

5 Газовая хроматография
Бурное развитие газовой хроматографии началось с 1952 года, после того как Мартин и Джеймс осуществили газожидкостной вариант, использовав смешанную неподвижную фазу, состоящую из стеариновой кислоты и полиметилфенилсилоксана. Это позволило сразу включить широкий набор жидкостей вместо ограниченного круга адсорбентов. Кроме того, линейность изотермы распределения анализируемых веществ (постоянство времени удерживания) позволило проводить надежную идентификацию даже очень сложных смесей.
Примерно в это же время целый ряд фирм начал серийный выпуск газовых хроматографов. Принципиальная схема хроматографической установки приведена на рис.7.
5.1 Газожидкостная хроматография.
Разделение анализируемой смеси в ГХ происходит за счет различия констант распределения K1 и K2, как это показано, например, на рис.1. При этом приведенный объем удерживания, V’R прямо зависит от количества жидкой фазы Vs в колонке.
Приведенный объем удерживания получается, если из объем удерживания VR вычесть мертвый объем VM.
VR’ = VR - VM = VS K , (21)
где K - коэффициентом распределения.
Объем удерживания VR определяется:
VR = uср tR , (22)
uср – объемная скорость газа-носителя.
Для сорбентов насадочных колонок количества жидкой фазы получило название степени пропитки и задается как процент жидкой фазы от веса всего сорбента. Выбор степени пропитки зависит от особенностей решаемых задач и твердого носителя. Наибольшее распространение получили твердые носители на основе «диатомитов», которые образовались в результате отмирания одноклеточных диатомитовых водорослей. Для анализа высококипящих соединений используется малый процент неподвижной фазы - 5% (а иногда 3 и даже 1%). Часто на упаковках с таким сорбентом ставится надпись: «для высокотемпературной хроматографии».
Вообще, можно нанести на твердый носитель практически любое количество жидкой фазы, но обычно степень пропитки составляет от 3 до 25%. Сам процесс приготовления сорбента для газожидкостной хроматографии предельно прост. Сначала взвешивают требуемое количество жидкой фазы и твердого носителя. Взвешенное количество жидкой фазы (обычно несколько грамм) растворяют в 50-100 мл низкокипящего растворителя. Готовый раствор выливают в «выпаривательную» чашку. Туда же помещают взвешенный твердый носитель так, чтобы полученный раствор полностью покрывал его поверхность. Затем при слабом нагревании и медленном перемешивании ждут полного испарения растворителя. Спустя некоторое время, после того как приготовленный сорбент вновь приобрел «сыпучесть», его можно засыпать в колонку.
Поверхность твердого носителя представляет собой набор узких и широких пор, заполненных жидкостью и участков поверхности, где жидкая фаза вовсе отсутствует (рис. 9).
Твердый носитель должен быть инертным, сохраняя при этом большую удельную поверхность. К сожалению, диатомитовые твердые носители содержат ряд примесей, которые приводят к образованию «хвостов» (смотри рис. 6) и сдвигу времен удерживания, характерному для выпуклой изотермы адсорбции.
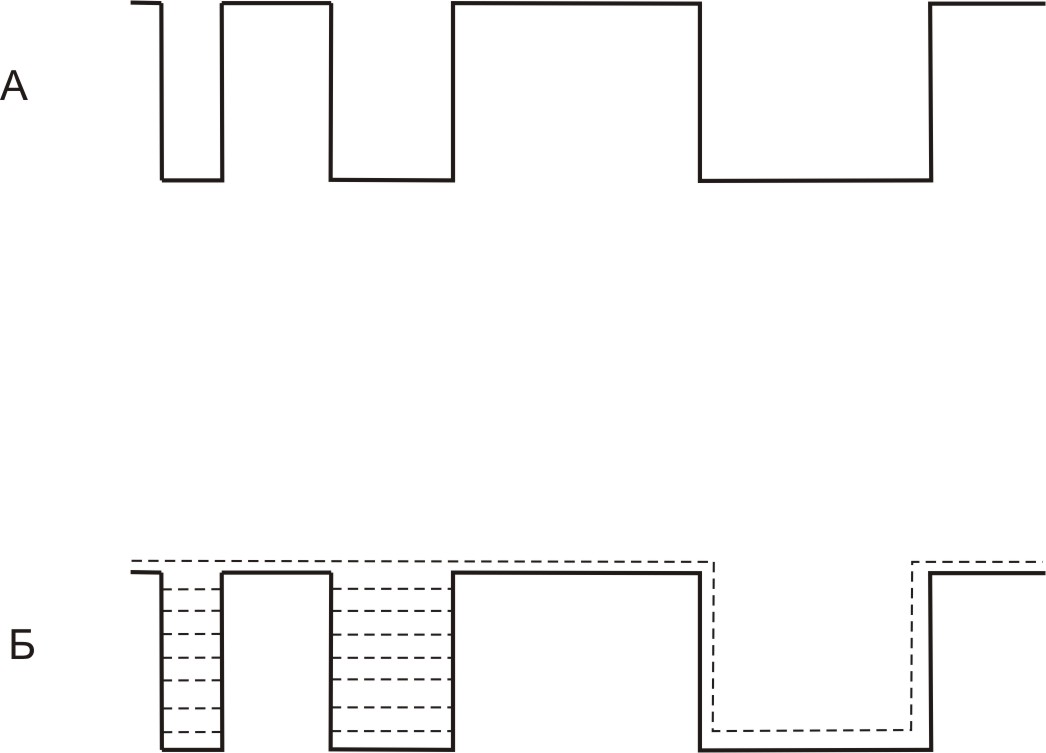
Рисунок 9. Модель заполнения твердого носителя неподвижной жидкой фазой. А – поверхность твердого носителя без неподвижной жидкой фазы. Б – поверхность твердого носителя, покрытая неподвижной жидкой фазой.
Для уменьшения влияния этого фактора диатомитовые твердые носители промывают кислотой и обрабатывают гексаметилдисиланом или диметилдихлорсиланом.
Понятно, что для различных твердых носителей, отличающихся по величине площади удельной поверхности, одинаковая степень пропитки приводит к различным временам удерживания. Чтобы иметь возможность сравнения сорбентов на различных твердых носителях, вводится концепция толщины слоя (пленки) неподвижной фазы. Толщины «пленки» рассчитывается делением объема жидкой фазы на площадь поверхности используемого твердого носителя.
Газовая хроматография позволяет установить, например, состав фракции нефти, содержащей несколько сотен компонентов. При этом температура кипения повышается от -161ºС для метана до более чем 800ºС для углеводорода C120H242.
Соответственно, возникает вопрос: как проанализировать все эти соединения одновременно в одних и тех же условиях?
Для этого рассмотрим влияние хроматографических параметров на объем удерживания.
Коэффициент распределения K зависит от величины свободной энергии растворения, F0 и температуры колонки, Т:
![]() ,
(23)
,
(23)
где R - универсальная газовая постоянная; C – безразмерная постоянная.
Если принять F Q - теплоте растворения, то, выражая теплоту растворения через температуру кипения по правилу Трутона (Q 21TK;
R = 1,992,0 кал/моль град), получим следующее соотношение
![]() (24)
(24)
Подставим значения приведенного удерживаемого объема из уравнения (21) в уравнение (22):
Vs K = uср t. (25)
t - приведенное время удерживания.
Теперь прологарифмируем это уравнение и подставим в левую часть значение K из соотношения (23) , и получим уравнение:
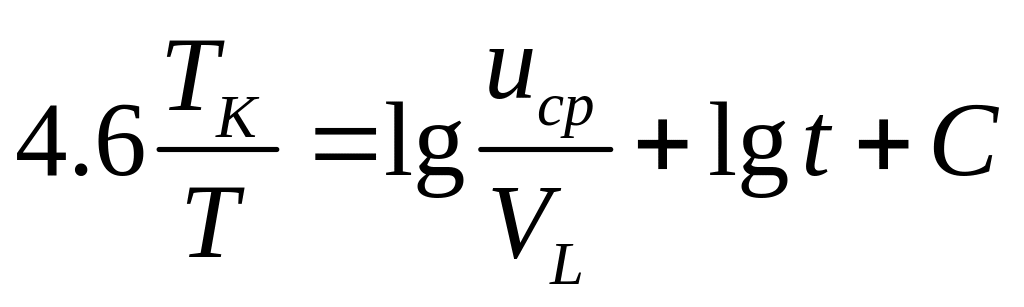 (26)
(26)
Если числитель и знаменатель отношения u/VL разделим на площадь поперечного сечения колонки S, то получим:
![]() (26a)
(26a)
где u – средняя линейная скорость газа-носителя; VL - объем жидкой фазы(VL = VS); L - длина колонки; - толщина слоя жидкой фазы; C – постоянная.
Это уравнение позволяет связать параметры хроматографического опыта с термодинамическими характеристиками вещества и, в частности с температурой кипения. Если анализируемые вещества низкокипящие, то необходимо либо понижать температуру колонки, либо увеличивать количество неподвижной фазы. В случае высококипящих веществ, для элюирования их при времени удерживания t, можно повысить температуру колонки, уменьшить процент неподвижной фазы, сократить длину колонки, увеличить линейную скорость газа-носителя. Кроме того, необходимо повысить температуру испарителя, детектора и т.д.
При повышении температуры колонки возникает проблема термической устойчивости анализируемых соединений и стационарных фаз, используемых в газожидкостной хроматографии. Причем, на термостойкость влияет материал испарителя и колонки, присутствие тех или иных примесей в твердом носителе и неподвижной фазе, состав анализируемой смеси. Кроме этих причин термической неустойчивости анализируемых веществ и стационарных фаз имеются принципиальные температурные ограничения. Так, при 350оС, исходя из расчета энергии связи Si – C, должен происходить отрыв органических радикалов. Экспериментально установлено, что при температуре 370оС наблюдается разрыв Si – O – Si связи.
Помимо силиконов , термическая устойчивость которых достаточно высока, в справочниках и проспектах ведущих фирм, предлагающих материалы для хроматографии, предлагаются стационарные фазы с температурным пределом от 120 до 250 оС. В этом случае температура колонки должна быть понижена.
Из уравнения (26) следует, что при этом значительно возрастет время удерживания. Очевидно, что анализ, на который надо затратить много часов, нецелесообразен. На практике время анализа всегда ограничено. Поэтому в хроматографии число теоретических тарелок n относят ко времени анализа. Соответственно RS (смотри уравнение 19) следует разделить на корень квадратный из времени анализа.
Хроматография обычно применяется при анализе сложных смесей, когда число компонентов в смеси значительно превышает количество пиков на хроматограмме. При этом неважно насколько хорошо делятся отдельные пики, а желательно оценить какое максимально-возможное число пиков TZ. (эффективное число пиков). TZ мы можем определить на данной колонке (с известным числом теоретических тарелок).
![]() ,
(27)
,
(27)
где 2 и 1 – ширина пиков на половине высоты.
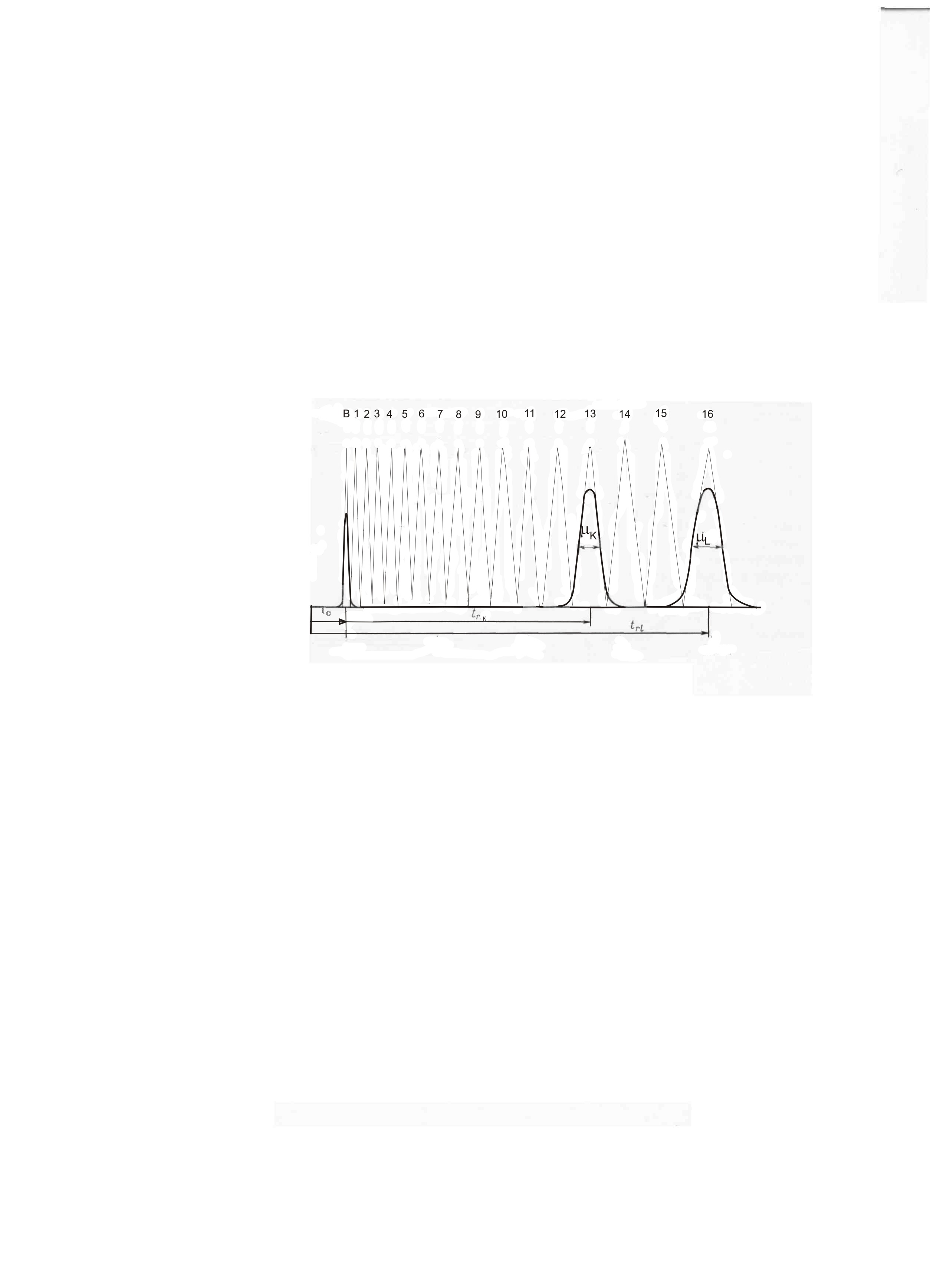
Рисунок 10. Оценка эффективности метода хроматографии числом разделений TZ, т.е. числом “условных” пиков, которое помещается между двумя пиками Для выделения высококипящих соединений в дистилляции используется вакуум. В газовой хроматографии можно понизить температуру анализа увеличением скорости газа-носителя, уменьшением толщины слоя неподвижной фазы, сокращением длины колонки. Это позволяет анализировать соединения с температурой кипения 700-800оС при температуре анализа 350-400оС.
Если интервал температур кипения смеси анализируемых веществ достигает более 100ºС, то время анализа резко возрастает. Из сравнения хроматограмм, приведенных на рис. 11 видно, что при изотермическом режиме пики низкокипящих компонентов “налезают” друг на друга. Пики высококипящих компонентов сильно размыты. Из рисунка видно, что некоторые из этих пиков в изотермическом режиме не выходят из колонки вовсе. При программировании температуры (повышении температуры в ходе анализа) элюируют все высококипящие компоненты. При этом эти пики становятся более узкими и симметричными (рис.11).