

22 Продолжение Синтоны и синтетические эквиваленты
Трансформации разъединения соответствуют конструктивным реакциям, главным образом реакциям, в результате которых образуется связь углерод-углерод. Связи углерод-углерод или углерод-гетероатом могут образовываться в результате следующих основных типов процессов:
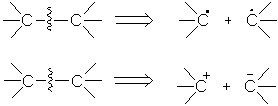
1)электроны для образования связи предоставляются обоими атомами, участвующими в реакции (радикальная реакция), например:
![]()
2) электроны для образования связи предоставляются одним из атомов (реакция нуклеофила и электрофила), например:
![]()
Операция противоположная конструктивной реакции - трансформация разъединения - может приводить к радикальным или ионным фрагментам, которые называют синтонами (радикальный, электрофильный, нуклеофильный):
Реальные химические соединения, имеющие остов синтона и соответствующим образом поляризованные, называются синтетическими эквивалентами данного синтона.
Продукт каждой конструктивной реакции может быть разъединен по образованной связи С-С на синтоны, синтетическими эквивалентами которых являются исходные соединениями реакции.
Реакция Манниха:

Синтонам, соответствующим реакции с гетеролитическим механизмом, можно произвольно приписать положительный или отрицательный заряд. Синтон, поляризация которого соответствует поляризации функционализированногополиена типа:
![]()
(где W - электроноакцепторная группа, а D - электронодонорная), называют синтоном с нормальной полярностью, синтон с противоположной поляризацией - синтоном с обращенной полярностью.
Очевидно, что при реакции синтетических эквивалентов, соответствующих синтонам с нормальной полярностью, могут быть получены лишь молекулы, в которых функциональные группы разделены цепочкой из нечетного числа атомов углерода. Следовательно, при разъединениях (соответствующих реакциям электрофилов и нуклеофилов) для молекул, в которых функциональные группы разделены цепочкой из четного числа атомов углерода, один из получающихся синтонов будет иметь обращенную полярность.
Простые правила, позволяющие отдать предпочтение тому или иному разъединению, заключаются в следующем:
1) разъединение должно базироваться на реальном механизме реакции образования соответствующей связи, что подразумевает в частности получение при разъединении:
а) синтонов с нормальной полярностью или синтонов с обращенной полярностью (с известным способом обращения), которым могут быть поставлены в соответствие реальные синтетические эквиваленты;
б) синтонов, которые соответствуют вероятным реакционным интермедиатам с наибольшей стабильностью.
2) разъединение должно приводить к максимальному упрощению, что может быть достигнуто:
а) разъединением у места наибольшего разветвления молекулы
б) разъединением, ведущим к синтонам с одинаковым остовом (учет симметрии).
В англоязычной литературе для таких (d1, a2, d3) синтонов используют термин "umpoledsynthons" от немецкого "dieUmpolung" - обращение полярности.
Так, например, в рассмотренном выше анализе ТМ4синтоны 4a, 5d, 6d и 7a являются естественными (логичными), а синтоны 4a, 5a, 6a и 7d - неестественными (нелогичными).

Конечно, такое подразделение синтонов условно, поскольку реально существуют реагенты, соответствующие "неестественным" (umpoled) синтонам [7-9]. Так, например, синтону 7d (ацил-анион) соответствует литиевая соль дитиана:
![]()
Теперь можно сформулировать принципы подхода к планированию синтеза из заданного исходного соединения:
1.Определите положение остова исходного соединения в ТМ.
2.Проведите расчленение связи, которая дает максимальное упрощение структуры. Лучше всего "малым укусом" (smallbite) разбить молекулу на два крупных "осколка".
3.Припишите продуктам расчленения заряды - при этом получатся синтоны.
4.Найдите реагенты, соответствующие этим синтонам.
5.Выберите наилучшую комбинацию синтонов с учетом соответствующих им реагентов.
6.Повторите процедуру до тех пор, пока не найдете способы построения всех интересующих Вас С-С связей.
7.Обратите ретросинтетическую процедуру и напишите схему синтеза.
Однако, в своей работе химик-синтетик сталкивается с другой постановкой проблемы: как правило, известна лишь структура молекулы целевого соединения (ТМ). В этом случае задача усложняется - в начале анализа не известно, к каким исходным соединениям мы придем. Ясно лишь, что эти исходные соединения должны быть доступными.
В этом случае решение также можно найти с помощью последовательных расчленений ТМ (disconnectionapproach). Именно такой подход является наиболее продуктивным. При этом очень важен правильный выбор расчленений и соответствующих трансформов.
В ходе анализа предпочтение следует отдавать таким трансформам, которые дают максимальное упрощение структуры молекулы. Этим мощным упрощающим трансформам соответствуют "мощные реакции" [2] синтеза. Ряд таких реакций приведен в таблице 1.
Эти реакции называются мощными, поскольку они в одну стадию приводят к значительному усложнению молекулы. При этом может произойти циклизация, либо существенная реорганизация молекулы. Относительно мало реакционноспособные функциональные группы могут превратиться в высоко реакционноспособные. О подобных мощных реакциях следует всегда помнить при конструировании дерева синтеза.
23. Карбкатионные реакции, приводящие к формированию углеводородного скелета. Понятие о стабильности карбкатионов. Получение карбкатионов из соединений различной природы (алкилгалогениды, алкены и т.д.).
КАРБКАТИОНЫ (карбокатионы), орг. катионы с четным числом электронов, строение которых м.б. представлено (по крайней мере, формально) структурой с электронодефицитным атомом углерода.имеющим вакантную орбиталь (С+). Реальный электронный дефицит этого атома (карбкатионного центра) м.б. очень мал из-за взаимод. с окружающими структурными фрагментами (делокализация заряда).карбкатионы подразделяют на: 1) классические ("карбениевые ионы") производные СН3+, которые характеризуются малой степенью взаимод. между карбкатионным центром и структурными фрагментами в b- или более удаленных положениях (напр., соед. ф-лы I), и 2) неклассические ("карбониевые ионы") - обычно производные СН5+ - со значит. степенью такого взаимод., например соед. II.
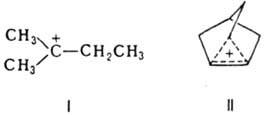
Термин "карбониевые ионы" ("ионы карбония") используют также для обозначения всех карбкатионыкарбкатионы играют важную роль в орг. химии как интермедиаты мн. гетеролитич. реакций, имеющих теоретич. и прикладное значение (электроф. замещение в ароматич. ряду, электроф. присоединение, нуклеоф. замещение в алифатич. ряду, мол. перегруппировки, катионная полимеризация. каталитич. крекинг и др.). карбкатионы обычно образуются при действии электроф. реагентов, например, протонных кислот, на орг. соединения (как правило, ненасыщенные; ур-ния 1 и 2), при гетеролитич. разрыве связи (3), при разложении диазониевых катионов (4):

В газовой фазе карбкатионы образуются, например, при ионизации электронным пучком в масс-спектрометре. Т. наз. долговечные карбкатионы генерируют обычно протонированием сравнительно слабоосновных орг. соед. в сверхкислых средах (HSO3F-SbF5, HF-SbF5 и др.) при низких температурах. Относит.устойчивость карбкатионы зависит от их строения и изменяется в очень широких пределах, увеличиваясь по мере увеличения степени делокализации заряда. Эффективно делокализуют заряд ненасыщ. фрагменты (двойная связь, ароматич. кольцо и т. п.), заместители с неподеленными парами электронов (OR, NR2 и др.) или содержащие атом переходного металла (ферроценил и др.).карбкатионы - реакционноспособные частицы, они взаимод. с нуклеофилами, присоединяя их (напр., ур-ния 5 и 6) или отщепляя протон (электрофил) (7):

Многие карбкатионы склонны к перегруппировкам (взаимод. с "внутр. нуклеофилом"), протекающим обычно путем 1,2-сдвига карбкатионы-л. мигранта (разрешенная сигматропная реакция согласно правилам Вудворда-Хофмана): Все виды химической продукции! Опт Спирты, соли, сода, аммиак, ацетон. При заказе от 100 тыс. руб. - скидка от 5%! centrsnab-b2bcompany.ru

Разновидность карбкатионы - карбоксоний-катионы; в этих ионах карбкатионный центр обычно непосредственно связан с одной или нескарбкатионы группами OR. Их строение м.б. описано двумя резонансными структурами:
![]()
где R - H, Alk, Ar и др. или часть циклич. системы, как, например, в пирилиевых катионах. К карбоксоний-катионам относятся и ацилий-катионы:
![]()
Бекмановский перегруппировкой 2-го рода (перегруппировкой Вернера, фрагментацией, или расщеплением, по Бекману) называют расщепление связи С—С в кетоксимах с образованием нитрилов. Это особенно характерно для оксимовгидроксикетонов:
![]()
Альдоксимы обычно также превращ. в нитрилы:
![]()
Аналогично оксимам перегруппировываются простые и сложные эфиры оксимов (перегруппировка Бекмана-Чепмена), напр.:

Б. п. родственна перегруппировкам Гофмана, Курциуса и Шмидта, в к-рых орг. радикал также мигрирует от С к N. Эта р-ция используется для пром. получениякапролактама из циклогексаноноксима, ацильных производных ароматич. аминов, а также для определения строения кетонов (путем превращ. их в амиды или нитрилы и далее в к-ты). Перегруппировка открыта Э. Бекманом в 1886.
Гофмана реакции, под этим названием известны три реакции, предложенные нем. химиком А. В. Гофманом. 1) Синтез первичных аминов действием брома и щёлочи на амиды карбоновых кислот. Образующиеся амины содержат на один атом углерода меньше, чем исходный амид:
RCONH2 + Br2 + OH- ® RNH2 + CO2 + H2O + Br -.
В результате этой реакции могут быть получены алифатические, жирноароматические, ароматические и гетероциклические амины. Осуществление Г. р. в спиртовой среде приводит к образованию уретанов
(RNHOCOR).
2) Синтез алифатических аминов действием алкилирующих реагентов (галогеналкилов, диалкилсульфатов и др.) на аммиак. В результате образуется (в виде солей) смесь первичного, вторичного и третичного амина и соль четвертичного аммониевого основания; например, при использовании CH3I получаются [CH3NH3]l; [(CH3)2NH2]I; [(CH3)3NH]I и [(CH3)4N]I соответственно.
3) Термическое расщепление гидроокисей четвертичных аммониевых оснований с образованием третичного амина и олефина:
Если атом азота в четвертичном основании связан с различными заместителями, образуется олефин, содержащий наименьшее число алкильных заместителей при двойной связи (правило Гофмана).
Важнейшая область применения Г. р. — исследование структуры алкалоидов.
Бекмана перегруппировка, химическая реакция превращения оксимов в амиды кислот под действием кислотных дегидратирующих агентов (PCI5, H2SO4, олеум и др.):
24. Перегруппировки карбкатионов. Факторы, влияющие на склонность катиона к перегруппировке. Способность различных групп к миграции. Другие перегруппировки электронодефицитных частиц (на примере перегруппировок Гофмана, Бекмана, Курциуса, Лоссена и т.д.), закономерности их протекания. Применение перегруппировок в органическом синтезе. Перегруппировка Бекмана в промышленном синтезе капролактама.
СЕКСТЕТНЫЕ ПЕРЕГРУППИРОВКИ (от лат. sextus-шестой), изомеризации, протекающие с 1,2-миграцией атома Н, групп алкил, арил, ацил и др. к положительно заряженному или нейтральному атому С, N или О с шестью электронами, т.е. секстетом, на внеш. электронной оболочке. С. п. могут происходить с участием соответствующих активных интермедиатов, содержащих (Вагнера-Меервейна перегруппировки ),(Вольфа перегруппировка ), =N+ (Бекмана перегруппировка , Шмидта реакция ),(Гофмана реакция , Курциуса реакция , Лоссена реакция ), —О+(Байера-Виллигера реакция и др.).
Наиб. распространены перегруппировки Вагнера-Меер-вейна с 1,2-миграцией к атому С (карбениевому центру). Среди них-ацилоиновая (см. Ацилоины ), Демьянова перегруппировка ,Пинаколиновая и ретропинаколиновая перегруппировки , Рупе перегруппировка, Тиффено реакция и др.
По сходным механизмам протекают перегруппировки Вольфа (см. также Арндта-Aйcmepma реакция)и Курциуса-с нуклеоф. 1,2-миграциями групп соотв. к двухкова-лентному нейтральному атому С и одноковалентному нейтральному атому N. Перегруппировки Гофмана и Лоссена отличаются от р-цииКурциуса способами генерирования нитреновой структуры; считают, что стадии этих изомеризации происходят согласованно. Примеры перегруппировок:
Кислотнокатализируемые перегруппировки Бекмана и Шмидта представляют собой разные способы превращения карбонильныхсоед. в амиды:
Подобно р-ции Шмидта протекает перегруппировка Байера-Виллигера, в к-рой участвуют ионы —О+.
Стереохим. исследованиями показано, что при С. п. конфигурация мигрирующего заместителя сохраняется, тогда как в начальном и конечном пунктах миграции преобладает инверсия.
БЕКМАНА ПЕРЕГРУППИРОВКА , изомеризация кетоксимов в N-замещенные амиды карбоновых к-т под действием кислотных агентов:

Р-цияэкзотермична. Кислотными агентами служат полифосфорные к-ты, пентахлорид и пентаоксид фосфора, хлорангидриды сульфокислот, карбоновых к-т и др. Процесс обычно проводят в пиридине, этиловом эфире или бензоле; скорость его сильно возрастает с ростом полярности р-рителя. Р-ция одинаково хорошо осуществляется с оксимамиалифатич. и ароматич. кетонов. Оксимыжирноароматич. кетонов всегда превращ. в ацильные производные ароматич. аминов. В случае циклич. кетонов происходит расширение цикла, напр.:

Б. п. стереоспецифична: всегда мигрирует радикал, находящийся в транс-положении к гидроксилу. Конфигурация атомов С в R' сохраняется. Установлено, что перегруппировываются не сами кетоксимы, а продукты их взаимод. с реагентами. Так, в случае ацилирующих агентов образуется ацильноепроизводное, последующий распад к-рого происходит с разрывом связи N—О и синхронной миграцией R'. Для жирноароматич. оксимов постулировано промежут. образование производных азиоина:


где R, R' — одинаковые или разные органические радикалы (СН3, C2H5, C6H5 и др.). Б. п. используют в промышленности для получения капрона (стадия превращения циклогексаноноксима в e-капролактам). Б. п. открыта в 1886 немецким химиком Э. О. Бекманом.
Курциуса 
Получение первичных аминов термической перегруппировкой азидов карбоновых кислот в изоцианаты с последующим гидролизом. Применима также к гетероциклическим и алициклическим азидам, содержащим различные функциональные группы. Азиды a-галогензамещенных и a,b-непредельных карбоновых кислот в этих условиях дают альдегиды или кетоны. Перегруппировка катализируется сильными кислотами. Метод широко применяется для синтеза изоцианатов, уретанов и N,N’-дизамещенных мочевин.
В промышленности капролактам получают
из бензола, фенола или толуола по схемам:
промышленности капролактам получают
из бензола, фенола или толуола по схемам:
В промышленности наибольшее распространение получил метод синтеза капролактама из бензола. Технологическая схема включает гидрирование бензола в циклогексан в присутствии Pt/Al2O3 или никель-хромового катализатора при 250-350 и 130-220 °С, соответственно. Жидкофазное окисление циклогексана в циклогексанон осуществляют при 140-160°С, 0,9-1,1 МПа в присутствии нафтената или стеарата Со. Получающийся в результате окисления циклогексанол превращают в циклогексанон путем дегидрирования на цинк-хромовых (360-400 °С), цинк-железных (400 °С) или медь-магниевых (260-300 °С) смешанных катализаторах. Превращение в оксим проводят действием избытка водного раствора сульфата гидроксиламина в присутствии щелочи или NH3 при 0-100°С. Завершающая стадия синтеза капролактама. - обработка циклогексаноноксима олеумом или конц. H2SO4 при 60-120 °С (перегруппировка Бекмана). Выход капролактама в расчете на бензол 66-68%. При фотохимическом методе синтеза капролактама из бензола циклогексан подвергают фотохимическомунитрозированию в оксим под действием NOCl при УФ облучении. Метод синтеза капролактама из фенола включает гидрирование последнего в циклогексанол в газовой фазе над Pd/Al2O3 при 120-140°С, 1-1,5 МПа, дегидрирование полученного продукта в циклогексанон и дальнейшую обработку как в методе синтеза из бензола. Выход 86-88%.