе жидких смесей могут быть использованы оба варианта хроматографического метода. Наибольшее распространение при исследовании жидких смесей, кипящих до 350 °С и не разлагающихся при этих температурах, получило хроматографирование их в газовой фазе (с предварительным испарением). Высокомолекулярные вещества с температурой кипения более 350°С хроматографируются в жидкой фазе.
Газовая хроматография в свою очередь разделяется на газоадсорбционную и газожидкостную.
В газоадсорбционной хроматографии подвижной фазой является инертный газ, а неподвижной — адсорбент — твердое пористое тело. Адсорбент должен обладать большой удельной поверхностью, которую выражают обычно в м2 на 1 г адсорбента. Высокой удельной поверхностью поглощения (сотни м2/г) отличается активный уголь (марок АГ-2 или АГ-3, СКТ, КАД, БАУ и др.), мелко- и крупнопористый сили-кагель (марок КСШ, АСК, АСМ, ШСК. и др.), активный оксид алюминия. Довольно широко используются и молекулярные сита — цеолиты марок NaA-4, CaA-5, NaX-9, СаХ-8 и др. В качестве подвижной фазы обычно используют следующие газы: гелий, азот, водород, аргон, углекислый газ.
Газоадсорбционная хроматография основана на различной склонности компонентов газовой смеси к адсорбции на данном адсорбенте. Во время перемещения анализируемого газа вдоль неподвижного слоя адсорбента беспорядочно движущиеся частицы газовой смеси как бы прилипают к активной поверхности твердого тела, а затем отделяются, улетают в окружающее пространство и снова возвращаются к поверхности адсорбента. С повышением температуры скорость движения частиц газа увеличивается, а адсорбция замедляется, так как при этом частицы газа легко отделяются от поверхности твердого вещества и диффундируют в газовую фазу. Наоборот, с увеличением давления адсорбция усиливается, так как частицы газа находятся ближе к активной поверхности адсорбента и чаще ее бомбардируют. Повышение температуры, снижение давления, введение в систему малоактивного газа — все это способствует уменьшению концентрации хорошо адсорбирующегося компонента газа на поверхности адсорбента и порождает десорбцию.
Не меньшее влияние на процесс адсорбции оказывает природа газа. Экспериментальным путем установлено, что, например, при температуре 20 °С и давлении 101,3 кПа (760 мм рт. ст.) газы по возрастающей склонности к адсорбции на активном угле располагаются в следующем подрядке: водород, азот, кислород, оксид углерода, метан, диоксид углерода.
При сопоставлении поглотительной способности активного угля с температурой кипения газов наблюдается определенная закономерность: чем выше температура кипения газа, тем лучше он адсорбируется углем. Например, адсорбционная способность углеводородных газов увеличивается в таком порядке: метан, этилен, этан, пропилен, пропан, изобутан, бутан, т. е. с повышением молекулярной массы. Опыт показывает также, что газообразные углеводороды с разветвленной цепью поглощаются несколько хуже их линейных изомеров.
В газожидкостной хроматографии, которая сейчас находит наибольшее применение на практике, неподвижной фазой служит нелетучая жидкость, распределенная по поверхности твердого носителя в виде жидкой пленки. Этот вид хроматографии основан на различной растворимости компонентов газовой смеси в жидкой неподвижной фазе, что приводит к различному давлению их пара над жидкой неподвижной пленкой. Ясно, что газ-носитель в первую очередь будет захватывать вещества с наибольшим давлением' пара и с наименьшей растворимостью в жидком сорбенте.
В зависимости от состава анализируемого газа применяют разнообразные полярные и неполярные жидкие поглотители. От правильного выбора жидкой фазы зависит четкость и полнота разделения компонентов. Жидкие поглотители должны быть особо чистыми, маловязкими, термостабильными и иметь минимальную летучесть при температуре хроматографирования (давление пара не выше 133 Па). Кроме того, они должны быть химически инертными к твердому носителю, компонентам разделяемой смеси и к газу-носителю. И самое главное — жидкий поглотитель должен селективно растворять компоненты анализируемой смеси.
Широкое применение в качестве неподвижной фазы нашли следующие соединения и смеси: неполярные — высокомолекулярные нефтяные и парафиновые углеводороды и их смеси (сквалан, гексадекан, вазелиновое масло); полярные — высокомолекулярные спирты, полиэтиленгликоли (молекулярной массы от 400 до 20000) и их эфиры, эфиры карбоновых кислот и алифатических спиртов (фталаты, адипинаты, себацинаты и др.), полиены, силиконовые масла и эластомеры. Для увеличения общей поверхности поглощения указанные жидкости наносят на крупнопористый носитель, не обладающий существенной адсорбционной активностью по отношению к компонентам газовой смеси.

Рис. 9. Общая схема хроматографа:
;—источник подвижной фазы; 2—дозатор; .5—колонка; 4—детектор; 5 — потенциометр; 6—термостат; 7—терморегулятор.
Носитель должен обладать достаточной удельной поверхностью, быть химически инертным к компонентам смеси и механически прочным, не разлагаться при температуре опыта и не оказывать большого сопротивления потоку газа-носителя. В газожидкостной хроматографии наибольшее применение нашли диатомитовые носители, такие, как инзенский кирпич (ИНЗ-600), сферохромы, порохромы, динохромы. В последнее время для разделения углеводородов и других нефтепродуктов начинают использовать пористые полимерные носители, по характеру взаимодействия с разделяемым веществом представляющие нечто среднее между твердым адсорбентом и высоковязкой неподвижной фазой. Наиболее распространен в отечественной практике полисорб-1 (сополимер стирола с n-дивинилбензолом). Хроматографирование производят либо непосредственно на полимерных носителях, либо их модифицируют небольшими добавками (1— 5 %) полярных и неполярных неподвижных фаз.
УСТРОЙСТВО ХРОМАТОГРАФА
Общая схема современного хроматографа представлена на рис. 9. Из источника подвижной фазы 1 очищенный от примесей газ-носитель через дроссель поступает в хроматографическую колонку 3. В дозирующее устройство 2 в газообразном или жидком состоянии вводится анализируемая смесь. Она подхватывается газом-носителем и также поступает в хроматографическую колонку. Разделенные компоненты анализируемой смеси вместе с газом-носителем выходят из колонки через детектор 4.
Детектором называется прибор, с помощью которого в газе-носителе обнаруживаются компоненты разделяемой смеси. Фиксируемые детектором те или иные физические параметры газа на выходе из колонки преобразуются в нем в электрические сигналы, которые регистрируются самопишущим потенциометром 5. На диаграммной бумаге потенциометра вычеркивается кривая, состоящая из чередующихся пиков. Эта кривая называется хроматограммой.
Для поддержания заданной постоянной температуры во время проведения анализа колонка, дозирующее устройство и детектор помещены в термостат 6, который обогревается электрической спиралью от терморегулятора 7.
Дозирующее устройство служит для ввода пробы в хроматографическую колонку. Для газообразной пробы используются краны-дозаторы, состоящие из калиброванной между двумя кранами трубки; вместимость такого дозатора обычно не более 5 мл. Жидкая проба в объеме тысячных долей миллилитра вводится с помощью специального шприца или микрошприца (работающего по принципу медицинского) через каучуковую мембрану.
Хроматографическая колонка изготовляется из инертного материала (стекла, нержавеющей стали и др.) и представляет собой трубку. В зависимости от внутреннего диаметра колонки разделяются на насадочные — до 6 мм, микронасадочные — в пределах 1 мм и капиллярные — около 0,25 мм. По форме колонки бывают прямые, U-образные, W-образные и спиральные, цельные или состоящие из отдельных секций. Диаметр и длина колонки определяются составом хроматографируемого вещества, объемом введенной пробы, природой и количеством неподвижной фазы, а также размерами частиц адсорбента или носителя жидкой фазы.
Детектор является важнейшей частью хроматографа. Одним из первых детекторов служила несколько измененная по конфигурации газовая бюретка с раствором щелочи (см. рис. 13). При попадании в бюретку смеси углекислого газа (газа-носителя) и выделяемого компонента углекислый газ поглощается раствором щелочи, а газовый компонент поднимается в верхнюю часть бюретки. Это и дает возможность количественно учесть его объем. По мере выхода из колонки газовых компонентов уровень раствора щелочи в бюретке будет снижаться до полного выхода пробы газа. Откладывая на вертикальной оси объемы выделившегося газа (в мл), а на горизонтальной — время их выделения (в с), получаем ступенчатую хроматограмму. Высота каждой ступени кривой характеризует относительный объем компонентов. Несмотря на простоту устройства этот волюмометрический детектор не обладает надлежащей точностью, в частности, во время определения на поверхности раствора щелочи образуется пена, что затрудняет измерение уровня, особенно при малых количествах газовых компонентов.
Подобного рода устройства относятся к детекторам интегрального типа. Они позволяют учитывать общее количество анализируемого вещества, прошедшее через детектор за время анализа. Благодаря этому хроматографы с интегральными детекторами не требуют предварительной калибровки для количественной расшифровки хроматограмм.
Развитию хроматографического метода анализа в значительной степени способствовало внедрение более чувствительных и точных дифференциальных детекторов. В них сравниваются физические свойства потока газа на выходе из колонки и чистого газа-носителя. К числу свойств газового потока, которые используются в этих детекторах, относятся теплопроводность, теплота сгорания, плотность, изменение ионного тока и др. Эти свойства в дальнейшем преобразуются в большинстве случаев в электрический сигнал, мгновенно фиксируемый регистратором. В дифференциальных детекторах, в отличие от интегральных, фиксируются и регистрируются на потенциометре мгновенные характеристики смеси газов, выходящей из колонки. Поэтому для расшифровки хроматограмм в этом случае необходима предварительная
 Рис. 10. Схема детектора
по теплопроводности;
Рис. 10. Схема детектора
по теплопроводности;
1, 2—камеры с термочувствительными элементами; 3 — потенциометр; 4, S — сопротивления; 6— милливольтметр; 7 — источник постоянного тока.
Рис. П. Схема ионизационно-пламенного детектора:
1 — электрод-коллектор; 2 — электрод-горелка; 3 — диффузор; 4—6—изоляторы электродов; 7—усилитель тока; 8 — потенциометр.
количественная калибровка. Среди детекторов этого типа широкое применение получили детектор по теплопроводности (катарометр) и ионизационно-пламенный.
При работе детектора по теплопроводности измеряется не абсолютная теплопроводность газа, а разность в теплопроводности газа-носителя и смеси газа-носителя с анализируемым компонентом. Чем эта разность больше, тем чувствительнее детектор. На практике в качестве газа-носителя наиболее широко применяется гелий, теплопроводность которого в несколько раз больше теплопроводности углеводородов и многих органических соединений. Хотя теплопроводность водорода выше, чем у гелия, но из-за взрывоопасное™ его применяют редко. Существенная часть детектора по теплопроводности (рис. 10) — два термочувствительных элемента, которые изготовлены из платиновых или вольфрамовых нитей, а иногда из полупроводникового материала (термистора). Каждый термочувствительный элемент помещен в камеру блока детектора. Через сравнительную камеру 1 непрерывно проходит газ-носитель, а через измерительную камеру 2 смесь газа-носителя с выделяемыми компонентами. Обе камеры вместе с сопротивлениями 3 и 4 образуют измерительный мост Уитстона. На мост подается постоянный ток напряжением 6—12 В, от которого нагреваются нити, а следовательно, и сам блок. Когда в обе камеры поступает только газ-носитель, температура элементов в них одинакова и разность потенциалов равна нулю. При изменении состава газа, проходящего через измерительную камеру, температура в ней изменяется вследствие передачи теплоты газовому потоку, обладающему иной теплопроводностью. Между точками А и Б возникает разность потенциалов, которая регистрируется в виде сигнала детектора.
Работа ионизационно-пламенного детектора (ДИП) основана на измерении электрической проводимости, возникающей в результате ионизации молекул газа при их поступлении в детектор. ДИП (рис. 11), применяемый при анализе горячих газов, состоит из камеры, в которую одновременно по отдельным каналам поступает водород, воздух и газ-носитель в смеси с компонентами анализируемого газа. В камеру помещена горелка для сжигания водорода. В начале опыта в горелке сгорает водород в присутствии только газа-носителя. В получающемся диффузионном пламени примеси, имеющиеся в воздухе, водороде и газе-носителе, ионизируются, образуется некоторое количество ионов, за счет которых возникает незначительная электрическая проводимость. Когда в пламя вместе с газом-носителем поступают горючие компоненты, выделяемые из колонки, количество ионов увеличивается и электрическая проводимость пламени резко возрастает. Механизм ионизации весьма сложен и до конца не установлен. Возникает как бы два электрода, одним из которых является сопло горелки, а другим — электродом-коллектором — колпачок 1. Образующийся ток через усилитель 7 направляется в потенциометр, на диаграммной бумаге которого будет вычерчиваться хроматограмма.
ДИП устойчив к некоторым колебаниям давления газа и окружающей температуры. Он обладает высокой чувствительностью. Величина его сигнала почти пропорциональна числу атомов углерода в углеводородах. Однако присутствие примесей в газе-носителе и водороде может несколько нарушить точность его показаний, так как ДИП обладает некоторой (правда, малой) чувствительностью к воде, воздуху, инертным и некоторым другим газам (H2S, SO2, CO2, CO, NH3 и др.).
Детекторы не являются универсальными приборами, и для каждой смеси хроматографируемых соединений в зависимости от их свойств выбирают соответствующий тип детектора. Поэтому очень часто во многих лабораторных хроматографах устанавливают несколько детекторов различных типов. Для каждой смеси подбирают сочетание колонок с разными сорбентами и комплексным использованием детекторов.
Широкое внедрение хроматографического метода анализа обеспечивается в настоящее время выпуском все новых и новых марок и систем хроматографов, оснащенных одновременно разнообразными детекторами и системами ввода веществ в жидком или газообразном состоянии. Различными детекторами комплектуются хроматографы «Цвет» (модели 1—6 и 100—150), ЛХМ (ЛХМ-8МД, ЛХМ-60, ЛХМ-72), ХЛ (1—4), УХ (1—2) и другие отечественные и зарубежные хроматографы, которые находят применение в лабораториях предприятий нефтеперерабатывающей и нефтехимической промышленности.
РАСШИФРОВКА ХРОМАТОГРАММ
На рис. 12 представлена типовая хроматограмма, состоящая из серии пиков. По форме пик напоминает треугольник, одна из сторон которого соответствует концентрации компонента, возрастающей до максимума (вершина треугольника), а другая — убывающей до минимума.
О тсчет
ведется от базовой (нулевой) прямой
линии 1,
параллельной
горизонтальной оси. Эта линия вычерчивается
при выходе из колонки чистого
газа-носителя. Пик 2
отображает
выход несорбирующегося компонента.
Пик 3
характеризует
период четкого выхода определенного
компонента или смеси неразделяемых
компонентов. Пик 4
по
сравнению с пиками типа 3
имеет
асимметричное строение. Такие пики
отображают неравномерный выход компонента
из колонки. Наконец, на практике
встречаются и накладывающиеся друг на
друга пики (5,
6), которые
объясняются близостью физико-химических
свойств разделяемых компонентов, а
также увеличением объема хроматографируемых
веществ, недостаточной инертностью
газа-носителя
тсчет
ведется от базовой (нулевой) прямой
линии 1,
параллельной
горизонтальной оси. Эта линия вычерчивается
при выходе из колонки чистого
газа-носителя. Пик 2
отображает
выход несорбирующегося компонента.
Пик 3
характеризует
период четкого выхода определенного
компонента или смеси неразделяемых
компонентов. Пик 4
по
сравнению с пиками типа 3
имеет
асимметричное строение. Такие пики
отображают неравномерный выход компонента
из колонки. Наконец, на практике
встречаются и накладывающиеся друг на
друга пики (5,
6), которые
объясняются близостью физико-химических
свойств разделяемых компонентов, а
также увеличением объема хроматографируемых
веществ, недостаточной инертностью
газа-носителя
Рис. 12. Типовая хроматограмма.
к разделяемым веществам и сорбенту, повышенными сорбционными свойствами твердого носителя, неточностью работы детектора. Встречаются и очень вытянутые в длину пики (7), площадь которых определить очень трудно. По месту расположения пиков на хроматограмме или, что то же самое, по времени выхода компонентов можно установить качественный состав анализируемой смеси, так как для каждого компонента при определенных постоянных условиях разделения время и порядок выхода из колонки всегда постоянны.
Для нахождения количественного состава анализуемой смеси газов используют зависимость между содержанием данного компонента в смеси и геометрическими размерами соответствующего ему пика на хроматограмме. Чаще всего количественную оценку хроматограммы производят по площади пиков S. Измерив площади пиков, относят их значения к сумме площадей всех пиков, умножают частное на 100 и таким образом находят содержание всех компонентов анализируемой смеси (в %).
Измерять площади пиков можно различными методами:
взвешиванием кусочков диаграммной бумаги, вырезанных по контуру каждого пика;
с помощью планиметра;
прямым подсчетом, путем умножения высоты пика на его ширину, измеренную на уровне половины высоты.
Рассмотрим последний случай определения площади каждого пика на примере хроматограммы, изображенной на рис. 12. Для пиков 3 и 4 высота h (в мм) измеряется от нулевой линии до максимума пика. На половине высоты пика проводят линию, параллельную базовой, и измеряют длину отрезка, ограниченного сторонами пика. Это и будет ширина пика а. Для пиков 5 и 6 прочеркивают пунктиром линии, недостающие для полной конфигурации пиков, какую они имели бы в случае нормального разделения компонентов. Затем обычным способом находят их высоту и ширину.
Площадь пика 7 измерить практически невозможно. Поэтому ее определяют методом взвешивания диаграммой бумаги на аналитических весах, относя массу диаграммной бумаги, вырезанной по контуру пика, к «массе всех пиков».
Иногда вместо площадей пиков используют их высоту, считая, что она пропорциональна площади.
Наконец, при хроматографировании смеси веществ, близких по химическому строению и свойствам, можно использовать еще один параметр — произведение высоты пика на время удерживания htR. Последнее может быть измерено на диаграммной бумаге по базовой линии от ввода пробы в колонку до максимума выхода компонента.
Выбор того или иного параметра (S, h или htR) зависит от многих факторов и прежде всего от состава смеси и эффективности ее разделения на данном хроматографе. Если подсчет площадей пиков не вызывает затруднений, то рекомендуется применять именно этот метод. Однако при использовании любого параметра для подсчета содержания компонентов в анализируемой смеси необходимо еще, как правило, вводить специальные коэффициенты, учитывающие чувствительность детекторов к отдельным компонентам газовой смеси. Только в отдельных, довольно редких случаях дифференциальные детекторы выдают импульсы, строго пропорциональные массовым количествам различных компонентов. В этих случаях расшифровку хроматограмм проводят методом простой нормировки, как это описано выше, т. е. по различным параметрам хроматографических пиков, принимая их сумму за 100.
Но чаще всего соотношение геометрических размеров пиков далеко не отражает истинного соотношения концентраций компонентов в смеси. Поэтому необходимо для каждой анализируемой смеси веществ и для каждого хроматографа проводить предварительную калибровку прибора на искусственных смесях данных веществ. Очевидно, что целью калибровки является установление зависимости выходного сигнала детектора (или, что то же самое, характера и параметров кривой на хроматограмме) в определенных условиях хроматографирования от количества того или иного вещества, присутствующего в анализируемой смеси. Следует особо отметить, что калибровку и последующие анализы необходимо проводить строго при одних и тех же условиях. Так, при использовании детектора по теплопроводности и подсчете результатов анализа по площадям пиков особое значение имеет постоянство скорости газа-носителя, так как ширина пиков обратно пропорциональна этой скорости. Существует несколько способов калибровки и расчета хроматограмм:
абсолютная калибровка;
метод внутренней нормализации (или метод нормировки);
метод внутреннего стандарта. Рассмотрим кратко сущность этих методов.
Задачей метода абсолютной калибровки является построение графиков зависимости высоты пиков, площади пиков или произведения высоты пиков на время удерживания (h, S или htR) от количества того или иного вещества, введенного в колонку. Такие графики необходимо иметь для каждого компонента анализируемой смеси. Для построения графика хроматографируют не менее пяти смесей, например, с воздухом точно заданного количества. Для получения искусственных смесей с разным содержанием данного компонента исходную калибровочную смесь можно разбавить газом-носителем. Каждый опыт повторяют не менее трех раз.
При хроматографировании исследуемых газовых смесей полученные хроматограммы расшифровывают с помощью этих калибровочных графиков, а зная количество пробы, введенной в колонку, рассчитывают и процентное содержание всех компонентов смеси.
Метод внутренней нормализации, или метод нормировки, с введением калибровочных коэффициентов заключается в том, что для расчета концентраций компонентов анализируемой смеси используют не просто высоты пиков, их площади или произведения высот на время удерживания (h, S или htR), как в методе абсолютной калибровки, а их значения, приведенные к величинам, пропорциональным концентрациям, т. е. произведения данного параметра пиков на калибровочный коэффициент. Использование калибровочных коэффициентов и дает возможность учесть чувствительность детектора ко всем компонентам смеси.
Калибровочные коэффициенты находят при хроматографировании искусственных смесей строго известного состава. Определение основано на пропорциональности концентраций компонентов в смеси С и параметров h, S или htR соответствующих пиков. Очевидно, что при равной чувствительности детектора к компонентам смеси будет иметь место соотношение:
![]()
При неодинаковой чувствительности детектора это соотношение нарушается, но может быть вновь восстановлено введением соответствующих коэффициентов. Для этого калибровочный коэффициент для одного из компонентов смеси принимают за единицу. Тогда коэффициенты для любого, т. е. i-го, компонента находят по формулам:
![]()
![]()
![]()
где hc, Sc, (htR)c и Cс относятся к компоненту, коэффициент которого принят за единицу.
При анализе углеводородных газов чаще всего приравнивают к единице калибровочный коэффициент для нормального бутана.
Зная все калибровочные коэффициенты, концентрацию любого компонента Ci находят по приведенным параметрам всех пиков:
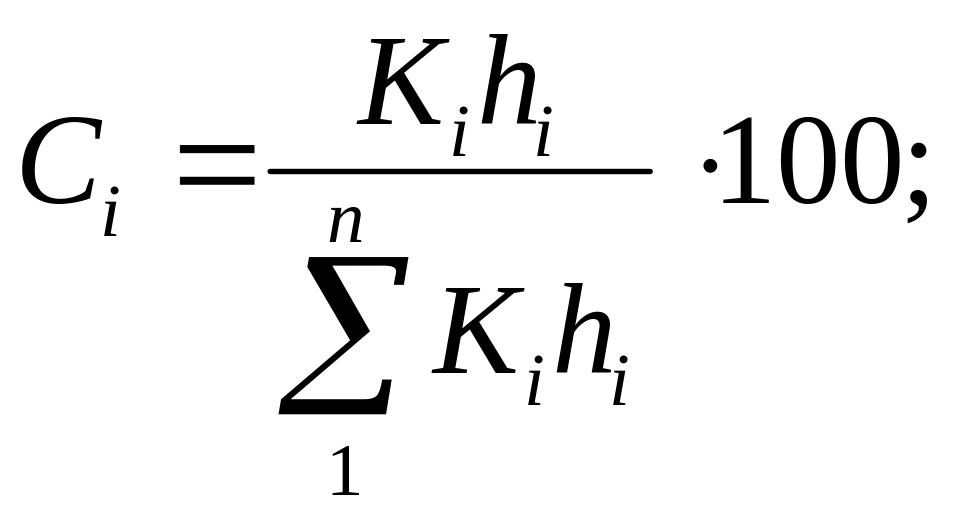
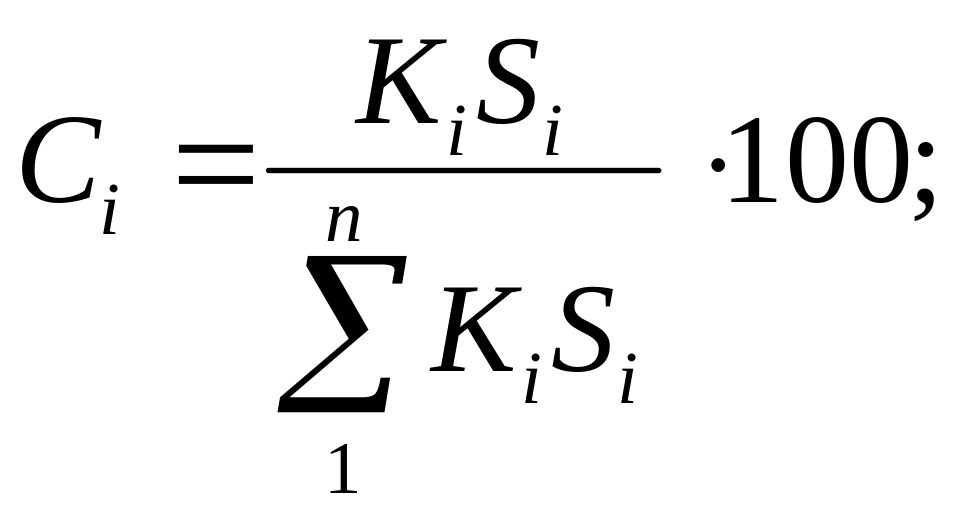

где п — число компонентов.
В этом методе необходимо точно определить выбранный параметр для всех пиков хроматограммы даже в тех случаях, когда интересуются только одним или несколькими компонентами или примесями. Однако метод удобен тем, что для многих веществ калибровочные коэффициенты известны и их можно принимать по литературным данным, а следовательно, не проводить предварительную калибровку прибора.
Метод внутреннего стандарта, или, как его иногда называют, «метод метки», основан на введении в анализируемую смесь точно известного количества какого-либо индивидуального вещества (стандарта). Для подсчета концентрации i-го компонента в анализируемой смеси используемая прямая зависимость между концентрациями искомого компонента Ci- и стандартного вещества Сст в анализируемой смеси и параметрами h и S соответствующих пиков. Необходимо только выбирать такое стандартное вещество, чтобы его пик на хроматограмме хорошо отделялся от остальных пиков. Так как чувствительность детектора к стандартному веществу может быть иной, чем к искомому компоненту, то в данном методе необходимо учитывать соответствующие поправочные коэффициенты Ki- и Kcт
Массовую долю искомого компонента Сi (в %) рассчитывают по формуле:
![]()
где Pi и Рcт — параметры пиков искомого и стандартного веществ; Сст — массовая доля стандартного вещества, %.
Метод удобен тем, что для подсчета количественного содержания того или иного компонента нет необходимости замерять параметры всех пиков.
В инструкциях по монтажу и эксплуатации различных хроматографов и в соответствующих ГОСТах обыкновенно приводятся подробные указания по расшифровке хроматограмм и поправочные коэффициенты, учитывающие чувствительность детектора.