

Журнал неврологии и психиатрии / 2007 / NEV_2007_10_01
.pdfОБЩИЕ ВОПРОСЫ НЕВРОЛОГИИ И ПСИХИАТРИИ
Глутаровая ацидурия тип 1: клиника, диагностика и лечение
С.В. МИХАЙЛОВА, Е.Ю. ЗАХАРОВА, М.Ю. БОБЫЛОВА, Е.С. ИЛЬИНА, А.В. БАНИН, И.В. РАССКАЗЧИКОВА, Г.В. БАЙДАКОВА, О.В. ШЕХТЕР, И.Б. БРЮСОВА, Г.И. ВОЛКОВА, А.С. ПЕТРУХИН
Glutaric aciduria type 1: clinical presentations, diagnostics and treatment
S.V. MIKHAILOVA, E.YU. ZAKHAROVA, M.YU. BOBYLOVA, E.S. IL’INA, A.V. BANIN, I.V. RASSKAZCHIKOVA, G.V. BAIDAKOVA, O.V. SHEKHTER, I.B. BRYUSOVA, G.I. VOLKOVA, A.S. PETRUKHIN
Кафедра нервных болезней педиатрического факультета Государственного образовательного учреждения высшего профессионального образования; Российский государственный медицинский университет Министерства здравоохранения РФ, Москва; Российская детская клиническая больница, Москва; ГУ Медико-генетический научный центр, Москва
Глутаровая ацидурия тип 1 — редкое аутосомно-рецессивное нейрометаболическое заболевание, обычно развивается на 1-м году жизни и характеризуется прогрессирующими экстрапирамидными расстройствами в результате повреждения базальных ганглиев. Описаны первые установленные в России случаи этого заболевания. Приводится сравнение клинических особенностей с данными литературы. Манифестация заболевания, как правило, возникает после перенесенных инфекций и характеризуется возникновением судорог, рвоты, метаболического ацидоза и угнетением сознания вплоть до комы. В результате перенесенных кризов развиваются некрозы базальных ганглиев, что приводит к развитию дистоний, дискинезий и хореоатетоза. Вторичными осложнениями заболевания являются трудности при кормлении, задержка речевого развития, хронический аспирационный синдром и грубая задержка двигательного развития. Диагностика заболевания основывается на анализе мочи и крови методами тандемной масс-спектрометрии и газовой хроматографии. Основным методом лечения являются низкобелковая диета с ограничением поступления лизина и дополнительное введение карнитина. Приводятся данные по лечению этого заболевания.
Ключевые слова: глутаровая ацидурия тип 1, органические ацидурии, судороги, дистония, задержка психомоторного развития, тандемная масс-спектрометрия и газовая хроматография.
Glutaric aciduria type I is a rare autosomic recessive neurometabolic disease, which develops in the first year of life and is characterized by progressive extrapyramidal disorders as a result of the basal ganglia damage. We describe first cases of this disease in Russian population. The clinical observations are compared to the literature data. The disease usually develops after infections and features by seizures, vomiting, metabolic acidosis and deprivation of consciousness up to coma. These crises lead to the development of necroses of the basal ganglia that results in dystonias, dyskinesias and choreoatethosis. The secondary complications of the disease are difficulties with feeding, speech delay, chronic aspiration syndrome and severe delay of movement development. Diagnostics of the disease is based on urine and blood tests using methods of tandem mass spectrometry and gas chromatography. Treatment is based on dietary lysine or protein restriction and supplementation with carnitine. The data on the treatment of this disease are presented.
Key words: Glutaric aciduria type I, organic acidurias, seizures, dystonia, psychomotor delay, tandem mass spectrometry, gas chromatography.
Глутаровая ацидурия тип 1 — аутосомно-рецес- |
имеются описания более 400 случаев этого заболева- |
||
сивное заболевание, обусловленное мутациями в гене, |
ния [29]. ГА1 относится к числу редких наследственных |
||
кодирующем фермент глутарил-КоА-дегидрогеназу |
болезней обмена веществ. Частота заболевания в стра- |
||
(GCDH). Дефицит данного фермента приводит к на- |
нах Западной Европы составляет в среднем 1:50 000 |
||
коплению в биологических жидкостях и тканях глу- |
живых новорожденных. Высокая частота встречаемо- |
||
таровой и 3-гидроксиглутаровой кислот, оказываю- |
сти ГА1 отмечена среди общин амишей в Америке и |
||
щих нейротоксическое действие [1, 2, 4, 45]. |
|
Канаде — 1:300 [48]. |
|
Глутаровая ацидурия тип 1 (глутаровая ацидемия |
Генетика, биохимия и патогенез |
||
тип 1) — ГА1 — впервые была описана S. Goodman и |
|||
|
|||
соавт. в 1975 г. На сегодняшний день в литературе |
Ген GCDH картирован на хромосоме 19p13.2. На |
||
|
|
сегодняшний день в гене GCDH описано более 150 |
|
|
|
различных мутаций, одной из самых распространен- |
|
© Коллектив авторов, 2007 |
|
ных является R402W, встречающаяся с частотой 12— |
|
|
40% в странах Западной Европы [21]. Некоторые му- |
||
|
|
||
Zh Nevrol Psikhiatr Im SS Korsakova 2007;107:10:4—12 |
тации характерны для определенных этнических групп |
||
4 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2007 |

èизолятов: мутации P248L и E365K в Турции, мутации IVS1+5G>T и A421V в общинах амишей [7].
GCDH участвует в метаболизме лизина, гидролизина, триптофана. Она локализована в матриксе митохондрий и представляет собой гомотетрамер. Каждая субъединица этого белка состоит из 438 аминокислот, N-терминальный остаток (44 аминокислоты) удаляется после транспортировки фермента в митохондрию [19, 23]. GCDH катализирует две последовательных реакции превращения глутарил-КоА в кро- тонил-КоА (реакции дегидрогенирования и декарбоксилирования). В результате блокирования данной ферментной реакции происходит накопление глутаровой
è3-гидроксиглутаровой кислот в биологических жидкостях и тканях. Мутантный фермент относится к одной из флавопротеиновых дегидрогеназ митохондриального матрикса, участвующих в переносе электронов к убихинону через электронпереносящий флавопротеин в дыхательной цепи митохондрий [1, 4, 45].
Механизмы патогенеза ГА1 не до конца изучены. Преимущественное поражение стриарной системы связывают с избирательной токсичностью глутаровой кислоты и/или ее производных. Также глутаровая кислота и ее метаболиты ингибируют декарбоксилазу глутаминовой кислоты (фермент, участвующий в метаболизме ГАМК), что вызывает снижение концентрации этого тормозного нейромедиатора. У больных с ГА1 выявлено значительное снижение декарбоксилазной активности в лобных отделах коры головного мозга, хвостатых ядрах и скорлупе и снижение концентрации ГАМК в спинномозговой жидкости (СМЖ) [38, 53]. Однако данные биохимические нарушения могут быть вторичными и возникать вследствие повреждений ГАМКергических нейронов мозга [14].
Механизмы патогенеза острых «энцефалопатиче- ских» кризов до конца не выяснены. Считают, что глутаровая и 3-гидроксиглутаровая кислоты, имеющие структурное сходство с глутаматом, взаимодействуют с N-метил-аспартатными рецепторами, для которых глутамат является естественным активатором, что вызывает чрезмерное накопление ионов Са2+
âпостсинаптических нейронах и приводит к гибели клеток [35, 36, 39]. Другим возможным нейротокси- ческим фактором считают накопление промежуточ- ного метаболита обмена триптофана и лизина — квинолиновой кислоты. В настоящее время до конца не распознана причина лобно-височной атрофии/гипоплазии и субдуральных кровоизлияний. Одним из возможных объяснений является поражение сосудов головного мозга. Считается, что во время эмбриогенеза 3-ОН-глутаровая кислота может нарушать формирование стенок сосудов, приводя к повышению их проницаемости [49, 50].
Патоморфология
ГЛУТАРОВАЯ АЦИДУРИЯ
ты, мозолистое тело, внутренняя капсула, глубокие отделы белого вещества мозжечка, ствола мозга. У ряда пациентов обнаруживают «губчатую» дегенерацию белого вещества, преимущественно в перивентрикулярных областях, реже в таламусе, бледном шаре и стволе головного мозга [1, 4, 45].
Клинические проявления
ГА1 обычно дебютирует в раннем детском возрасте — от 6 до 18 мес [2, 3, 20, 24, 45].
Â75% случаев наблюдается «энцефалитоподобный» вариант заболевания. У таких пациентов внезапно появляются частые срыгивания, неукротимая рвота, судороги, развивается диффузная мышечная гипотония, быстро трансформирующаяся в мышеч- ную ригидность/спастичность, возникают различные виды гиперкинезов (орофациальные, хореиформные, хореоатетоидные, баллистические), часто происходит угнетение сознания до сопора и комы [40]. Нередко «провоцирующими» факторами развития заболевания являются черепно-мозговая травма, хирургические вмешательства, инфекции или вакцинация [45].
Как правило, заболевание носит волнообразный характер: после перенесенных метаболических кризов происходит медленное, но неполное восстановление неврологических нарушений [27—29]. У неле- ченых больных смертельный исход наступает в тече- ние 1-го десятилетия жизни на фоне тяжелого метаболического криза или развития Рейе-подобного синдрома [3, 8, 9, 11, 14, 23, 46].
Â25% случаев заболевание имеет менее острое, доброкачественное течение. На 1-м году жизни у детей наблюдаются задержка психомоторного развития,
àв дальнейшем постепенная утрата ранее приобретенных навыков, присоединяются различные виды гиперкинезов. Дистонические гиперкинезы приводят к нарушению ходьбы, письма и речевых функций. Многие больные длительное время наблюдаются у неврологов со спастико-гиперкинетической формой детского церебрального паралича (ДЦП) [3, 47]. У большинства пациентов интеллект не страдает или наблюдаются легкие когнитивные нарушения [1—3, 20, 45, 46]. У 75—80% больных макроцефалия может быть первым появлением болезни, которое отмеча- ется с рождения или развивается в первые месяцы жизни [3, 11, 31, 33, 46].
Другими частыми симптомами являются профузное потоотделение, эпизоды немотивированной лихорадки. Среди офтальмологических нарушений нередко обнаруживают кровоизлияния в сетчатку, ее пигментную дегенерацию, катаракту, офтальмопарез, косоглазие [3, 32]. Крайне редко заболевание встре- чается у взрослых [5, 15]. При семейном скрининге, проводимом в общинах амишей, в 5% случаев выявлены бессимптомные больные [4, 48].
Нейрорадиологические особенности
При аутопсии у всех пациентов с ГА1 выявляют выраженную субкортикальную и кортикальную атрофию головного мозга, атрофическую вентрикуломегалию. В большинстве случаев визуализируют некротические изменения в области скорлупы и головки хвостатого ядра. Реже поражаются зрительные трак-
Наиболее частыми нейрорадиологическими признаками при ГА1 являются лобно-теменная гипоплазия/атрофия, вентрикуломегалия, субдуральные гематомы, задержка миелинизации/демиелинизация и некроз базальных ганглиев [10, 33, 34]. У пациентов эти изменения могут встречаться как изолированно,
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2007 |
5 |

ОБЩИЕ ВОПРОСЫ НЕВРОЛОГИИ И ПСИХИАТРИИ
так и практически в любом сочетании. По данным литературы [10, 16, 52], наиболее характерной нейрорадиологической особенностью ГА1 (встречается у 95% больных) является симметричное расширение сильвиевых щелей с формированием «эффекта надкушенного яблока» или «крыльев летучей мыши». До настоящего времени нет единого мнения о генезе выявляемых изменений лобно-височной и заднебазальной областей головного мозга. Согласно одной из гипотез, их причиной является врожденная гипоплазия. У большинства нелеченых пациентов выявлено расширение субарахноидальных пространств даже на пресимптоматической стадии заболевания, не претерпевающих никаких изменений при проведении контрольных магнитно-резонансных томографий (МРТ) головного мозга и сочетающихся с нормальной толщиной коры головного мозга и нормально развитыми извилинами [16, 52]. С другой стороны, у некоторых новорожденных с точно установленным диагнозом при проведении компьютерной томографии (КТ)/МРТ головного мозга повреждение лобно- височно-теменных областей обнаруживают только через несколько месяцев после рождения, что является аргументом в пользу кортикальной атрофии большого мозга [10, 14].
У большинства пациентов субдуральные гигромы/ гематомы часто располагаются билатерально, реже имеют одностороннюю локализацию [34]. Некоторые авторы описывают образование битемпоральных арахноидальных или субэпендимальных кист. Изменения белого вещества головного мозга описаны у многих больных с ГА1 и часто сочетаются с некрозом базальных ганглиев. Поражение белого вещества, как правило, симметричное, преимущественно в перивентрикулярных зонах, в области передних и задних рогов боковых желудочков и в области семиовального центра, в некоторых случаях обнаруживают демиелинизацию субкортикальных U-волокон. Не совсем понятно, представляют ли данные области очаги демиелинизации или гипомиелинизации. У некоторых больных описано поражение мозолистого тела. По данным Е. Twomey и соавт. [52], у 3 из 14 обследованных ими пациентов выявлены изменения в области колена мозолистого тела, но другие авторы не находили данных нарушений при сходных условиях исследования [51].
Лабораторная диагностика и профилактика ГА1
Основными методами подтверждающей диагностики ГА1 являются биохимические. В биологиче- ских жидкостях (моча и кровь) определяют концентрацию органических кислот и/или ацилкарнитинов (глутарилкарнитина). При данном заболевании повышаются концентрации глутаровой, 3-ОН-глутаро- вой кислот и глутарилкарнитина в десятки раз по сравнению с нормой. Органические кислоты определяют методом хромато-масс-спектрометрии, ацилкарнитины — методом тандемной масс-спектромет- рии [6].
У некоторых больных с ГА1 концентрация глутаровой кислоты в моче и крови может быть в пределах нормы, в таких случаях диагностически значимым маркером является повышение концентрации 3-OH-
глутаровой кислоты, что позволяет предположить диагноз [11, 23].
ДНК-диагностика и определение активности глу- тарил-КоА-дегидрогеназы (в лейкоцитах и фибробластах кожи) необходимы как для верификации диагноза, так и для последующей пренатальной диагностики [7, 22, 24, 42, 54].
В последние годы в связи с широким внедрением тандемной масс-спектрометрии (ТМС) во многих странах проведены пилотные исследования по неонатальному скринингу на ГА1. С помощью ТМС определяют концентрации глутарилкарнитина в пятнах высушенной крови. Повышение уровня этого метаболита при ГА1 происходит до появления клиниче- ских признаков заболевания. Многие исследователи считают, что включение этого заболевания в программы массового скрининга оправдано, так как раннее начало лечения позволяет значительно улучшить ка- чество жизни больных [43, 44].
Лечение
Всем больным с установленным диагнозом ГА1 назначают низкобелковую диету (1,5—2,0 г/кг белка
âдень) с низким содержанием триптофана (17—20 мг/кг/день) и лизина (80—100 мг/кг/день) [1, 2, 4, 20, 45]. Ограничение поступления лизина является главным принципом диетотерапии. В настоящее время разработаны специальные смеси аминокислот (глутаридон, фирма «Нутриция»).
Основным методом метаболической коррекции является прием L-карнитина в дозе от 50—100 мг/кг
âдень длительно, постоянно. L-карнитин связывает глутаровую кислоту и обеспечивает ее выведение из организма в виде глутарилкарнитина [20].
Наряду со специфической терапией проводится симптоматическое лечение: назначают препараты для коррекции мышечного тонуса, гиперкинетических расстройств, судорог. Препаратом выбора для коррекции мышечной дистонии является баклофен, для купирования судорог — вигабатрин, суксилеп, топамакс [29, 30]. Не рекомендуется назначать препараты солей вальпроевой кислоты, так как вальпроаты выводятся из организма в виде вальпроилкарнитина, что истощает резерв свободного карнитина. Кроме того, вальпроевая кислота угнетает работу дыхательной цепи митохондрий [30, 37].
Крайне редко встречается рибофлавинчувствительная форма ГА1. У пациентов с данной формой заболевания при назначении рибофлавина в дозе 100— 200 мг/сут отмечается улучшение состояния в виде уменьшения степени выраженности неврологических нарушений [13]. Остается дискуссионным вопрос о проведении вентрикулоперитонеального шунтирования и эвакуации субдуральных скоплений в мозге при прогрессирующей макроцефалии. Существует мнение, что экстренное нейрохирургическое вмешательство во время острых метаболических кризов может ухудшить состояние пациента и привести к серьезным неврологическим нарушениям [25].
Токсико-метаболические кризы могут возникать во время инфекционных заболеваний, после проведенной иммунизации и хирургического вмешательства. Основными принципами лечения во время ост-
6 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2007 |
рых состояний являются восстановление кислотнощелочного равновесия в организме и дезинтоксикационная терапия. Раннее начало лечения позволяет предотвратить развитие тяжелых неврологических нарушений и метаболических кризов у большинства пациентов. При позднем начале терапии полностью купировать неврологические расстройства не удается, однако специфическое лечение позволяет улуч- шить качество жизни пациента (снизить частоту эпилептических приступов, уменьшить степень выраженности экстрапирамидных расстройств, предотвратить развитие токсико-метаболических кризов).
Материал и методы
Под нашим наблюдением находятся 4 пациента из 3 семей с точно установленным диагнозом ГА1. Всем больным были проведены клиническое и неврологи- ческое обследования, включающие физикальный осмотр, клинические и биохимические анализы крови и мочи, проведенные по стандартным методикам. Инструментальные методы обследования включали проведение электроэнцефалографии, МРТ, КТ и ультразвуковое исследование внутренних органов. Уровень глутаровой кислоты определяли методом хромато-масс-спектрометрии, уровень ацилкарнитинов в крови — методом тандемной масс-спектромет- рии. Молекулярно-генетические исследования для выявления распространенных мутаций гена ГА1 проводились с использованием стандартных методов мо- лекулярно-генетического анализа. Клинические данные приведены в табл. 1.
Результаты и обсуждение
Дифференциальная диагностика ГА1 довольно сложна. Иногда проходит несколько лет после дебюта болезни, прежде чем будет установлен правильный диагноз. Трудности диагностики ГА1 связаны с редкостью данной патологии, клиническим полиморфизмом заболевания, особенностями течения. В отличие от других органических ацидурий при ГА1 обычно не возникает метаболический ацидоз, редко встречаются гипогликемия, тромбоцитопения, лейкопения [3, 17, 27]. Пограничные, а иногда и нормальные концентрации метаболитов также затрудняют диагностику этого заболевания [19].
Среди обследованных нами больных средний возраст манифестации основных клинических симптомов приходился на 4—5-й месяцы жизни. Самое раннее начало — 1-й месяц жизни — наблюдалось у пациента О.А., что, вероятно, связано с неблагоприятным перинатальным анамнезом (острая гипоксия в родах, экстренное родоразрешение путем операции кесарева сечения, низкая оценка по шкале Апгар). У больных Р.Р. и С.Д. до появления первых клиниче- ских симптомов психомоторное развитие соответствовало возрасту, а у больного С.С. наблюдалась задержка двигательного развития. У всех пациентов отмечалось увеличение окружности головы свыше 90 центилей, поэтому дети наблюдались у невролога с диагнозом «последствия перинатального поражения, гидроцефальный синдром» и систематически полу-
ГЛУТАРОВАЯ АЦИДУРИЯ
чали дегидратационную терапию. Патологический прирост окружности головы является типичным для ряда других наследственных болезней: болезнь Канавана, Gm2-ганглиозидозы, мукополисахаридозы. При болезни Канавана в клинической картине доминирует поражение пирамидных путей, при Gm2-ганглио- зидозе, помимо прогрессирующих неврологических расстройств, выявляется гепатолиенальный синдром, при мукополисахаридозах — «черты гаргоилизма»[3].
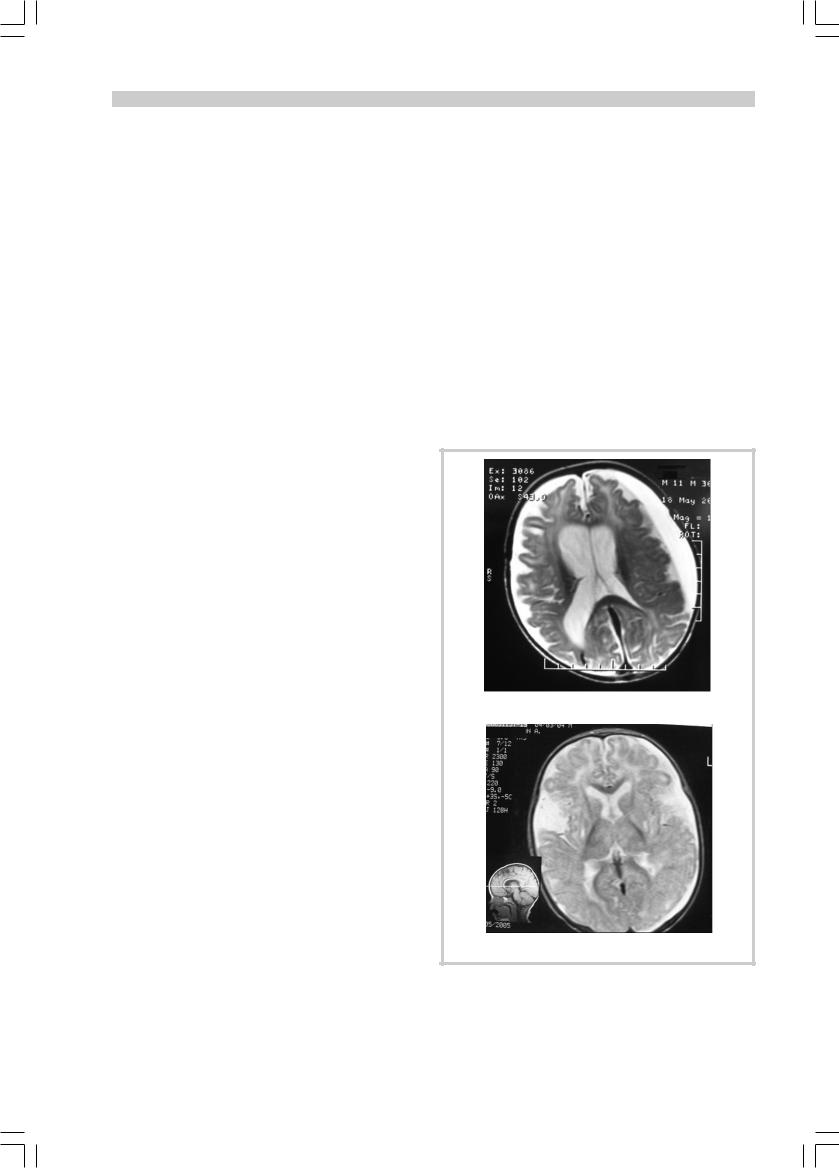
У 3 наблюдаемых нами пациентов заболевание дебютировало внезапно с развития острых «энцефалопатических» кризов. Так, у больных Р.Р. и С.Д. на 2-е сутки после перенесенной черепно-мозговой травмы (ЧМТ) в возрасте 7 и 9 мес соответственно появились угнетение сознания, судороги, повторная рвота; при МРТ/КТ головного мозга на 2-й день болезни были выявлены хронические субдуральные гематомы и расширение желудочковой системы (рис. 1). Дифференциальный диагноз проводился с заболеваниями, протекающими с нарушениями гемостаза, ЧМТ [25].
à
á
Рис. 1. МРТ головного мозга на 2-й день болезни.
а — больной Р., 9 мес; б — больной С., 9 мес.
Грубая кортикальная и субкортикальная атрофия головного мозга, атрофическая вентрикуломегалия, хроническая субдуральная гематома.
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2007 |
7 |

ОБЩИЕ ВОПРОСЫ НЕВРОЛОГИИ И ПСИХИАТРИИ
Таблица 1. Клинический полиморфизм глутаровой ацидурии тип 1 у детей раннего возраста
Показатель |
Î.À. |
Ð.Ð. |
Ñ.Ñ. |
Ñ.Ä. |
|
|
|
|
|
Возраст пациента |
3 ãîäà 8 ìåñ |
2 ãîäà 2 ìåñ |
7,5 ëåò |
1 ãîä |
Возраст дебюта заболевания |
1 ìåñ |
7 ìåñ |
1,5 ìåñ |
9 ìåñ |
Ïîë |
Ì |
Ì |
Ì |
Ì |
Диагноз при поступлении |
Врожденная |
Последствия ЧМТ |
ÄÖÏ |
Последствия ЧМТ |
в стационар |
гидроцефалия |
|
|
|
Возраст установления диагноза |
2,8 ëåò |
2,2 ãîäà |
7,5 ëåò |
9 ìåñ |
Характер манифестации |
Острое |
Острое |
Постепенное |
Острое |
заболевания |
|
|
|
|
Возможные провоцирующие |
— |
×ÌÒ |
— |
×ÌÒ |
факторы |
|
|
|
|
Макрокрания |
+++ |
+++ |
+++ |
+++ |
Гиперкинез: |
|
|
|
|
орофациальный |
+++ |
+ |
+++ |
+ |
хореиформный |
+++ |
+ |
+++ |
+ |
баллистический |
+++ |
+ |
+++ |
+ |
торсионный |
+++ |
+ |
+++ |
+ |
Тортиколиз мышц шеи |
++ |
— |
+++ |
— |
Судороги |
++ |
+ |
— |
++ |
Мышечная дистония |
+++ |
++ |
+++ |
++ |
Спастический тетрапарез |
+++ |
++ |
+++ |
+ |
Задержка двигательного развития |
+++ |
+ |
+++ |
+ |
Задержка речевого развития |
+++ |
+ |
++ |
+ |
Интеллектуальное развитие |
++ |
+ |
+ |
— |
Сопор/кома |
+++ |
+ |
— |
+ |
Уровень свободного карнитина в |
77,8/— |
20,3/69,1 |
4,79/74,3 |
49,9/71,7 |
крови (норма 10—90) до /после |
|
|
|
|
лечения |
|
|
|
|
Уровень глутарилкарнитина в |
4,12/— |
0,827/0,675 |
1,08/1,38 |
0,644/1,13 |
крови (норма до 0,24 усл.ед.) |
|
|
|
|
до/после лечения |
|
|
|
|
Генотип |
R402W/R402W |
R402W/? |
R402W/? |
R402W/? |
Улучшение состояния после |
+ |
++ |
+++ |
++ |
назначения L-карнитина |
|
|
|
|
Kатамнез на фоне специфического |
Грубый |
Задержка |
Грубый |
Задержка |
лечения |
неврологический |
темпов моторного |
неврологический |
темпов моторного |
|
дефицит |
развития |
дефицит |
развития |
Примечание. Степень проявления признаков: + — умеренная, ++ — выраженная, +++ — ярко выраженная. |
|
|||
При исследовании показателей крови ни у одно- |
периодами стабилизации. Внезапно на фоне полного |
го пациента гемостатических изменений в коагуло- |
благополучия появлялись ярко выраженные общемоз- |
грамме крови выявлено не было. Исключить полно- |
говые симптомы: сонливость, срыгивания, рвоты, |
стью последствия ЧМТ не представлялось возмож- |
генерализованные судороги. После 3-го «энцефало- |
ным, так как дебют ГА1 во многом был с ней сходен. |
патического» криза у ребенка развился спастико-ги- |
При ЧМТ после травмирующего фактора остро раз- |
перкинетический и псевдобульбарный синдром. При |
виваются общемозговые симптомы в виде угнетения |
МРТ/КТ головного мозга в динамике визуализиро- |
сознания, тошноты, рвоты, судорог, однако выра- |
вались прогрессирующая атрофическая вентрикуло- |
женные атрофические изменения и хронические суб- |
мегалия и расширение субарахноидальных про- |
дуральные гематомы, обнаруженные при МРТ/КТ |
странств (рис. 2). До установления правильного диа- |
головного мозга в ранний период после травмы, ста- |
гноза ребенок длительное время наблюдался в различ- |
вили диагноз ЧМТ под сомнение [12]. У пациента О.А. |
ных стационарах с подозрением на вялотекущий вен- |
заболевание дебютировало остро и протекало под |
трикулит, врожденную гидроцефалию. Однако харак- |
маской хронической нейроинфекции: эпизоды ухуд- |
терных для вентрикулита изменений в СМЖ у наше- |
шения состояния (1, 6, 12, 18-й месяцы) сменялись |
го пациента обнаружено не было. Врожденная гидро- |
8 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2007 |

Рис. 2. МРТ головного мозга больного О., 3 года.
Атрофическая вентрикуломегалия и кортикальная атрофия, преимущественно лобно-височно-теменных областей, демиелинизации в перивентрикулярных областях и субкортикальных U-волокон.
цефалия может манифестировать в раннем неонатальном периоде тремором, беспокойством, симптомом Грефе, выбуханием большого родничка, расхождением черепных швов и патологическим приростом окружности головы. У нашего пациента не выявлялись симптомы, характерные для гидроцефалии, и при КТ/МРТ головного мозга были обнаружены атрофическая вентрикуломегалия и атрофия кортикаль- но-субкортикальных отделов мозга, что практически сразу отвергло диагноз врожденной гидроцефалии.
Экстрапирамидные нарушения (различные виды гиперкинезов, мышечная дистония) могут быть ведущими не только при наследственных заболеваниях нервной системы, но и при последствиях гипоксиче- ски-ишемических, инфекционных заболеваний мозга. Среди наследственных заболеваний, дебютирующих на 1-м году жизни, дифференциальный диагноз ГА1 должен проводиться с заболеваниями из группы нарушений нейромедиаторного обмена, синдромом Леша—Нихена, митохондриальной патологией, другими органическими ацидуриями и аминоацидопатиями [1—3, 20, 26, 41, 45]. Для ГА1 характерным является развитие орофациальных и торсионных дискинезий различной степени выраженности, имеющих прогредиентное течение, несмотря на проводимую терапию. Заболевания из группы нейротрансмиттерного обмена характеризуются ранним дебютом, а основным клиническим синдромом является гиперто- нически-гипокинетический: брадикинезия, гипомимия, мышечная гипертония; при болезни Ли клини- ческий фенотип в виде прогрессирующей мышечной дистонии, тремора, миоклоний [1, 20]. При класси- ческой форме болезни Леша—Нихена первые симптомы могут отмечаться с рождения: синдром повышенной нервно-рефлекторной возбудимости, мышеч- ная гипотония, нарушения вскармливания. К 3—6-му месяцу жизни становится явной задержка психомо-
ГЛУТАРОВАЯ АЦИДУРИЯ
торного развития, мышечная гипотония сменяется мышечным гипертонусом, в дальнейшем присоединяются подкорковые нарушения (атетоидные движения рук, хореоатетоз, торсионно-дистонические гиперкинезы) [1, 4, 20]. В отличие от ГА1 при болезни Леша—Нихена не наблюдается макроцефалия, заболевание имеет медленно прогрессирующее течение [41].
Как и в наших наблюдениях, ГА1 может протекать под маской гиперкинетической формы ДЦП. При этой форме ДЦП двигательные расстройства представлены гиперкинезами — атетозом, хореоатетозом, торсионной дистонией. В период новорожденности у большинства детей с формирующейся картиной гиперкинетической формы ДЦП характерны бедность активных движений, вялость, снижение мышечного тонуса. Ребенок слабо сосет, часто срыгивает, у него нарушена координация сосания, глотания и дыхания. В возрасте 2—3 мес появляются «дистонические атаки», характеризующиеся внезапным повышением мышечного тонуса во время движения. Они протекают по типу рефлекса Моро, асимметричного шейного тонического рефлекса или в форме внезапного запрокидывания головы с переразгибанием всего тела. Все врожденные автоматизмы ярко выражены, сухожильные рефлексы повышены. Гиперкинезы появляются к 1—1,5 годам и с возрастом становятся более выраженными. Чаще других встречаются атетоз, хореические гиперкинезы. Торсионные гиперкинезы встречаются реже. Все гиперкинезы минимальны в покое, исчезают во сне, усиливаются при произвольных движениях, провоцируются эмоциями. У больного С.С. клиническая картина заболевания была сходна с основными проявлениями гиперкинетической формы ДЦП, однако, помимо эктрапирамидных расстройств, у ребенка выявлялась прогрессирующая макроцефалия, прогрессирующая утрата двигательных навыков и нарастание экстрапирамидных расстройств, несмотря на проводимую реабилитационную терапию. Состояние ребенка ухудшалось после перенесенных интеркуррентных заболеваний.
У 3 наблюдаемых нами пациентов отмечались легкие когнитивные нарушения, что совпадает с данными литературы. У больного О.А. — грубая задержка психоречевого развития, что можно объяснить не только тяжелым течением основного заболевания (в анамнезе несколько «энцефалопатических» кризов), поздним началом метаболической терапии, но и отягощенным акушерским и перинатальным анамнезом.
Выявляемые изменения при МРТ/КТ головного мозга при ГА1 в виде атрофической вентрикуломегалии и атрофии лобно-теменных отделов головного мозга являются высокоспецифичными, что и было обнаружено у всех обследуемых пациентов. Хотелось бы отметить, что подобная МР-картина может наблюдаться при гипоксически-ишемических, инфекционных повреждениях головного мозга или при наследственной патологии. Например, спустя несколько месяцев после перенесенной микоплазменной пневмонии у детей появляются экстрапирамидные расстройства в виде синдрома паркинсонизма, и при МРТ/КТ головного мозга обнаруживают билатеральные повреждения базальных ганглиев. Хореические
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2007 |
9 |

ОБЩИЕ ВОПРОСЫ НЕВРОЛОГИИ И ПСИХИАТРИИ
Таблица 2. Диагностические критерии, позволяющие предположить ГА1 [29]
Вероятность |
Низкая |
Умеренная |
Высокая |
|
диагноза |
||||
|
|
|
||
|
|
|
|
|
Kлинические |
Макроцефалия; выступающие |
Рейеподобный эпизод (острая |
Острая энцефалопатия; |
|
критерии |
лобные бугры; нарушения |
метаболическая энцефалопатия); |
прогрессирующие |
|
|
вскармливания; задержка |
энцефалитоподобные эпизоды; |
экстрапирамидные |
|
|
двигательного развития |
гиперкинетическая форма |
нарушения; орофациальные |
|
|
|
детского церебрального паралича; |
дискинезии |
|
|
|
периодическая атаксия |
|
Семейный анамнез |
— |
Синдром внезапной смерти или |
|
|
смертельный исход от |
|
|
некупируемых судорог в семье |
Наличие в семье больного с подтвержденным диагнозом ГА1, наличие смертельного исхода в семье от острого симметричного некроза базальных ганглиев
Нейрорадиологи- |
Атрофия/гипоплазия лобных |
Субдуральные скопления; |
Лобно-височная атрофия |
ческие критерии |
отделов; субэпендимальные |
атрофия/гипоплазия височных |
головного мозга; |
|
псевдокисты; задержка |
отделов; лейкоэнцефалопатия |
изолированные |
|
миелинизации; |
(подростки, взрослые); |
симметричные некрозы |
|
вентрикуломегалия; расширение |
изолированное поражение |
базальных ганглиев |
|
субарахноидальных пространств |
бледного шара |
(бледного шара и хвостатых |
|
|
|
ÿäåð) |
|
|
|
|
гиперкинезы в сочетании с поражениями подкорковых структур могут наблюдаться при ревматической хорее, после перенесенной острой гипоксии, отравлении угарным газом или тяжелой ЧМТ.
При назначении специфической терапии у всех больных наблюдалось улучшение как по неврологи- ческому, так и по биохимическому статусу.
Таким образом, диагноз ГА1 может быть заподозрен у пациентов, имеющих в клинической картине макроцефалию, экстрапирамидные нарушения с дебютом в раннем детском возрасте и характерные нейрорадиологические изменения (табл. 2).
В качестве иллюстрации приводим выписки из истории болезни семейного случая, которые иллюстрируют выраженный внутрисемейный клинический полиморфизм ГА1 [20, 23].
Больной С.С., 7 лет, жалобы при поступлении на навяз- чивые движения, задержку двигательного и речевого развития.
Из анамнеза известно, что ребенок от 1-й беременности, протекавшей на фоне угрозы прерывания во 2-й половине, анемии; роды в срок, самостоятельные; вес при рождении 4250 г, рост 58 см, окружность головы 40,5 см (макрокрания), с оценкой по шкале Апгар 8/9 баллов. Выписан из роддома на 5-е сутки жизни в удовлетворительном состоянии. С рождения наблюдалась задержка психомоторного развития: голову держит с 1 года, сидит с 1 года 2 мес, с 1 года 7 мес встает с поддержкой и ходит за обе руки. С 1,5 до 2 мес жизни наблюдались периодические рвоты, которые изначально расценивали как проявления пилороспазма. Ежемесячное увеличение размеров головы на 1-м году жизни составляло в среднем 2,0—2,5 см, поэтому ребенок наблюдался у невролога с гидроцефальным синдромом на фоне перинатального поражения нервной системы.
Несмотря на проводимые курсы дегидратационной и нейротрофической терапии, его состояние прогрессивно ухудшалось. В 10 мес ребенок перенес кишечную инфекцию, сопровождавшуюся субфебрильной температурой, слабостью и повторной рвотой. После выздоровления родители отметили утрату двигательных и речевых навыков, а через некоторое время появились хореиформные и дистонические гиперкинезы. В дальнейшем был установлен диагноз: гипер-
кинетическая форма ДЦП. В 1 год 9 мес после перенесенной острой респираторной вирусной инфекции у ребенка произошло значительное ухудшение: развился правосторонний гемипарез и наросли гиперкинетические нарушения. При обследовании в лаборатории наследственных болезней обмена веществ Медико-генетического центра РАМН в 7 лет было выявлено повышение уровня глутаровой кислоты в моче до 635 мМ/Мкреатинина (патология от 500 мМ/Мкреатинина).
При осмотре в 7 лет общемозговых и менингеальных симптомов нет. Макрокрания (окружность головы 54,5 см при норме 49—54 см). Функции черепных нервов (ЧН): глазные щели симметричные, движения глазных яблок в полном объеме, зрачки симметричные, фотореакция сохранена, лицо симметричное, глоточные и небные рефлексы высокие, поперхивается при приеме пищи, саливация усилена, дизартрия псевдобульбарная и подкорковая, язык в полости рта по средней линии, гиперкинезы мышц языка, напряжение и утолщение грудиноключично-сосцевидной мышцы (больше справа). Голову держит, переворачивается, самостоятельно садится, не ходит. Мышечный тонус изменен по экстрапирамидно-пирамидному типу, хуже справа. Умеренная гипотрофия конечностей. Спастический тетрапарез, больше справа. Сухожильные рефлексы оживлены, больше справа. Патологический симптом Бабинского с двух сторон. Ретракции голеностопных суставов, больше справа. Грубые орофациальные, баллистические, торсионные гиперкинезы конечностей, мышц шеи и туловища, усиливающиеся при эмоциональном напряжении и произвольных движениях и проходящие во сне. Мелкоамплитудный тремор в руках. Дистонические установки конечностей, туловища. В сознании, на осмотр реагирует адекватно, контактен, говорит фразами, смотрит мультфильмы, играет в компьютерные игры, занимается конструктором, рисует красками, односложно отвечает на вопросы, знает буквы алфавита, пытается читать по слогам.
При динамическом осмотре через 1,5 мес на фоне специфической терапии (низкобелковая диета с ограничением в суточном рационе лизина и триптофана, L-карнитин, баклофен, рибофлавин, фенибут) отмечается положительная динамика в виде уменьшения гиперкинетических расстройств, снижения мышечного тонуса, повысился эмоциональный фон, произошло улучшение тонкой моторики, ребенок стал лучше рисовать (рис. 3). При контрольной МРТ
10 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2007 |

à
á
Рис. 3. Рисунки пациента С.С., 7 лет.
а — до начала терапии, б — через 2 мес после начала терапии.
головного мозга улучшилось состояние белого вещества больших полушарий, стало менее выражено расширение конвекситальных субарахноидальных пространств.
Больной С.Д., 1 год (младший сибс), жалобы при поступлении на ограничение движений в левых конечностях, повышенную сонливость, утрату приобретенных ранее двигательных навыков.
Из анамнеза известно, что ребенок от 3-й беременности, протекавшей на фоне хронической герпетической и цитомегаловирусной инфекции, угрозы прерывания и токсикоза, 2-х срочных родов путем экстренного родоразрешения, вес при рождении 4900 г, рост 53 см, окружность головы 40,0 см (макрокрания), с оценкой по шкале Апгар 8/9 баллов. Выписан из роддома на 10-е сутки жизни в удовлетворительном состоянии. До появления первых симптомов заболевания развитие ребенка соответствовало возрасту: голову держит с 1 мес, переворачивается с 4 мес, самостоятельно сидит и встает с 7 мес, ходит за одну ручку с 8 мес. В 1,5 мес отмечалось расширение желудочковой системы, ребенок наблюдался у невролога с субкомпенсирован-
ГЛУТАРОВАЯ АЦИДУРИЯ
ной формой гидроцефального синдрома. Сходные случаи манифестации заболевания описаны в литературе [18]. Дебют заболевания в 9 мес, после перенесенной легкой ЧМТ развилась общемозговая симптоматика в виде сонливости. Через 1 ч появились многократная рвота, тонические судороги и левосторонний гемипарез. Ребенок был госпитализирован в отделение реанимации: уровень сознания — сопор, фокальные клонические приступы, при КТ головного мозга были обнаружены субдуральные гематомы и выраженное расширение желудочковой системы головного мозга, что явилось основанием для установления длительного наружного дренажа. Состояние стабилизировалось через 1,5 нед. В последующем при сборе анамнеза было выявлено, что у старшего брата отмечалось повышение уровня глутаровой кислоты в моче. Биологический материал (кровь и моча) ребенка был отправлен в лабораторию наследственных болезней обмена ГУ МГНЦ РАМН, где был установлен диагноз ГА1.
При осмотре в 1 год: общемозговых и менингеальных симптомов нет. Макрокрания (окружность головы 51,5 см при норме 44,0—49,5 см). ЧН: глазные щели симметричные, зрачки симметричные, фотореакция сохранена, движения глазных яблок в полном объеме, легкая сглаженность левой носогубной складки справа, глоточный и небные рефлексы живые, язык в полости рта по средней линии. Голову держит, переворачивается, ползает, садится самостоятельно, но сидит неустойчиво, ходит с поддержкой за обе руки. Мышечная дистония. Левосторонний гемипарез до 4,5 баллов. Хореиформные гиперкинезы в конечностях и негрубые орофациальные гиперкинезы. Корковая атаксия. Захват игрушки без интенции. В сознании, на осмотр реагирует адекватно, игрушками интересуется, активный лепет, дифференцирует окружающих, говорит слогами, гипердинамиче- ский синдром.
При осмотре в динамике через 1,5 мес после назначения метаболической терапии (низкобелковая диета с ограниче- нием в суточном рационе лизина, L-карнитин, баклофен, рибофлавин, фенибут, топамакс) отмечена некоторая положительная динамика в виде регресса экстрапирамидных расстройств, улучшения общего состояния, уменьшения степени гемипареза до 4,8 баллов, стал больше интересоваться окружающим и сидеть более устойчиво. Повторная МРТ головного мозга без существенной динамики.
Заключение
Впервые в России установлен диагноз глутаровой ацидурии тип 1, подтвержденный биохимическими и молекулярно-генетическими методами, и проведена метаболическая коррекция данной наследственной патологии. Число наследственных болезней обмена веществ, для которых разработаны эффективные методы терапии, растет с каждым годом, однако эффективность лечения во многом зависит от сроков установления правильного диагноза и назначения патогенетической терапии. Таким образом, приведенные наблюдения, как нам представляется, способствуют своевременной диагностике и рациональной терапии ГА1.
ЛИТЕРАТУРА
1.Краснопольская К.Д. Наследственные болезни обмена веществ. 3. Феничел Дж.М. Педиатрическая неврология. Основы клиниче-
Справочное пособие для врачей. М 2005. |
ской диагностики: перевод с англ. М: Медицина 2004. |
2.Темин П.А., Казанцева Л.З. Наследственные нарушения нерв- 4. Aicardi J. Diseases of the Nervous System in Childhood. 2-nd Edi-
но-психического развития у детей. М: Медицина 2001; 159— |
tion. 1998. |
174. |
|
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2007 |
11 |

ОБЩИЕ ВОПРОСЫ НЕВРОЛОГИИ И ПСИХИАТРИИ
5.Bahr O., Mader I., Zschocke J. et al. Adult onset glutaric aciduria type I presenting with a leukoencephalopathy. Neurology 2002; 59: 1802— 1804.
6.Baric I., Wagner P., Feyh P. et al. Sensitivity and specificity of free and total glutaric acid and 3-hydroxyglutaric acid measurements by stable-isotope dilution assays for the diagnosis of glutaric aciduria type I. J Inher Metab Dis 1999; 22: 867—882.
7.Biery B.J., Stein D.E., Morton D.H., Goodman S.I. Gene Structure and Mutations of Glutaryl-Coenzyme A Dehydrogenase: Impaired Association of Enzyme Subunits That is Due to an A421V Substitution Causes Glutaric Acidemia Type I in the Amish. Am J Hum Genet 1996; 59: 1006—1011.
8.Bjugstad K.B., Goodman S.I., Freed C.R. Age at symptom onset predicts severity of motor impairment and clinical outcome of glutaric acidemia type I. J Pediat 2000; 137: 681—686.
9.Brandt N.J. Symptoms and Signs in Organic Acidurias. J Inher Metab Dis 1984; 7: 23—27.
10.Brismar J., Ozand P.T. CT and MR of the brain in glutaric acidemia type I: a review of 59 published cases and a report of 5 new patients. Am J Neuroradiol 1995; 16: 675—683.
11.Busquets C., Merinero B., Christensen E. et al. Glutaryl-CoA Dehydrogenase Deficiency in Spain: Evidence of Two Groups of Patients, Genetically, and Biochemically Distinct. Pediat Res 2000;
48:315—322.
12.Caffey J. The whiplash shaken infant syndrome: manual shaking by the extremities with whiplash-induced intracranial and intraocular bleedings, linked with residual permanent brain damage and mental retardation. Pediatrics 1974; 54: 396—403.
13.Chalmers R.A., Bain M.D., Zschocke J. Riboflavin-responsive glutaryl CoA dehydrogenase deficiency. Mol Genet Metab 2006; 88: 29—37.
14.Chow C.W., Haan E.A., Goodman S.I. et al. Neuropathology in glutaric acidaemia type 1. Acta Neuropathol 1988; 76: 590—594.
15.Corral I., Martinez C.J.C., Martinez-Pardo M., Gimeno A. Glutaric aciduria type 1: diagnosis in adulthood and phenotypic variability. Neurologia 2001; 16: 377—380.
16.Desai N.K., Runge V.M., Crisp D.E. Magnetic resonance imaging of the brain in glutaric acidemia type I: a review of the literature and a report of four new cases with attention to the basal ganglia and imaging technique. Invest Radiol 2003; 38: 489—496.
17.Dunger D.B., Snodgrass G.J.A.I. Glutaric Aciduria Type I Presenting with Hypoglycaemia. J Inher Metab Di. 1984; 7: 122—124.
18.Forstner R., Hoffman G.F., Gassner I. et al. Glutaric aciduria type 1: ultrasonographic demonstrations of early signs. Pediat Radiol 1999;
29:138—143.
19.Funk C.B., Prasad A.N., Frosk P. et al. Neuropathological, biochemical and molecular findings in a glutaric acidemia type 1 cohort. Brain 2005; 128: 711—722.
20.Gilles L., Raymond D. Adams., Edwin H. Kolodny. Neurology of Herediatary Metabolic Diseases of Children, 2nd ed. New York 1996.
21.Goodman S.I., Stein D.E., Schlesinger S. et al. Glutaryl-CoA Dehydrogenase Mutations in Glutaric Acidemia (Type I): Review and Report of Thirty Novel Mutations. Hum Mutat 1998; 12: 141—144.
22.Goodman S.I. Prenatal diagnosis of glutaric acidemias. Prenat Diagn 2001; 21: 1167—1168.
23.Goodman S.I., Frerman F.E. Organic Acidemia Due to Defects in Lysine Oxidation: 2-ketoadipic Acidemiaand Glutaric Acidemia.The Metabolic and Molecular Basis of Inherited Disease. Eighth ed., McGraw-Hill. New York 2001; 2195—2204.
24.Greenberg C.R., Prasad A.N., Dilling L.A. et al. Outcome of the First 3-Years of a DNA-Based Neonatal Screening Program for Glutaric Acidemia Type 1 in Manitoba and Northwestern Ontario, Canada. Mol Genet Metab 2002; 75: 70—78.
25.Hartley L.M., Khwaja O.S., Verity C.M. Glutaric Aciduria Type 1 and Nonaccidental Head Injury. Pediatria 2001; 107: 174—176.
30.Hoffmann G.F., Zschocke J. Glutaric aciduria type I: From clinical, biochemical and molecular diversity to successful therapy. J Inher Metab Dis 1999; 22: 381—391.
31.Iafolla A.K., Kahler S.G. Megalencephaly in the neonatal period as the initial manifestation of glutaric aciduria type I. J Pediat 1989;
114:1004—1006.
32.Kafil-Hussain N.A., Monavari A., Bowell R. et al. Ocular Findings in Glutaric Aciduria Type 1. J Pediat Opth & Strabismus 2000; 3: 289— 293.
33.Kimura S., Hara M., Nezu A. et al. Two cases of glutaric aciduria type I: clinical and neuropathological findings. J Neurol Sci 1994; 123: 38—43.
34.Knapp J.F., Soden S.E., Dasouki M.J., Walsh I.R. A 9-month-old baby with subdural hematomas, retinal hemorrhages, and developmental delay. Pediat Emerg Care 2002; 18: 44—47.
35.Kolker S., Ahlemeyer A., Krieglstein J., Hoffmann G.F. 3-Hydroxyglu- taric and glutaric acids are neurotoxic through NMDA receptors in vitro. J Inher Metab Dis 1999; 22: 259—262.
36.Kolker S., Ahlemeyer A., Krieglstein J., Hoffmann G.F. MaturationDependent Neurotoxicity of 3-Hydroxyglutaric and Glutaric Acids In Vitro: A New Pathophysiologic Approach to Glutaryl-CoA Dehydrogenase Deficiency. Pediat Res 2000; 47: 495—503.
37.Kolker S., Greenberg C.R., Lindner M. et al. Emergency treatment in glutaryl-CoA dehydrogenase deficiency. J Inher Metab Dis 2004;
27:893—902.
38.Kolker S., Koeller D.M., Okun J.G., Hoffmann G.F. Pathomechanisms of neurodegeneration in glutaryl-CoA dehydrogenase deficiency. Ann Neurol 2004; 55: 7—12.
39.Kolker S., Koeller D.M., Sauer S. et al. Excitotoxicity and bioenergetics in glutaryl-CoA dehydrogenase deficiency. J Inher Metab Dis 2004; 27: 805—812.
40.Kyllerman M., Skjeldal O.H., Lundberg M. et al. Dystonia and dyskinesia in glutaric aciduria type I: clinical heterogeneity and therapeutic considerations. Mov Disord 1994; 9: 22—30.
41.Leibel R.L., Shih V., Goodman S.I. et al. Glutaric acidemia: A metabolic disorder causing progressive choreoathetosis. Neurology 1980;
30:1163.
42.Lin S.K., Hsu S.G., Ho S.C. et al. Novel mutation and prenatal sonographic findings of glutaric aciduria (type I) in two Taiwanese families. Prenat Diagn 2002; 22: 725—729.
43.Lindner M., Kolker S., Schulze A. Neonatal screening for glutarylCoA dehydrogenase deficiency. J Inher Metab Dis 2004; 27: 851— 859.
44.Lindner M., Ho S., Fang-Hoffmann J. et al. Neonatal screening for glutaric aciduria type I: Strategies to proceed. J Inher Metab Dis 2006; 29: 378—382.
45.Menkes J.H. Textbook of Child Neurology, 3 nd Ed. Philadelphia Lea & Febiger 1985; 91—98.
46.Merinero B., Perez-Cerda C., Font L.M. et al. Variable clinical and Biochemical Presentation of Seven Spanish Cases with GlutarylCoA Dehydrogenase Deficiency. Neuropediat 1995; 26: 238—242.
47.Monavari A.A., Naughten E.R. Prevention of cerebral palsy in glutaric aciduria type 1 by dietary management. Arch Dis Child 2000; 82: 67—70.
48.Morton D.H., Bennett M.J., Seargeant L.E. et al. Glutaric aciduria type I: a common cause of episodic encephalopathy and spastic paralysis in the Amish of Lancaster County, Pennsylvania. Am J Med Genet 1991; 41: 89—95.
49.Muhlhausen C., Ergun S., Strauss K.A. Vascular dysfunction as an additional pathomechanism in glutaric aciduria type I. J Inher Metab Dis 2004; 27: 829—834.
50.Muhlhausen C., Ott N., Chalajour F. Endothelial effects of 3-hydrox- yglutaric acid: implications for glutaric aciduria type I. Pediat Res 2006; 59: 196—202.
26.Heidenriech R., Natowicz M., Hainline B.E. et al. Acute extrapyrami51. Strauss K.A., Puffenberger E.G., Robinson D.L. et al. Type I glutaric
dal syndrome in methylmalonic acidemia: “Metabolic stroke” involving the globus pallidus. J Pediat 1988; 113: 1022.
aciduria, part 1: natural history of 77 patients. Am J Med Genet C Semin Med Genet 2003; 15: 38—52.
27.Hoffmann G.F., Gibson K.M., Nyhan W.L. et al. Neurological mani52. Twomey E.L., Naughten E.R., Donoghue V.B. Neuroimaging findings
festations of organic acid disorders. Eur J Pediat 1994; 153: 94— 100.
28.Hoffmann G.F., Bohles H.J., Burlina A. et al. Early signs and course of disease of glutaryl-CoA dehydrogenase deficiency. J Inher Metab Dis 1995; 18: 173—176.
29.Hoffmann G.F., Athanassopoulos S., Burlina A.B. et al. Clinical Course, Early Diagnosis, Treatment, and Prevention of Disease in Glutar- yl-CoA Dehydrogenase Deficiency. Neuropediat 1996; 27: 115—123.
in glutaric aciduria type 1. Pediat Radiol 2003; 33: 823—830.
53.Wajner M., Kolker S., Souza D.O. et al. Modulation of glutamatergic and GABAergic neurotransmission in glutaryl-CoA dehydrogenase deficiency. J Inher Metab Dis 2004; 27: 825—828.
54.Zschocke J., Quak E., Guldberg P., Hoffmann G.F. Mutation analysis in glutaric aciduria type I. J Med Genet 2000; 37: 177—181.
12 |
ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ, 10, 2007 |