

The Toll-like Receptor 4 and Signalling through Ubiquitylation
factor complex comprising RelA (p65) and NFB1 (p50) (Figure 15.3). The founding member of this family of transcription factors is v-rel (p58), a viral gene responsible for reticuloendotheliosis in chickens. Mammalian REL-containing proteins include RelB (p60), c-Rel (p66) (involved in lymphopoiesis), and NFB2 (p100/p52).
Dorsal and NF- B also share the characteristic of being sequestered in the cytoplasm by inhibitory proteins, cactus in Drosophila and I B in mammals. These inhibitors are destroyed by an ubiquitylation-based process that permits their translocation to the nucleus where they bind to the B sequence motif (see page 493).
Lipopolysaccharides: shield and signal
Bacteria with no outer membrane have cell walls composed of a thick layer of peptidoglycan, lipoteichoic acid and S-layer proteins. Bacteria with a second, outer membrane have a thin peptidoglycan layer between the two (Fig 15.4). The outermost membrane surface is almost entirely lipopolysaccharide (LPS). The thicker peptidoglycan layers may be detected by the Gram test (staining with crystal violet and iodine), initially devised to discriminate between pneumococci (gram positive) and Klebsiella pneumoniae (gram negative).26 Mycobacteria form a separate group. They have a cell wall made of glycolipids and mycolic acid and only a thin peptidoglycan layer. They are able to survive because, inhabiting the intracellular environment, they are subject to little osmotic stress.
Because the innate immune system lacks any means of instigating receptor diversity, all its targets are highly conserved structural components of micro-organisms. One such is lipopolysaccacharide.27 LPS, shed from the outer membrane of gram-negative bacteria (Table 15.3), are detected at picomolar concentrations and serve to alert the host (Figure 15.4). Of the Tolllike receptors, TLR4 has emerged as a specific conduit for the innate immune response to bacterial LPS.28,29 We focus on LPS-mediated signalling because it is one of the most potent immunostimulants.
LPS and endotoxin. At the start of the 20th century, long before its chemical composition was realized, Richard Pfeiffer identified LPS as endotoxin. It represents the major fever-evoking component of gram-negative bacteria and, when applied systemically, it causes shock.29 We now recognize the lipid A-KDO2 moiety of LPS as being responsible for most of the toxic effects of gram-negative organisms (see Figure 15.4). The immediate pyrogenic effects of endotoxin are due to signalling through TLR-4 that elevates the expression of PLA2 and cyclo-oxygenase2 (COX2) and the production of prostaglandin E2 (PGE2).31
The cross-linking of the peptidoglycan is targeted by penicillin, which causes rapid lysis of microbes.
Virulence of Yersinia pestis and acylation of LPS. On entering a host having a body temperature of 37°C, the plague bacterium
Yersinia pestis switches production of LPS to a form that lacks secondary acylations. This fails
to activate TLR4 and antagonizes the binding of highly acylated LPS. Forced expression of an E. coli gene responsible for secondary acylations renders Y. pestis completely avirulent. Resistance to disease requires TLR4 and the adaptor protein MyD88. Both innate and adaptive responses are required for immunity against the modified strain. By evading TLR4 activation in this way, the altered
LPS may contribute to the virulence of various gramnegative bacteria.30
457

Signal Transduction
The outer leaflet of the outer membrane of gram-negative bacteria is made up almost entirely of LPS. These have a basic structure composed of lipid A, consisting of 4–6 acyl chains covalently bound to two diaminodideoxy- d-glucose residues,
and a hydrophilic heteropolysaccharide component. The inner component of the polysaccharide, the inner core, is composed of heptose and keto- 3-deoxyoctonic acid (KDO). To this, a long chain of polysaccharides is attached, the outer core, of which, the O-antigen, is repeated many times. The long polysaccharide chain is not essential for bacterial survival, but the lipid-
A moiety is vital. The polysaccharides play a role in pathogenesis
and serve as an antigen for the generation of protective antibodies.
Table 15.3 Examples of Gram-negative bacteria
Bacteria |
Diseases caused |
|
|
Cyanobacteria |
photosynthesis (3.8 billion years old) |
|
|
Enterobacter cloacae |
urinary and pulmonary tract infections |
|
|
Escherichia coli |
commensal in intestine, urinary tract |
|
infection, septicemia |
|
|
Green sulfur and green non-sulfur |
|
bacteria |
|
|
|
Helicobacter pylori |
peptic ulcer |
|
|
Haemophilus influenzae (Pfeiffer |
lung infections and meningitis |
baccillus) |
|
|
|
Klebsiella pneumoniae |
pneumonia, hospital-acquired urinary tract |
|
and wound infections |
|
|
Legionells pneumophila |
legionellosis (Legionnaires’ disease) |
|
|
Leptospiractero haemorrhagiae |
Weil’s disease |
|
|
Moraxella catarrhalis |
bronchitis, sinusitis, laryngitis and otitis |
|
media |
|
|
Neisseria gonorrhoeae |
gonorrhoea |
|
|
Neisseria meningitidis |
meningitis |
|
|
Proteus mirabilis |
urinary tract infections |
|
|
Pseudomonas aeruginosa |
pulmonary tract, urinary tract, burns, |
|
wounds infections |
|
|
Salmonell enteridis |
gastroenteritis |
|
|
Salmonella typhi |
typhoid fever |
|
|
Serratia marcescens |
conjunctivitis, keratitis endophthalmitis |
|
|
Signalling through the TLR4 receptor
The TLR4 differs from other TLRs because it requires the collaboration of other receptors, most notably CD14 and MD-2, though how these interact with LPS and how they interact with each other remains uncertain. LPS, bound
to LPS-binding protein (LBP, a trace ‘acute phase’ plasma protein involved in the early stages of septic shock)32 interacts with CD14.33 This presents LPS to MD-2, linked to the membrane via TLR4 (Figure 15.5).34 Two major signalling pathways are activated. A signalling complex is formed with a
458

The Toll-like Receptor 4 and Signalling through Ubiquitylation
FIG 15.4 Composition of the cell wall of bacteria and molecular detail of LPS.
Bacteria are protected from the environment by a cell wall. In the case of gram-positive bacteria, this is composed of a lipid membrane surrounded by
a thick layer composed of peptidoglycan, lipoteichoic acid, and a coating of S-layer proteins. Gram-negative bacteria are surrounded by two membranes separated by the peptidoglycan. Mycobacteria have a single membrane, a relatively thin peptidoglycan, and a thick outer layer composed of mannosephosphatidylinositol, lipoarabinomannan, and mycolic acid. Adapted from Akira et al.6
FIG 15.5 Signalling from the TLR4 receptor.
LPS binds different receptors, first CD14, then MD-2, and finally TLR4. Ligand-mediated dimerization (or multimerization) of TLR4 initiates the assembly of receptor signalling complexes through the intermediate of adaptors that harbour the TIR protein-interaction domain. One complex is formed by association of TRAM with TRIF which, through a death domain (DD), recruits the adaptor/effector TRAF3 (ubiquitin ligase). This causes activation of the transcription factor IRF3 and results in the expression of inflammatory cytokines IFN- , IL-10, and RANTES. The other complex is formed by association of the adaptors TIRAP and MyD88 which, through a death domain, recruit the kinase IRAK4. This pathway reaches the transcription factors NF- B, ATF2, c-Jun, and IRF5 and plays an important role in the expression of IL-1 , IL-6, IL-12, and TNF- .
459

Signal Transduction
Discovery of TLR4, the LPS receptor. The earliest indication that LPS might interact with specific receptors came from the discovery of LPS-resistant mice.35 The search for the receptor then commenced
in earnest with the realization of two substrains both having defective responses to LPS. Four years of fine mapping of the Lps locus by several research groups culminated in the discovery of LTR4
as the sole candidate.29 Although LPS interacts directly with TLR4, the interaction also requires one or more coreceptors, among which CD14, a glycosylphosphoinositolanchored membrane protein expressed on leukocytes.18,33
LPS and mucosa. The form of LPS having secondary acylations (Figure 15.5) allows bacteria to persist in the host mucosa.36–38 They are tolerated in this external environment but as
soon as they traverse the epithelial barrier, as
occurs in trauma, they are effectively eliminated.
number of adaptors and assembly then proceeds through the association of proteins via the TIR domain. Many components of the pathway also operate downstream of the TNF, Toll, or TGF -receptors (which explains why so many of the acronyms begin with the letter T). One downstream pathway of TLR4 requires the adaptors TIRAP and MyD88 and culminates in the production of pro-inflammatory cytokines (IL-1 , IL-12, TNF- , IL-6). These signal to the transcription factor complexes NF- B, ATF2/c-Jun (AP-1), and IRF-5 (Fig15.5 and pages 464 and 525). The other pathway requires the adaptors TRAM and
TRIF and culminates in the production of IFN- , IL-10, and RANTES. It signals to the transcription factor IRF-3 and acts in cooperation with NK- B.
The TIRAP/MyD88 pathway
Oligomerization of the TLR4 receptor provokes the formation of an adaptor complex through the assembly of components TIRAP and MyD88, carrying TIR domains. How the oligomerization creates homophilic TIR-TIR binding sites remains to be elucidated. The death domain of MyD88 recruits the kinase IRAK4 which phosphorylates IRAK139,40 (see Figure 15.6). This draws TRAF6 to the receptor complex which now acts as a docking platform on to which another large signalling complex develops. The new docking sites are created, not by phosphorylation as is the case for growth factor receptors, but by polyubiquitylation of TRAF6, in the so-called K63-type reaction (i.e. they are interconnected through lysine-63 (see Figure 15.13 and page 468). TRAF6 is an E3-ubiquitin ligase that by association with other factors (Ubc13/ Mms2) constitutes the conjugation system that supplies activated ubiquitin
monomers. Importantly, TRAF6, decorated with its polyubiquitin chains, brings together two kinase complexes that act in series to ensure phosphorylation of I B (inhibitor of NF- B) and activation of the JNK and p38 pathways.
The significance of IRAK4 is clearly illustrated in patients who either fail to express it, or have a mutation in its kinase domain which renders the enzyme inactive. Because they have ineffective innate immunity, these people are particularly susceptible to infection with Streptococcus pneumoniae, suffering recurrent pyrogenic infections and bacteraemia.41
From TRAF6 to activation of NF- B
The first complex recruited by TRAF6 is composed of the serine/threonine kinase TAK1 with its associated adaptor TAB2 (or TAB3) and its regulatory subunit TAB1 (both TAK1/TAB1/TAB2 and TAK1/TAB1/TAB3 complexes have been detected).42,43 It is the TAB2 (or TAB3) that binds K63-ubiquitylated TRAF6 through a CUE domain44 (Figure 15.7 illustrates the domain architecture of these proteins). This activates TAK1 through autophosphorylation.45 The
460
The Toll-like Receptor 4 and Signalling through Ubiquitylation
FIG 15.6 TRAF6-mediated signalling complex formation.
Recruitment of IRAK4 brings IRAK1 and TRAF6 to the receptor signalling complex (1). IRAK1 is phosphorylated by IRAK4 (2) and this induces catalytic activity the complex comprising TRAF6 (E3-ubiquitin ligase), Ubc13 and Mms2 (3). Autoubiquitylation of TRAF6 follows (4). The K63-linked ubiquitin chain acts as a docking site for two kinase complexes. One of these comprises TAB2 coupled to the serine/threonine kinase TAK1 with its regulatory subunit TAB1
(5). The other comprises NEMO attached to I B-kinases- and - (IKKand - ). Recruitment of TAB2/TAB1/TAK1 results in autophosphorylation of the kinase (7) and subsequent phosphorylation and activation of IKK- (8).
FIG 15.7 Domain architecture of proteins involved the TRAF6-mediated activation of IKK .
The phosphorylation sites in the kinase domain of IKK and TAK1 are situated in the activation segment. The TAK1 regulatory subunit TAB1 has a phosphatase fold similar to PP2C but is inactive. TAB2 binds K63-linked ubiquitin through its CUE domain. TRAF6 possesses an N-terminal RING domain with which it binds to the E2-conjugation complex (Ubc13/Mms2).
second complex is composed of the serine/threonine kinases IKK and IKK , linked to the ubiquitin-binding protein that acts as the sensor for K63-linked polyubiquitylated TRAF6 (sometimes referred to as IKK , IKK signifying I B
461
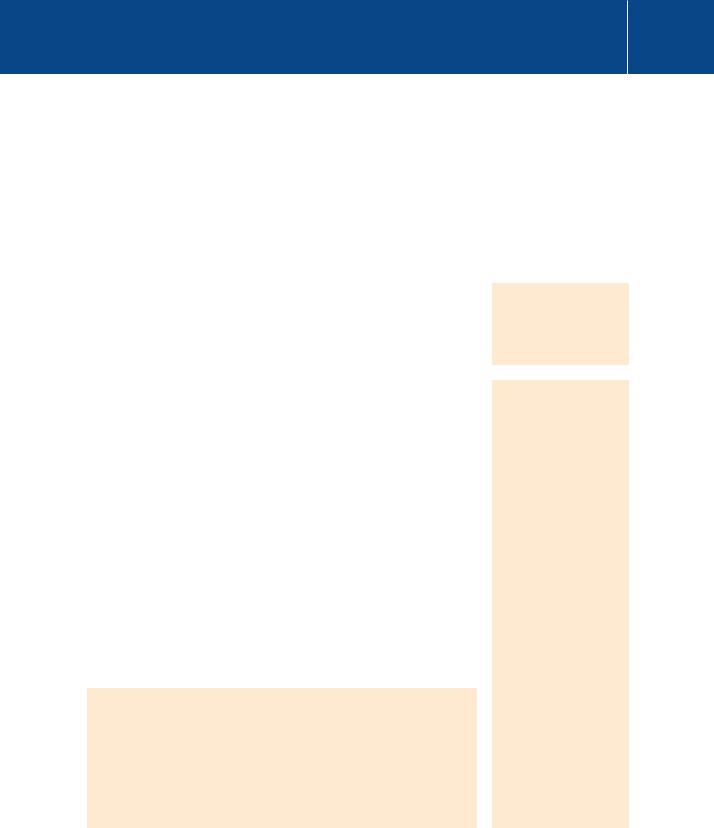
Signal Transduction
FIG 15.8 IKK -mediated activation of the RelA/NF- B complex. Phosphorylated/activated IKK phosphorylates I B on two serine residues in its N-terminal domain
(1, 2) allowing its recognition by the SCF Tcrp ubiquitin-ligase complex (3). Following (K48-type) ubiquitylation (4), I B is marked for destruction by the proteasome (5), liberating RelA (p65) and NFB1 (p50) and exposing their nuclear localization signal (NLS). They enter the nucleus (6), there to bind DNA at the B element, driving expression of inflammatory cytokines.
kinase).46, 47 Upon binding to TRAF6, TAK1 phosphorylates and activates IKK which then phosphorylates I B in the ‘destruction box’ (Figure 15.8). The phosphorylated I B is detected by the receptor component ( -TrCP) of an E3-ligase complex and subsequent polyubiquitylation (K48-type) allows detection by the proteasome followed by its degradation. Finally, IKK is also activated and this plays a role in the initiation of the ‘non-canonical’ pathway. Finally, NEMO too is ubiquitylated by TRAF6 and serves to recruit more
TAB2/3-TAB1-TAK1 into the complex and enhance the output of the signalling pathway (not shown in Figure 15.6).
On destruction of I B , the NF- B dimer, (comprising proteins of the Relfamily p65 and p50) is recognized by the - and -importins at the nuclear pore and enters the nucleus.48 This pathway of activation of NK- B, through destruction of I B, is referred to as the ‘canonical’ NF- B pathway. We have given here the example I B , but the other inhibitor proteins, I B and I B , can also bind NF- B thereby blocking the nuclear localization signal and thus transcriptional activity.25
Alternative activation pathways for NF- B
These pathways involve unprocessed (precursor) members of the Rel family, NFB1 and NFB2. To complicate matters further, these proteins qualify as members of both the Rel and I B families. They are composed of two regions, the one resembling Rel and the other I B (ankyrin repeats). They bind to other members of the Rel family and, having an intrinsic inhibitory region that masks the nuclear localization signal, they render the complexes inactive with respect to their transcriptional function25 (see Figure 15.3).
462
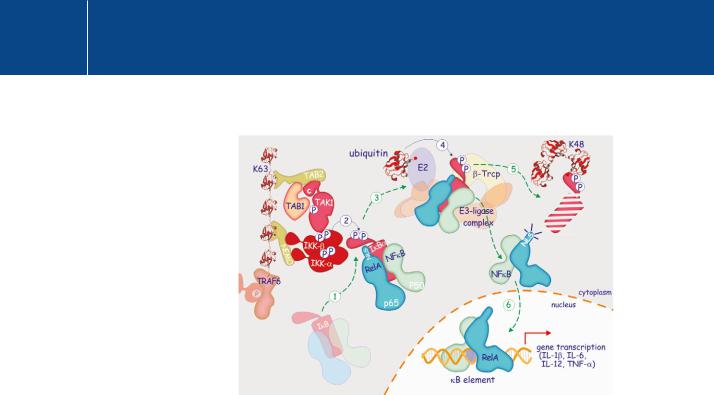
The Toll-like Receptor 4 and Signalling through Ubiquitylation
FIG 15.9 TAK1-mediated activation of p38 and JNK1.
(a) TAK1 phosphorylates MEK3 and MEK4 (MAP2K) (1). These in turn phosphorylate and activate p38 (2) and JNK1 respectively (4). P38 phosphorylates and activates MK2 (3) which enhances expression of inflammatory cytokines through stabilization of their mRNAs. It also promotes secretion. JNK1 enters the nucleus and phosphorylates the transcription factors ATF2 and c-Jun (5). These form heterodimers that bind the AP-1 response element (6) and drive expression of numerous inflammatory cytokines. (b) Domain architecture of ATF2 (human) and c-Jun. The N-terminal DNA binding regions of the proteins are phosphorylated by JNK1. They dimerize through a basic leucine zipper (bZIP) in the c-terminal segment.
The p105 pathway involves the unprocessed NFB1 (p105) which preferentially binds to a homodimer of processed NFB1. The complex is inactive and ubiquitylation of p105 followed by total destruction is required for liberation of the processed NF- B dimer.
In the non-canonical (or alternative) pathway, NFB2 is bound to RelB and is phosphorylated in its I B-like segment by IKK . Poly-ubiquitylation is then followed by only partial proteolysis through the proteasome. This processed (or mature) form of NFB2 translocates, in association with Rel-B, to the
nucleus. This pathway operates mainly in B cells in response to a subset of TNF receptors, including BAFF, LT , LT and CD40.49
From TRAF to activation of p38 and JNK1
In parallel with the activation of NF- B, two other pathways contribute to the transcriptional regulation of inflammatory mediators.50 TAK1 phosphorylates the activation segments of MEK3 (or 6) and MEK4 (or 7) which then phosphorylate and activate p38 and JNK1 (respectively) (Figure 15.9). These members of the Big MAP kinase family are involved in the responses to cellular stress and inflammation (see page 349 and Figure 22, page 351).
463