

Короткий виклад навчального матеріалу
Спадкові хвороби - це хвороби, які виникають внаслідок пошкодження генетичної інформації.
Синдром (або хвороба) Дауна (СД) - найбільш вивчена хромосомна хвороба. Її частота 1:700 - 1:800 (описана в 1866 р.), в Києві частота складала 1,16 на 1000 серед живонароджених (Галаган В.О. та співав., 2005). Частота народження залежить від віку матері і, в меншій мірі, від віку батька. У віці після 45 років частота патології може зростати до 1:12. Цитогенетичні варіанти СД різноманітні. У 95% це просто повна трисомія 21, при цьому вклад материнського нерозходження 80% і 20% батьківського. Біля 2% дітей мають мозаїчну форму (47+21/46), в 4% хворі мають транслокаційну форму трисомії (транслокацію 21-ї хромосоми на одну із груп хромосом D). При цьому майже 50% транслокаційних форм успадковується від батьків - носіїв.
Клінічна картина синдрому Дауна різноманітна: це і вроджені вади розвитку, і порушення постнатального розвитку нервової системи, і вторинний імунодефіцит тощо. У перінатальному періоді діагностика синдрому Дауна ускладнена. Діти народжуються в строк з помірно вираженою гіпоплазією (на 8-10% нижче середніх величин).

Новонароджений із синдромом Дауна

Рис. 6.1 Каріотип при синдромі Дауна (трисомія 21)
Симптоми помітні уже в пологовому будинку, де діагноз виставляють у 90% хворих. У дітей відмічається монголоїдний розріз очей, кругле сплощене обличчя, пласка спинка носа, епікант (вертикальна складка шкіри біля внутрішнього кутка ока), великий (зазвичай висунутий) язик (макроглосія), брахіцефалія (збільшення поперечного розміру голови), вкорочення передньо-заднього розміру, скошена потилиця, “надлишок” шкіри на шиї, деформовані вушні раковини. Наявна м’язова гіпотонія та збільшення об’єму рухів у суглобах. Характерні зміни дерматогліфіки (чотирипальцева або мавпяча складка на долоні, дві шкірні складки замість трьох на мізинці, вкорочення мізинця, сандалевидна щілина, високе положення трирадіусу та ін.). Часто зустрічаються вроджені вади серця (в 53,2%), також вади шлунково-кишкового тракту (15,3%). Жоден з перерахованих симптомів в 100% випадків не зустрічається. Велике значення для діагностики має динаміка фізичного та розумового розвитку дитини. Зріст дорослих хворих на 20 см нижче середнього. Затримка в розумовому розвитку може досягати імбецильності. Діти зазвичай ласкаві, уважні, слухняні, терплячі при навчання. Реакція цих дітей на навколишнє середовище низька, знижені компенсаторні можливості всіх органів і систем.
Вроджені вади розвитку часто приводять до летального результату в перші 5 років життя. У таких дітей часто (1%) зустрічається лейкоз.
Захворювання необхідно диференціювати з вродженим гіпотиреозом (зустрічається з частотою 1:3500, характерна затяжна жовтяниця за рахунок прямого білірубіну, на першому місяці погіршення апетиту, млявість, блювання, закрепи, блідість, гіпотонія, зниження шкірної температури, після 6-го тижня розвивається одутлість повік. У дітей жорстке волосся, великий язик, який не поміщається в ротовій порожнині, збільшення живота, порушення росту та розвитку, підвищення рівня ТТГ та зниження рівня Т3, Т4).
Лікування багатопланове та неспецифічне. Здійснюється загальнозміцнююча терапія, повноцінне харчування, вітамінотерапія, ретельний догляд. Проводиться корекція вроджених вад розвитку. Важливим є спеціальні методи навчання (педагогічна корекція). Тривалість життя залежить від догляду за хворим та наявності вроджених вад розвитку серця, шлунково-кишкового тракту. Дорослі жінки з хворобою Дауна можуть мати потомство (частота здорового потомства при цьму 50%).
Розробляються методики лікування хворих із трисомією в утробі матері (введення білків, що виробляються нейрогліальними клітинами, які регулюють розвиток нейронів). З’явилися обнадійливі результати використання таких методик у тварин.
Синдром Патау (трисомія 13) –зустрічається з частотою 1:5000 - 1:7000. Проста трисомія, як результат нерозходження хромосом в мейозі в одного із батьків (головним чином у матері), зустрічається у 85% хворих. Решта випадків - в основному за рахунок передачі додаткової хромосоми (точніше довгого плеча) в транслокаціях (pобертсонівська транслокація D/13).
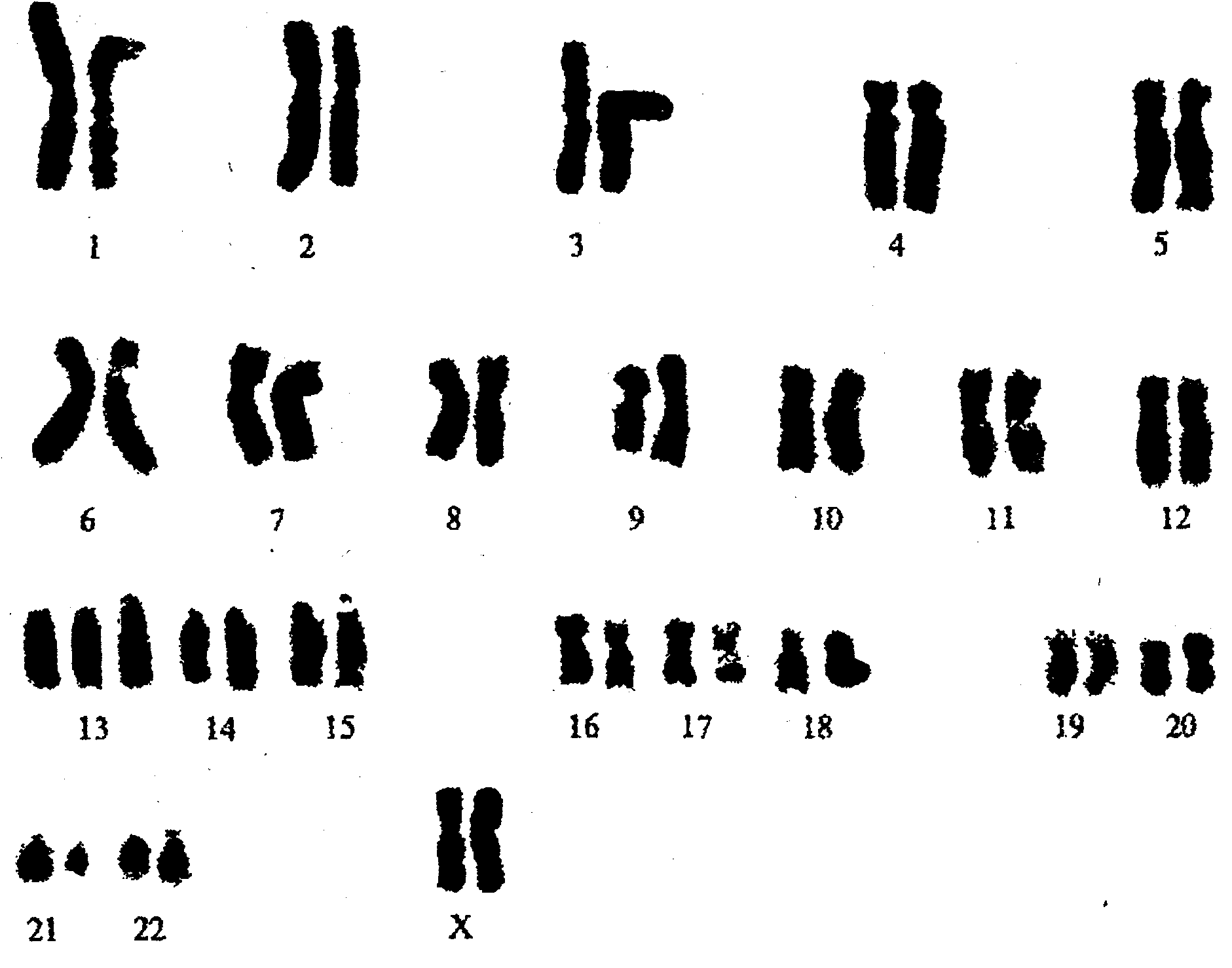
Рис. 6.2. Каріотип при синдромі Патау. Проста повна трисомія 13 (47, ХХ, +13)
Мозаїцизм та ізохромосома зустрічаються рідко. Клініка простих трисомних форм та транслокаційних не відрізняються. Співвідношення хлопчиків та дівчаток 1:1. Діти народжуються з пренатальною гіпоплазією (25-30% нижче середніх величин). Характерним ускладненням є багатоводдя, що зустрічається в половині випадків. Для синдрому Патау характерні множинні вади розвитку мозку та обличчя, кістково-м’язової системи та очей. Це патогенетично єдина група ранніх порушень формування головного мозку, очних яблук, кісток мозкової та лицевої частин черепа. Окружність черепа зазвичай зменшена, зустрічається і тригоноцефалія (розширення в потиличній та звуження в лобній частині). Лоб скошений, низький, очні щілини вузькі, перенісся запале, вушні раковини низько розташовані та деформовані. Типова ознака - розщілини верхньої губи та піднебіння (двосторонні). Завжди наявні вади розвитку декількох внутрішніх органів (дефекти перетинок серця, незавершений поворот кишечника, кісти нирок, аномалії внутрішніх статевих органів, дефекти підшлункової залози). Характерна колобома, катаракта райдужки. Як правило, наявна полідактилія (частіше двобічна на руках), флексорне положення кистей, описують мавпячу складку, аксіальний трирадіус t’, t’’. При підозрі на синдром Патау показано ультразвукове обстеження всіх внутрішніх органів для виявлення вроджених аномалій.


Полідактилія Розщілина верхньої губи та піднебіння
У зв’язку з наявністю грубої вродженої патології 95% дітей помирає протягом першого року життя. Відмічені випадки життя дітей до 10 років (2-3%). Клінічно до цього синдрому за окремими ознаками подібні синдроми Меккелі і Мора, тригоноцефалія Опітца. Вирішальним фактором в діагностиці є дослідження хромосом. Діти із синдромом Патау завжди мають затримку психічного розвитку до ступеня ідіотії.
Лікування: симптоматичне, хірургічна корекція вад, загальнозміцнююча терапія, догляд, профілактика простудних хвороб.
Синдром Едвардса (трисомія 18) – у більшості випадків обумовлений простою трисомією, зустрічаються і мозаїчні форми. Траслокаційні форми - зустрічаються вкрай рідко і клінічних відмінностей не мають. Частота серед новонароджених складає 1:5000 - 1:7000. Співвідношення хлопчиків та дівчаток 1:3. Основними симптомами захворювання є зміни мозкового черепа та обличчя, вади опорно-рухового апарату, вади розвитку серцево-судинної системи. Для синдрому Едвардса (СЕ) характерна різка затримка росту, гіпоплазія м’язової тканини та підшкірно-жирового шару, різке недорозвинення та множинні вади розвитку лицевої частини черепу, серця, кісткової системи та статевих органів.
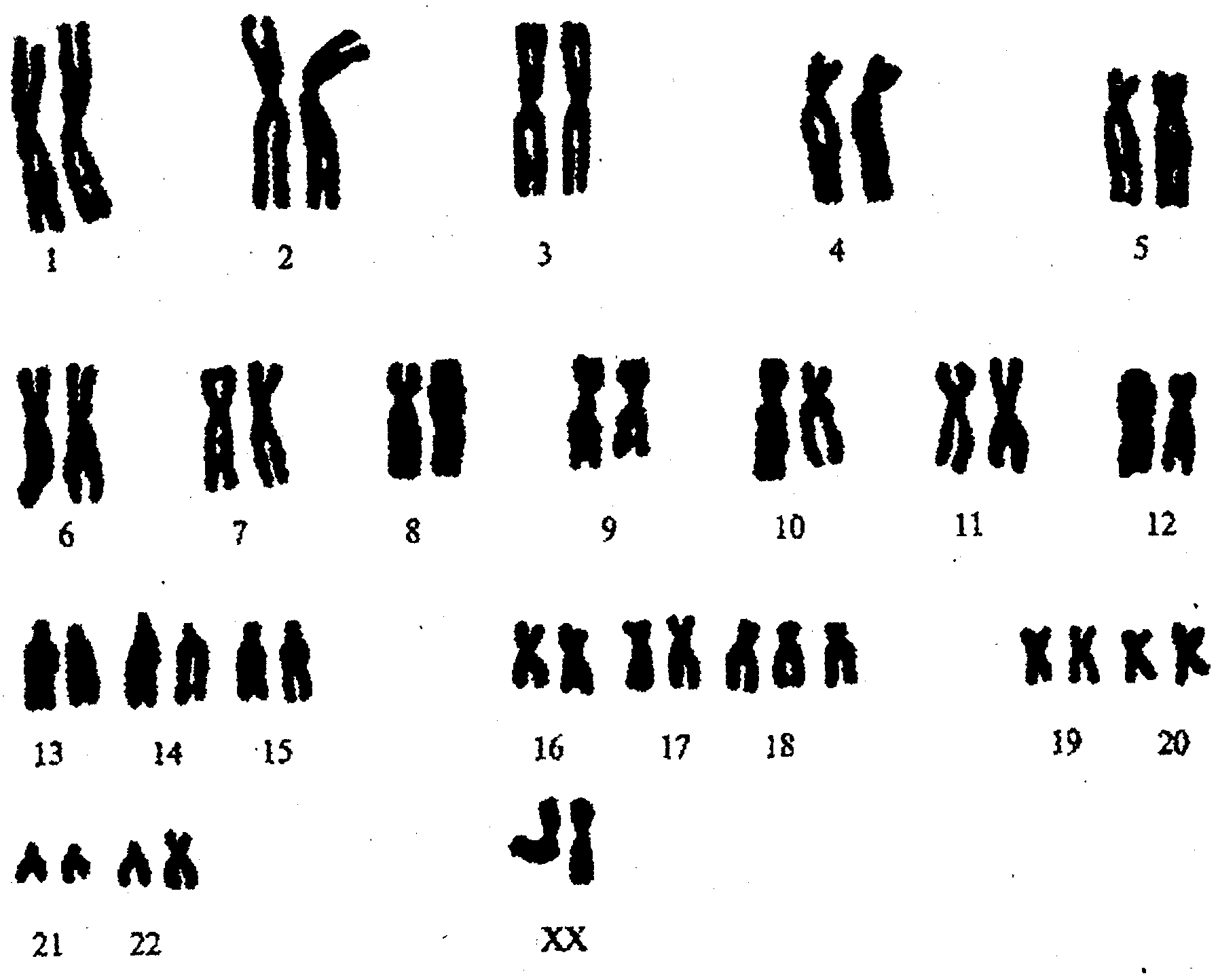
Рис. 6.3. Каріотип при синдромі Едвардса. Проста повна трисомія 18 (47,ХХ, +18)
Череп доліхоцефалітичної форми, нижня щелепа та ротовий отвір маленькі, очні щілини вузькі та короткі, низько розташовані вушні раковини. Перенісся тонке, виступаюче. Відмічається флексорне положення кистей, аномально розвинута стопа, 1-й палець стопи коротший за 2-й, збільшення дуг на пальцях. У 90% діти гинуть у віці до 1 року від ускладнень грубої патології. Лікування - симптоматичне.
Трисомія - 8 зустрічається з частотою 1:5000, переважають хворі хлопчики (5:2). В основному (90%) синдром відноситься до мозаїчних форм. Повні трисомії-8, як правило, летальні. Основними симптомами є розумова відсталість. Важкість клінічних форм варіабельна. Діти народжуються недоношеними.
У дітей констатуються: особливості в будові обличчя (виступаючий лоб, епікант, глибоко посаджені очі, гіпертелоризм очей та сосків, товсті губи та вивернута нижня губа, великі вушні раковини), вади опорно-рухливого апарату – аномалії суглобів (контрактури суглобів, аплазія наколінника), аномалії хребта (додаткові хребці, неповне закриття хребетного каналу), аномалії ребер, сечостатевої системи (зокрема гідронефроз), аномалії ануса. Прогноз несприятливий, хоча описані випадки тривалості життя окремих осіб до 17 років. З часом наростає розумова відсталість, гідроцефалія, контрактури, аплазія мозолистого тіла, зміни з боку скелета. Методи лікування не розроблені.
Трисомія 9. При даній трисомії, а також мозаїцизмі і трисомії 9pter-9q32/33, у випадку живонародження у дитини виражена гіпоплазія, множинні порушення лицьового і мозкового відділу черепа, опорно-рухової, серцево-судинної, сечової систем та вади розвитку головного мозку. Ці аномалії можуть бути значно згладжені при мозаїцизмі. При трисомії 9 діти маложиттєздатні, а при мозаїцизмі живуть кілька місяців (описані випадки, коли діти доживали до 8 років). Лікування не розроблене.
Трисомія 22. Природа цієї аномалії була встановлена тільки після впровадження диференціального забарвлення хромосом. Для синдрому характерна мікроцефалія, наявність розщілини піднебіння, низько розташовані вушні раковини, гіпотонія м’язів, косоокість, гіпоплазія 1-го пальця, аномалії серця, нирок, статевих органів. Усі хворі глибокі олігофрени. Лікування не розроблено.
Синдром Вольфа-Хіршхорна (описаний в 1965 році групою німецьких генетиків на чолі з U.Wolf, американськими дослідниками на чолі з K.Hirschhorn). Частота 1:100000. Для захворювання характерна втрата частини матеріалу коротких плеч 4 хромосоми. Крім того описані випадки синдрому в результаті транслокацій, інверсій, інерцій.
Основними клінічними ознаками синдрому є груба затримка психомоторного та фізичного розвитку, диспластичний синдром з мікроцефалією асиметрією черепа, гіпертелоризмом, дзьобоподібним носом. Цитогенетично підтверджується –4р-.
Діти з синдромом Вольфа-Хоршхорна зазвичай народжуються у молодих батьків від вагітності з фізіологічним перебігом (хоча описані випадки народження передчасно). У періоді новонародженості відмічається пригнічення безумовних рефлексів та зниження м’язового тонусу, а згодом проявляється затримка фізичного та психомоторного розвитку, часто спостерігаються судоми. Характерний зовнішній вигляд дитини: наявна асиметрія черепа (100%), мікроцефалія (92%), гіпертелоризм (90%), виступаюче надперенісся (68%), дзьобоподібний ніс (67%), мікрогенія (7%), деформовані та низько розташовані вушні раковини (86%), наявність деформацій скелета, гіпоспадія та крипторхізм. Часто спостерігаються вроджені вади серця, нирок, інколи – вади розвитку шлунково-кишкового тракту та центральної нервової системи (гіпоплазія мозочка, агенезія або гіпоплазія мозолистого тіла).
Лікування симптоматичне.