

Министерство образования и науки, молодежи и спорта Украины
Высшее учебное заведение
Донецкий национальный технический университет
Кафедра “Прикладной экологии и охраны окружающей среды”
Самостоятельна работа
по дисциплине “Материаловедение”
вариант 10
Выполнил студент гр. ТТМ - 10 Лубин Б.С.
Проверил: профессор Прилипко Ю.С.
Донецк-2012
РЕФЕРАТ
Страниц – 67,таблицы – 3,рисунка – 22,источников – 11
Цель работы:Изучить состав,структуру,свойства и технологию получения функциональных материалов.
В работе рассмотрены способы получения монокристаллов,ферритов,описаны пьезоэлектрики и область их применения.
МОНОКРИСТАЛЛЫ, ТВЕРДОФАЗНЫЕ РЕАКЦИИ, ФЕРРИТОВЫЕ ПОРОШКИ, КРИСТАЛЛОХИМИЯ, ПЬЕЗОЭЛЕКТРИКИ.
VORTAG
Seite - 67 Tabelle - 3 Vorlagen - 22 Quellenmaterial - 11
Arbeitziel: Elternen Gemisch, Struktur, Erscheinungsbild und Technologie Empfang funktional Verrauchsabweichung
In Tatigkeit betrachten Methode Empfang Einkristall, Ferrit, schildem Piezoelektrikum, ihr Charakteristik und Reichweite
EINKRISTALL, HART REAKTION, FERRIT PULVER, KRISTALLCHEMIE
СОДЕРЖАНИЕ
ВВЕДЕНИЕ…………………………………………………………………………...6
1.СПОСОБЫ ПОЛУЧЕНИЯ МОНОКРИСТАЛЛОВ………………………….....7
1.1Метод Стогбаргера……………………………………………………………...7
1.2.Метод Чохральского…………………………………………………………....8
1.3.Метод Вернейля………………………………………………………………...9
1.4.Метод зонной плавки…………………………………………………………..10
1.5.Метод выращивания из раствора………………………………………….......11
1.6.Гидротермальный синтез………………………………………………………12
1.7.Метод кристаллизации из газовой фазы……………………………………...13
2.ТВЕРДОФАЗНЫЕ РЕАКЦИИ…………………………………………………….15
2.1.Физико-химические факторы,определяющие механизм твердофазных
реакций……………………………………………………………………………..16
2.2.Методы исследования механизма твердофазных реакций………………….18
2.3.Теория твердофазного взаимодействия………………………………………20
2.3.1.Теория Вагнера…………………………………………………………...20
2.3.2.Механизм реакции двойного обмена по Иосту и Вагнеру……………22
3.ПОЛУЧЕНИЕ ФЕРРИТОВЫХ ПОРОШКОВ……………………………………28
3.1.Технология ферритов по методу растворной химии………………………..28
3.1.1.Метод осаждения солей или гидроксидов……………………………...30
3.1.2.Метод термического разложения солей………………………………...31
3.2.Керамическая технология получения ферритов……………………………32
3.2.1.Подготовка сырьевых материалов………………………………………32
3.2.2.Приготовление шихты…………………………………………………...32
3.2.3.Ферритизация шихты…………………………………………………….34
3.2.4.Помол ферритизованной шихты………………………………………...35
3.2.5.Методы синтеза ферритовых порошков………………………………..37
3.2.5.1.Метод изотермического и изоконцентрационного снятия………..39
3.2.5.2.Метод изотермического и изоконцентрационного испарения……39
3.2.5.3.Методы соосаждения……………………………………………….40
3.2.5.4.Метод замены растворителя………………………………………..40
3.2.5.5.Распылительная сушка……………………………………………...41
3.2.5.6.Криохимический метод……………………………………………..42
3.2.5.7.Электрохимический метод………………………………………….43
3.2.5.8.Оксалатный метод…………………………………………………...44
4.КРИСТАЛЛОХИМИЯ ФЕРРИТОВ……………………………………………...47
4.1.Типы структур………………………………………………………………….47
4.2.Катионное распределение……………………………………………………..48
4.3.Энергия стабилизации…………………………………………………………54
5.ПЬЕЗОЭЛЕКТРИКИ.ХАРАКТЕРИСТИКА.…………………………………….58
6.ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ ТЕОРЕТИЧЕСКИХ ЗНАНИЙ…………..64
ВЫВОДЫ……………………………………………………………………………..66
СПИСОК ЛИТЕРАТУРЫ…………..………………………………………………..67
ВВЕДЕНИЕ
Материаловедение изучает состав, структуру, свойства и поведение материалов в зависимости от воздействия окружающей среды. Повышение требований качества изделий из различных материалов, в частности ферритовых изделий, привело к нахождению способов управления их параметрами. Повышение качества является результатом тщательного изучения физико-химической природы вещества.
Среди магнитных, полупроводниковых и технически ценных материалов важное место занимают ферриты. Особенно магнитомягкие марганец-цинковые ферриты, которые являются основой для разработки изделий специального назначения, для деталей телевизионной, СВЧ-техники и др. Результаты исследований составляют физико-химические основы управляемого обжига и позволяют получать ферритовые изделия с наперед заданными свойствами.
Структура и свойства сегнетоэлектриков в значительной степени зависят от физико-химических свойств исходных компонентов: их дисперсности, формы частиц, наличия примесей, фазового состава, дефектности кристаллической решетки и др. Поэтому для разработки научно обоснованных принципов управления свойствами порошков и получаемых из них материалов требовались физико-химические исследования процессов производства исходных компонентов и их технологическая реализация. Возможности оптимизации керамической технологии связаны также с увеличением однородности, дисперсности шихты, с ростом активности готового материала за счет интенсификации предшествующих стадий технологического процесса и создания необходимой поверхности для повышения скорости гетерофазных процессов при последующем спекании. Рассмотрение этих вопросов является важной и актуальной задачей технологии сегнетоэлектрических материалов.
1.СПОСОБЫ ПОЛУЧЕНИЯ МОНОКРИСТАЛЛОВ
Монокристалл - отдельный однородный кристалл, имеющий непрерывную кристаллическую решётку и характеризующийся анизотропией свойств. Внешняя форма монокристалла обусловлена его атомнокристаллической структурой и условиями кристаллизации. Часто монокристалл приобретает хорошо выраженную естественную огранку, в неравновесных условиях кристаллизации огранка проявляется слабо. Примерами огранённых природных монокристаллов могут служить монокристаллы кварца, каменной соли, исландского шпата, алмаза, топаза. От монокристаллов отличают поликристаллы и поликристаллические агрегаты, состоящие из множества различно ориентированных мелких монокристаллов.
Монокристаллы ценны как материал, обладающий особыми физическими свойствами. Например, алмаз и боразон предельно тверды, флюорит прозрачен для широкого диапазона длин волн, кварц — пьезоэлектрик. Монокристаллы способны менять свои свойства под влиянием внешних воздействий (света, механических напряжений, электрического и магнитного полей, радиации, температуры, давления). Поэтому изделия и элементы, изготовленные из монокристаллов, применяются в качестве различных преобразователей в радиоэлектронике, квантовой электронике, акустике, вычислительной технике и др. Первоначально в технике использовались природные монокристаллы, однако их запасы ограничены, а качество не всегда достаточно высоко. В то же время многие ценные свойства были найдены только у синтетических кристаллов. Поэтому появилась необходимость искусственного выращивания монокристаллов Исходное вещество для выращивания монокристаллов может быть в твёрдом (в частности, в порошкообразном), жидком (расплавы и растворы) и газообразном состояниях[1].
1.1 Метод Стокбаргера
В методе Стокбаргера: тигель с расплавом 1 перемещают вдоль печи 3 в вертикальном направлении со скоростью 1—20 мм/ч (рис. 1.1). Температура в плоскости диафрагмы 6 поддерживается равной температуре кристаллизации вещества. Так как тигель имеет коническое дно, то при его медленном опускании расплав в конусе оказывается при температуре ниже температуры кристаллизации, и в нём происходит образование (зарождение) мельчайших кристалликов, из которых в дальнейшем благодаря геометрическому отбору выживает лишь один. Отбор связан главным образом с анизотропией скоростей роста граней монокристаллов. Этот метод широко используется в промышленном производстве крупных монокристаллов флюорита, фтористого лития, сернистого кадмия и др [2]

Рисунок 1.1. Схема аппарата для выращивания монокристаллов по методу Стокбаргера
1 – тигель с расплавом; 2 – кристалл; 3 – печь; 4 – холодильник; 5 – термопара; 6 – диафрагма
1.2 Метод Чохральского
В методе Чохральского монокристалл медленно вытягивается из расплава (рис.1.2). Скорость вытягивания 1—20 мм/ч.
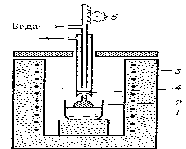
Рисунок 1.2 – Схема аппарата для выращивания монокристаллов по методу Чохральского
1 – тигель с расплавом; 2 – кристалл; 3 – печь; 4 – холодильник;5 – механизм вытягивания
Метод позволяет получать монокристаллы заданной кристаллографической ориентации. Метод Чохральского применяется при выращивании монокристаллов иттриево-алюминиевого граната, ниобата лития и полупроводниковых монокристаллов. А.В.Степанов создал на основе этого метода способ для выращивания монокристаллов с сечением заданной формы, который используется для производства полупроводниковых монокристаллов.[3]
1.3 Метод Вернейля
Метод Вернейля - бестигельный. Вещество в виде порошка (размер частиц 2—100 мкм) из бункера 1 (рис. 1.3) через кислородно-водородное пламя подаётся на верхний оплавленный торец затравочного монокристалла 2, медленно опускающегося с помощью механизма 5. Метод Вернейля — основной промышленный метод производства тугоплавких монокристаллов: рубина, шпинелей, рутила и др. [4].
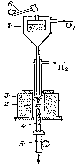
Рисунок 1.3 – Схема аппарата для выращивания монокристаллов по методу Вернейля
1 – бункер; 2 – кристалл; 3 – печь; 4 – свеча; 5 – механизм опускания;
6 – механизм встряхивания.
1.4 Метод зонной плавки
В методе зонной плавки создаётся весьма ограниченная по ширине область расплава. Затем благодаря последовательному проплавлению всего слитка получают монокристалл. Метод зонного проплавления получил широкое распространение в производстве полупроводниковых монокристаллов, а также тугоплавких металлических монокристаллов молибден, вольфрам и др.[5]
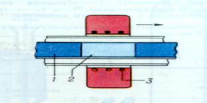
Рисунок 1.4.Схема устройства для зонной плавки:
1 - твердая фаза,2 - расплав,3 – нагреватель (стрелкой показано направление
движения нагревателя).
1.5 Метод выращивания из раствора
Методы выращивания из раствора включают 3 способа:
низкотемпературный (растворители: вода, спирты, кислоты и др.);
высокотемпературный (растворители: расплавленные соли и др.);
гидротермальный.
Низкотемпературный кристаллизатор представляет собой сосуд с раствором 1, в котором создаётся пересыщение, необходимое для роста кристаллов 2 путём медленного снижения температуры, реже испарением растворителя (рис. 1.5). Этот методиспользуется для получения крупных монокристаллов сегнетовой соли, дигидрофосфата калия (КDР), нафталина и др[10].

Рисунок 1.5 – Схема низкотемпературного кристаллизатора
1 – раствор; 2 – кристалл; 3 – печь; 4 – термостат; 5 – мешалка; 6 – контактный термометр; 7 – терморегулятор.
Высокотемпературный кристаллизатор (рис. 1.6) содержит тигель с растворителем и кристаллизуемым соединением, помещённый в печь. Кристаллизуемое соединение выпадает из растворителя при медленном снижении температуры (раствор - расплавная кристаллизация). Метод применяется для получения монокристаллов железоиттриевых гранатов, слюды, а также различных полупроводниковых плёнок [10].
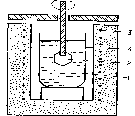
Рисунок 1.6 – Схема высокотемпературного кристаллизатора
1 – раствор; 2 – кристалл; 3 – печь; 4 – тигель.
1.6 Гидротермальный синтез
Гидротермальный синтез монокристаллов основан на зависимости растворимости вещества в водных растворах кислот и щелочей от давления и температуры. Необходимые для образования монокристаллов концентрация вещества в растворе и пересыщение создаются за счёт высокого давления (до 300Мн/м2 или 3000 кгс/см2) и перепадом температуры между верхней (Т1~250оС) и нижней (Т2~500°С) частями автоклава (рис. 1.7). Перенос вещества осуществляется конвективным перемешиванием.Гидротермальный синтез является основным процессом производства монокристаллов
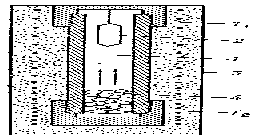
Рисунок 1.7 – Схема автоклава для гидротермального синтеза
1 – раствор; 2 – кристалл; 3 – печь; 4 – вещество для кристаллизации.
Методы выращивания монокристаллов из газообразного вещества:
испарение исходного вещества в вакууме с последующим осаждением пара на кристалл, причём осаждение поддерживается определенным перепадом температуры Т ;
испарение в газе (обычно инертном), перенос кристаллизуемого вещества осуществляется направленным потоком газа ;
осаждение продуктов химической реакций, исходящих на поверхности затравочного монокристалла[10].
1.7 Метод кристаллизации из газовой фазы
Широко используется для получения монокристальных пленок и микрокристаллов для интегральных схем и др. целей.
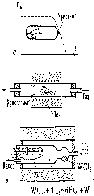
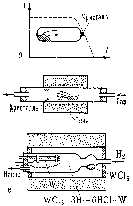
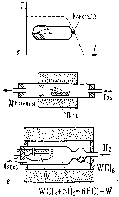
Рисунок 1.8 – Схема установки для кристаллизации из газовой фазы (Пунктиром показано распределение температуры вдоль печи.)
Выбор метода выращивания монокристаллов определяется требованием к качеству монокристаллов (количество и характер присущих монокристаллов дефектов). Различают макроскопические дефекты (инородные включения, блоки, напряжения) и микроскопические (дислокации, примеси, вакансии).Существуют специальные методы уменьшения числа дефектов в монокристаллах (отжиг, выращивание монокристалла на бездефектных затравочных кристаллах и др.).
При выращивании монокристаллов используются различные способы нагревания: омический, высокочастотный, газопламенный, реже плазменный, электроннолучевой, радиационный (в т. ч. лазерный) и электродуговой.
2.ТВЕРДОФАЗНЫЕ РЕАКЦИИ
Твердофазные реакции - это реакции с участием твёрдых реагентов или продуктов реакции.
Особенности: реакции протекают в гетерогенных системах; количество частиц участвующих в реакции очень велико; реакции характеризуются большим количеством промежуточных стадий; малые изменения энтальпии и энтропии реакции; реакции имеют ярко выраженный топохимический характер.
Типы твёрдофазных реакций:
1) Т1→Т2;
2) Т1+Т2→Т3 - синтез вещества с заданными свойствами;
3) Т1+Т2→Т3+Т4 - синтез различных гетероструктур[11].
Одна из характеристик описывающих твёрдофазные вещества – это степень превращения:
x=(m0-m)/m0 ,где(2.1)
m0 – количество исходного реагента,
m – количество реагента в момент времени.
Основные положения в теории твердофазных процессов:
- скорость реакции лимитируется диффузией растворов через слой готового продукта;
- слой готового продукта является достаточно компактным, находящиеся в нём дефекты не влияют на скорость реакции;
- реакции на границах фаз протекают быстрее, чем в объёме готового продукта.
На границе устанавливаются локальные термодинамические равновесия. Рассмотрим подробнее лимитирующую стадию процесса, т.е. перенос реагирующего материала через слой готового продукта[11].
Возможны 2 варианта переноса частиц через слой:
- перенос через нормальную кристаллическую решётку;
- перенос через дефекты в решётке образующего продукта.
Данные механизмы протекают параллельно. В общем случае скорость взаимодействия зависит от размера частиц, от размера решётки и от количества дефектов в ней.
Методы управления твёрдофазными реакциями основаны на изменении условий проведения реакции и на изменении исходных характеристик твёрдых реагентов [11].
2.1 Физико-химические факторы, определяющие механизм твердофазных реакций
Твердофазное взаимодействие, в отличие от реакций в жидкой или газовой среде, складывается из двух фундаментальных процессов: собственно химической реакции и переноса вещества к реакционной зоне. Так как массоперенос осуществляется путем диффузии, а диффузионная подвижность частиц твердого тела зависит от дефектности его структуры, можно ожидать существенного влияния дефектов на механизм и кинетику твердофазных реакции. Это и наблюдается в действителъности.
Диффузия в твердых телах
Известно, что в твердых телах диффузия может протекать по различным механизмам, включая вакансионный, междоузельный и эстафетный. Первый предусматривает перемещение атомов или ионов благодаря их переходу в соседние вакантные узлы (рис. 2.1, в), во втором случае ионы переходят из одних междоузельных позиций в другие (рис. 2.1, а). В случае эстафетного механизма диффузии, ионы перемещаются из одних регулярных узлов в другие через междоузлия (рис. 2.1, б) [5].

Рисунок 2.1 – Различные механизмы диффузии
а – диффузия по междоузлиям; б – эстафетная диффузия; в – диффузия по вакансионному механизму.
В соответствии с законом Фика направленная диффузия возможна лишь при наличии градиента химического потенциала компонентов, составляющих кристалл
Ii = -Di(∂ci/∂x), (2.2)
где Ii – поток i-того компонента в направлении х через единичную площадку, перпендикулярную х;
Di – коэффициент диффузии;
∂ci/∂x – градиент концентрации i-того компонента в направлении х.
Если диффузия осуществляется по вакансионному механизму, то
D=αao2ω[V]=αao2kν exp(ΔSV+ΔSпер)/Rexp(-(ΔHV+ΔHпер)/RT), (2.3)
где α – коэффициент, зависящий от геометрии кристалла;
a – постоянная решетки;
ω – частота перескока атомов из регулярных в соседние вакантные узлы;
[V] – концентрация вакансий;
k – трансмиссионный коэффициент, характеризующий вероятность того, что атом с достаточной для скачка энергией действительно совершит перескок;
ν – частота такого перескока;
ΔHV и ΔSV – энтальпия и энтропия образования вакансий;
ΔHпер и ΔSпер – энтальпия и энтропия активации переноса
Соотношение(2.3) показывает, что интенсивность диффузионного массопереноса в объеме кристалла зависит от легкости образования в нем вакансий и становится тем больше, чем выше концентрация последних. Однако выражаемая этим уравнением простейшая взаимосвязь между коэффициентом диффузии и концентрацией вакансий справедлива лишь при условии, что концентрация вакансий сравнительно невелика, а перескоки атомов в вакантные узлы решетки взаимонезависимы. В противном случае, следует учитывать, что коэффициент диффузии пропорционален величине, выражающей вероятность заполнения соседнего с вакансией узла. Поэтому для нестехиометрического кристалла МХ1-γ, в котором дефицит компонента X вызван образованием вакансий в Х-подрешетке, коэффициент диффузии атомов X:
DX=DVγ(1-γ),где DV – коэффициент диффузии вакансий.
Допустим теперь, что между вакансиями действуют отталкивающие силы, так что при некоторой концентрации γ’ вакансии полностью упорядочиваются, образуя новое соединение МХ1-γ. В этом случае:
DX=DVγ(1-(γ/γ’))
При у = у' коэффициент DV= 0, т. е. вакансии становятся неподвижными и теряют способность участвовать в массопереносе компонента X (во всяком случае по вакансионному механизму).
Рассмотрим, как отсутствие беспорядка в распределении вакансий влияет на соотношение между коэффициентами диффузии, измеренными различными методами. Коэффициент диффузии можно определить, измеряя частоту перескоков атомов методам внутреннего трения, или нейтронной спектроскопии. Кроме того, можно использовать методы, основанные на измерении потока вещества в концентрационном поле. В последнем случае имеет место взаимная диффузия обоих компонентов, протекающая с различной скоростью и сопровождаемая смещением первоначальной поверхности между составляющими диффузионной пары (эффект Киркендаля) [11].
2.2 Методы исследования механизма твердофазных реакций
Для изучения механизма твердофазных реакций применяют различные методы, среди которых наибольшую популярность получили метод Тубанда— Вагнера и метод меченых граничных поверхностей, предложенный Бенгсоном и Ягичем. В первом из них о направлении массопереноса в ходе реакции судят по изменению массы отдельных реагентов и продукта.
Идея метода меченой поверхности заключается в том, что между реагентами помещается инертная метка, например тонкий слой Pt, Mo или W, и по ее положению после прохождения реакции судят о возможных механизмах, оценивая направление и скорость миграционных процессов. На рис. (2.2) представлены ситуации, возникающие в системе АО—В2О3, у которой массоперенос осуществляется противодиффузией катионов (а), односторонним перемещением составных частей реагента АО (б) и односторонним перемещением составных частей реагента В2О3 (в). К сожалению, метод меченых поверхностей не лишен недостатков. К ним следует отнести, в первую очередь, трудность определения истинного положения метки и возможность ее смещения за счет возникновения напряжения роста, а главное, за счет проявления эффекта Киркендаля. Последний, отсутствует лишь тогда, когда реакция осуществляется путем противодиффузии катионов. К тому же при высоких температурах метка недостаточно инертна по отношению к реагентам и продуктам взаимодействия.

Рисунок 2.2 – Смещение инертной метки, первоначально разделяющей реагенты, в результате взаимодействия А2++ О2-+В2О3=АВ2О4.
Указанные недостатки отсутствуют в методе свободной поверхности, в котором в качестве указателя исходной границы раздела фаз используют плоскость одной из таблеток, имеющей меньший размер, чем таблетка второго реагента. Иначе говоря, в этом случае искусственная метка заменена естественной. Схема возможных изменений в системе АО—В2О3 при различных механизмах образования продуктов представлена на рис. 2.3 Большим преимуществом последнего метода является возможность изучения механизма реакции при минимальной толщине слоя продуктов. Вместе с тем, метод свободной поверхности, как и метод инертной метки не позволяет указать точный механизм при одностороннем перемещении одного из реагентов[9].
 Рисунок
2.3 – Взаимное расположение реагентов
и продукта реакции при изучении механизма
твердофазных реакций методом свободной
поверхности.
Рисунок
2.3 – Взаимное расположение реагентов
и продукта реакции при изучении механизма
твердофазных реакций методом свободной
поверхности.
Всё большее применение находит метод микрорентгеноспектрального анализа (локальный микроанализатор), идея которого заключается в следующем. Образец с хорошо отполированной поверхностью облучается узким сканирующим электронным пучком диаметром около 1 мкм. Характеристическое рентгеновское излучение, испускаемое при облучении атомами образца, анализируется электронным логическим блоком. Подвергая такому исследованию поперечные сечения образцов, содержащие реагенты и продукт взаимодействия, удается изучить распределение компонентов и фаз в зоне реакции, установить геометрию фронта реакции и найти характер и последовательность превращений при твердофазном взаимодействии. В ряде случаев полезную информацию о механизме реакции дает применение метода гамма-резонансной спектроскопии (эффект Мессбауэра), а также методов инфракрасной и эмиссионной микроскопии [5].
Нередко о механизме твердофазных реакций судят лишь на основании того, что экспериментальные данные о степени взаимодействия как функции времени описываются лучше всего какой-либо конкретной кинетической моделью и соответствующим уравнением формальной кинетики. Такой подход может привести к неверным выводам, если отсутствуют полученные независимым методом дополнительные доказательства в пользу предполагаемого механизма реакции.
Наиболее достоверные сведения о механизме твердофазных реакций получают при комплексном исследовании, позволяющем одновременно наблюдать несколько параметров реагирующей системы, включая, например, фазовый состав, тепловые эффекты изменения массы, размеров, электропроводности и теплопроводности.
2.3 Теория твердофазного взаимодействия
Термодинамическая теория твердофазных реакций была предложена Вагнером и в дальнейшем развита Шмальцридом на примере реакции присоединения
А2++ О2-+В2О3=АВ2О4.
2.2.1 Теория Вагнера
1) скорость реакции лимитируется диффузией ионов через слой образующегося продукта;
2) слой продукта реакции является компактным, и содержащиеся в нем неравновесные дефекты (дислокации, границы зерен) не вносят определяющего вклада в подвижность ионов;
3) реакции на границе фаз протекают значительно быстрее, чем процессы диффузии через слой продукта, и поэтому на границах фаз устанавливается локальное термодинамическое равновесие;
4) отдельные ионы движутся в реакционном слое независимо друг от друга; в любом поперечном сечении продукта сохраняется условие электронейтральности.
На рис. 2.4 представлены наиболее типичные схемы массопереноса для реакцииА2++ О2-+В2О3=АВ2О4. В первом случае (рис. 2.4 а) наиболее подвижными в слое продукта являются ионы А2+ и О2-; они переносятся в эквивалентных количествах к границе раздела АВ2О4—В2О3 и взаимодействуют там с образованием продукта. Во втором случае (рис. 2.4 б) более подвижные ионы В3+ и О2- взаимодействуют, проходя через слой продукта к границе АО—АВ2О4. Наконец, в третьем случае (рис. 2.4 в) образование продукта реакции происходит на обеих границах в результате противодиффузии катионов [7].

Рисунок 2.4. Схемы массопереноса в реакцииА2++ О2-+В2О3=АВ2О4.
Следует упомянуть еще один механизм образования тройных оксидных кристаллов, который весьма вероятен при проведении реакции в атмосфере кислорода. Если продукт реакции имеет достаточно высокую проводимость, катионы вместе с эквивалентным потоком электронов перемещаются в одном и том же направлении, тогда как кислород переносится через газовую фазу [11].
Связь между степенью разупорядочения решетки продукта и интенсивностью массопереноса не всегда является однозначной, даже если реакция осуществляется при высоких температурах, когда в любой части системы устанавливается локальное равновесие. Доминирующий в решетке тип катионных дефектов лимитирует скорость массопереноса, если перенос кислорода происходит с высокой скоростью через газовую фазу и межкристаллитные раницы [11]. Такая ситуация имеет место, например, при образовании из оксидов молибдата меди, в котором вакансии в молибденовой подрешетке являются дефектами, одновременно доминирующими в решетке и лимитирующими скорость массопереноса. Если же перенос компонентов через газовую фазу исключен или сильно ограничен, то скорость твердофазного взаимодействия лимитируется движением недоминирующих, наименее подвижных в решетке дефектов, как это имеет место при образовании из оксидов ортоферрита иттрия[11].
2.3.2 Механизм реакций двойного обмена по Иосту и Вагнеру
Твердофазные реакции типа
AX + BY = AY + BX
с участием реагентов, содержащих различные катионы и анионы. В зависимости от относительной подвижности ионов и способности реагентов и продуктов к образованию твердых растворов можно выделить несколько типов взаимодействия. Для случая пренебрежимо малой взаимной растворимости АХ, BY, AY и ВХ и более высокой подвижности катионов, чем анионов, Иостом и Вагнером предложено два различных механизма, иллюстрируемые схемами на рис. 2.5.

Рисунок 2.5. Механизм реакций двойного обмена по Иосту (а) и Вагнеру (б).
Согласно модели Иоста, исходные вещества АХ и BY разделены продуктами реакции AY и ВХ, возникающими за счет катионной противодиффузии в практически неподвижном анионном каркасе. Само твердофазное взаимодействие предполагает существование пусть очень малой, но отличной от нуля растворимости ионов А3+ в кристалле ВХ и ионов В+ в кристалле AY, делающей возможным движение этих ионов в соответствующих кристаллах. Для количественного описания процесса необходимо фиксировать Р, Т и парциальные давления летучих компонентов в газовой фазе. Согласно модели Вагнера, из-за незначительной растворимости и подвижности ионов А+ в кристаллах ВХ и ионов В+ в кристаллах AY твердофазный процесс осуществляется только благодаря диффузии собственных катионов, которая совершается в частицах реагентов взаимосогласованным образом. На рис. 2.6. представлена схема твердофазного взаимодействия AgCl + NaI=AgI + NaCl по Вагнеру [10]. Процесс начинается с того, что на поверхности соприкосновения исходных кристаллов AgCl и Nal образуются зародыши кристалла Agl. При этом в кристаллах AgCl возникает дефицит серебра, а в кристаллах Nal — дефицит йода. Благодаря этому в другой точке соприкосновения AgCl и Nal образуются зародыши фазы NaCl. Процесс повторяется до тех пор, пока не наступит стационарное равновесное состояние, определяемое соотношением скоростей образования и роста кристаллических зародышей. В конечном счете образуется конгломерат продуктов расположенных между кристаллами реагентов. Одинаково заряженные ионы, содержащиеся в различных фазах, перемещаются таким образом, что создается ток, протекающий по замкнутому контуру, как оказано на рис. 2.6.
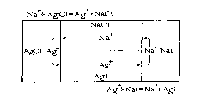
Рисунок 2.6. Схема твердофазного взаимодействия AgCl+NaCl=AgI +NaCl пo
Вагнеру.
Схема (2.6) является идеализацией действительно протекающего процесса. Из-за накопления вещества в одних частях реакционной смеси и потери его в других создаются сильные внутренние напряжения, приводящие к пластической деформации и изменению положения кристаллитов. Несмотря на это, модельные представления Вагнера были подтверждены при сопоставлении экспериментально наблюдаемой скорости реакции с рассчитанной из э. д. с. соответствующих цепей и значений электропроводности.
Предложенная Вагнером схема короткозамкнутых локальных элементов применима и для описания реакций двойного обмена в порошкообразных смесях (рис.2.7). В этом случае процесс интенсифицируется благодаря облегченной диффузии ионов по поверхности частиц, границам зерен и протяженных дефектов.
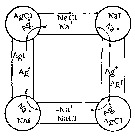
Рисунок 2.7. Схема короткозамкнутых локальных элементов в применении к реакции двойного обмена AgCl + NaI=AgI + NaCl в порошкообразных смесях.
При среднем размере частиц, равном 10-4 см, во многих порошкообразных системах реакция двойного обмена должна завершаться в течение нескольких минут. Этого не наблюдают в действительности, по-видимому, из-за того, что скорость процессов в целом лимитируется скоростью образования зародышей или взаимодействия на границах фаз [10].
Большой интерес представляет выяснение механизма обменных твердофазных реакций, ведущих к образованию нестехиометрических соединений с упорядоченными вакансиями. Основной химический процесс можно представить уравнением :
2Li+ М2+
+ V,
М2+
+ V,
где М2+ — двухвалентный катион; V—вакансия.
Например,
при нагревании оксидной шпинели
LiZn0,5Ge1,5O4с
сульфатом цинка при 400 °С имеет место
взаимодействие, прикотором катионы,
заключенные в квадратные скобки, занимают
октаэдрические, а катионы перед скобками
— тетраэдрические узлы шпинельной
структуры. В исходной шпинели октаэдрическая
подрешетка характеризуется упорядоченным
расположением ионов Li+
и Ge4+,
а в продукте реакции — ионов Ge4+
и катионных вакансий (сверхструктура
типа 3:1). Благодаря сходству структур
реагента и продукта реакция протекает
по топотактическому
механизму. Образующиеся при замещении
вакансии структуры усиливают диффузию
катионов и делают возможным завершить
взаимодействие при сравнительно
невысоких температурах. Обмен может
быть осуществлен, если энергия Гиббса
образования соли двухвалентного металла
заметно меньше, чем у соли лития, благодаря
чему значение
 G
обменной реакции отрицательно [11].
G
обменной реакции отрицательно [11].
Такая реакция была использована для получения большого числа дефектных шпинелей. Установлено, что относительно устойчивый продукт можно получить, если размер иона, замещающего литий, не превышает 0,097 нм. Непременной стадией любой твердофазной реакции является перенос вещества через границу фаз, образуемую реагентами и твердым продуктом. Так как граница фаз создает сопротивление потоку частиц из одного реагента в другой, то на ней возникает скачок химического потенциала компонентов. С увеличением толщины реакционного слоя его диффузионное сопротивление растет по сравнению с сопротивлением границы потока частиц, скачок химического потенциала на границе уменьшается и с наступлением локального равновесия полностью исчезает. Напротив, при наличии очень тонкого слоя продукта его диффузионным сопротивлением можно пренебречь; скачок химического потенциала на границе фаз будет иметь максимально допустимое термодинамикой значение и скорость реакции будет определяться только сопротивлением границы. Так как это сопротивление практически постоянно, то и продукт реакции будет накапливаться с постоянной скоростью. Линейный закон роста наблюдается на начальной стадии образования шпинелей и силикатов (рис. 2.8)
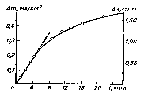
Рисунок 2.8. Кривая накопления массы т и толщины слоя продукта х в реакции 2ZnO + SiО2 = Zn2SiО4 на воздухе при 1348 °С.
Если исходить из существования идеального контакта между частицами реагентов и продуктов, можно выделить следующие случаи:
1. Одна или несколько подрешеток исходного вещества остаются неизменными в ходе рекации. Например, на границе MgO—MgCr2O4 возникающей при взаимодействии оксидов MgO и Сr2О3, реагент и продукт имеют сходную структуру, образующуюся в результате плотной гранецентрированной упаковки ионов О2-. Сама реакция практически заключается в переходе ионов из октаэдрических в тетраэдрические узлы жесткого кислородного каркаса, вследствие чего часто наблюдается эпитаксиальный рост продуктов.
2. Одна или несколько подрешеток реагента превращаются в подрешетки продукта благодаря кооперативным процессам. Например, на границе А12О3—СоА12О4, возникающей при взаимодействии корунда с закисью кобальта, происходит следующий процесс: гексагональная плотная упаковка ионов О2-, характерная для корунда, превращается в результате кооперативного смещения
частиц в кубическую упаковку шпинели. Подвижность катионов при этом настолько велика, что их движение, сопутствующее движению кислородных ионов, не лимитирует процесса. В этом случае также может происходить эпитаксиальный рост: гексагональная С-ось (001) корунда параллельна пространственной диагонали <111> шпинели, так что плоскости кислородных ионов с плотнейшей упаковкой остаются без изменений при переходе от решетки реагентов к продукту реакции.
3. Структуры реагентов и продукта не имеют общих лементов— тогда фазовая граница разупорядочена и структурная взаимосвязь между реагентами и продуктом полностью отсутствует.
В отсутствие идеального контакта реагентов порошкообразных систем перенос вещества через поры осуществляется благодаря испарению; скорость процесса может лимитироваться скоростью испарения, конденсации или газового переноса от одной твердой границы к другой [11].
3. Получение ферритовых порошков
3.1 Технология ферритов по методу растворной химии
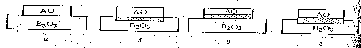
Основная задача создания оптимальной технологии ферритов состоит в получении материалов с воспроизводимыми, однородными, заданными магнитными и электрическими функциями при минимальных затратах на оборудование, сырьё, электроэнергию и рабочую силу при максимальном выходе годных изделий.
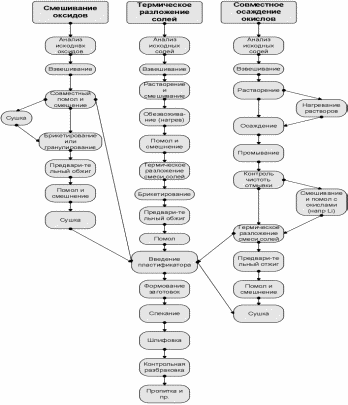
Рисунок 3.1.Схема технологического процесса получения ферритов
Технологический процесс является многооперационным и длительным.
При производстве ферритов применяют три основных метода приготовления шихты:
1) метод смешения и помола порошкообразных оксидов (керамический метод);
2) метод термического разложения смеси солей соответствующих металлов до оксидов; 3)метод совместного осаждения углекислых солей или гидрооксидов с последующим термическим разложением их до оксидов [6].
Преимуществами двух последних (химических) методов являются, получение высокогомогенной смеси без помола и смешения, а также обеспечение высокой воспроизводимости ее физико-химических и структурных характеристик.
Недостатком химических методов является необходимость переработки большого количества сырых материалов; кроме того, отходы производства при использовании этих методов загрязняют окружающую среду. При совместном осаждении гидроксидов осадок адсорбирует находящиеся в растворе соли, трудно удаляемые последующей отмывкой и обжигом.
Наиболее распространен метод приготовления шихты из оксидов. К его достоинствам относятся: возможность точного соблюдения заданного состава; отсутствие отходов и соответственно переработка меньших количеств сырья; отсутствие вредных выделений, загрязняющих атмосферу; относительная простота технологической схемы производства.
Недостатком этого метода является необходимость тщательного измельчения и смешения оксидов с целью получения смеси высокой однородности[7].Последовательность операций при изготовлении ферритов указанными методами указана на рис. 3.1.
Любым из упомянутых методов можно получить ферриты с близкими магнитными свойствами,компенсируя меньшую активность шихты,полученной смешением порошкообразных оксидов,более высокой температурой и продолжительностью обжига[6].
3.1.1 Метод осаждения солей или гидроксидов
При использовании метода осаждения можно получать продукты реакции в виде солей или гидроксидов. При этом используется раствор солей (сернокислых, азотнокислых) в соотношении, требуемом для получения феррита определенного химического состава. В раствор добавляют осадитель (щелочь, аммиак, углекислый аммоний), в результате чего происходит совместное осаждение продуктов реакции (~50° С), а полученный пастообразный осадок отмывают, отжимают и сушат при 120° С.
Осадок прокаливают при 300 — 800°С; при этой температуре он разлагается на оксиды. Полученную массу размалывают и т. д. Применяют и раздельное осаждение компонентов шихты, осаждение части компонентов или совместное осаждение. Последнее не всегда возможно, так как для полного осаждения каждого гидроксида требуется среда с определенной кислотностью. Полное смешение возможно, когда соосажденные компоненты образуют твердые растворы (в противном случае в осадке образуется механическая смесь кристаллов и степень смешения на молекулярном уровне не может быть достигнута) и когда катионы равномерно распределены по объему кристаллов (это условие невыполнимо, если процесс кристаллизации идет не одновременно).
Совместное и полное осаждение смесей солей невозможно при различной растворимости осажденных компонентов, зависящей от дисперсности осадка и концентрации образующихся растворимых солей, а также из-за различных скоростей кристаллизации отдельных солевых компонентов и образования комплексных соединений, а также неизоморфности соосаждаемых соединений.
Осадки раздельно осажденных компонентов высушивают, взвешивают в требуемой пропорции и тщательно перемешивают. При осаждении части компонентов мокрый осадок смешивают с недостающими оксидами и размалывают. При совместном осаждении всех компонентов осадок высушивают и нагревают до температуры, при которой разлагается наиболее термостойкая составная часть смеси. В результате разложения получается смесь оксидов. Полученные этим методом ферритовые порошки, как правило, являются мелкодисперсными и однородными по составу[3]
3.1.2 Метод термического разложения солей
Материалы, полученные этим методом, в отечественной практике получили название оксиферов. Сырьем служат соли металлов (преимущественно сернокислые), содержащие кристаллизационную воду. В смесь солей, взятых в необходимых количествах для получения феррита заданного состава, добавляют небольшое количество воды. При нагревании смесь расплавляется (60—70° С), кипит (100—120° С) и после выпаривания воды соли затвердевают (300° С). При дальнейшем нагревании (950—1100° С) обезвоженная смесь солей разлагается на оксиды, а образовавшиеся газы улетучиваются. Оставшуюся массу оксидов размалывают.
Для этого метода характерна высокая однородность смешения компонентов шихты. В то же время полная однородность в распределении компонентов по объему практически не достигается, так как растворимость солей разных металлов неодинакова; (при выпаривании из пересыщенного раствора в первую очередь будут выпадать менее растворимые соли) [3].
Технологический процесс получения ферритовых порошков включает следующие основные стадии:
Подготовка производства.
2. Подготовка сырьевых материалов.
3. Приготовление шихты (взвешивание сырьевых материалов и помол шихты).
4. Ферритизация шихты.
5.Помол ферритизованной шихты.
3.2 Керамическая технология получения ферритов
3.2.1 Подготовка сырьевых материалов
В качестве исходного сырья для производства марганец-цинковых ферритовых порошков применяют железо (ІІІ) оксид, марганца (ІІ, ІІІ) оксид, белила цинковые.
Железо (ІІІ) оксид поступает в цеховой склад в одноразовых мягких контейнерах типа МКР-1. во избежение случайного увлажнения хранимого в цеховом складе сырья, последнее должно храниться на поддонах.
Оксид марганца и цинковые белила поступают в 25-30 кг полиэтиленовых мешках.
Из поступающего в цеховой склад сырья лаборант цеха специальным пробоотборником отбирает пробы от каждой партии сырья для определения массовой доли основного вещества и химического анализа согласно техническим условиям на сырьё.
Оксид марганца разных партий перед подачей на шихтовку подвергают усреднению в барабанном смесителе типа с рабочей поверхностью 1,0 м3/поз.1/. смеситель представляет собой цилиндрический корпус, расположенный под наклоном 300 к штативу, на котором закреплён двигатель. Двигатель соединён с барабаном при помощи ременного привода. Таким образом, при работе двигателя, барабан вращается в виде гироскопа. Такое вращение барабана обеспечивает равномерное по всему объёму порока перемешивание. Процесс усреднения длится от 1 до 2 часов. Усреднённый после анализа на содержание основного вещества выгружают в бумажные барабаны [4].
3.2.2 Приготовление шихты
Взвешивание сырьевых компонентов осуществляют на платформенных весах, пределы взвешивания 0,5-1000кг. Для подачи мешков и барабанов с сырьём на платформу весов используют электротельфер. Для удобства взвешивания оксида железа его пересыпают из мягкого контейнера в барабаны по 30кг. При взвешивании определяют массу нетто по разности масс заполненных и опорожненных барабанов с сырьём.
Количество компонентов шихты (кг) рассчитывают, исходя из количества марганец-цинкового ферритового порошка, который необходимо получить как конечный продукт, по следующим формулам:
а) масса оксида железа в шихте рассчитывается по формуле:
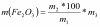 ,
,
где m1 - масса марганец-цинкового ферритового порошка, кг.;
m2 - массовая доля Fe2O3 в составе феррита, %;
m3 - массовая доля основного вещества в исходном оксиде железа по анализу,%
б) масса оксида марганца (ІІ, ІІІ) в шихте рассчитывается по формуле:
 ,
,
где m1 - масса марганец-цинкового ферритового порошка, кг;
m2 - массовая доля MnO в составе феррита, %;
m3 - массовая доля MnO в исходном оксиде марганца по анализу, %.
в) масса цинковых белил в шихте рассчитывается по формуле:
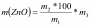 ,
,
где m1 - масса марганец-цинкового ферритового порошка, кг;
m2 - массовая доля ZnO в составе феррита, %;
m3 - массовая доля основного вещества в исходных цинковых белилах поанализу, %.
Для
смешения и помола шихты используют
вибромельницу типа М-400 с максимальной
загрузкой 400 кг. Мощность мельницы 45
кВт. Мельница представляет собой
горизонтальный стальной барабан
диаметром 1м и длиной 2м. внутри камеры
мельницы находятся стальные шары
диаметром от 10 до 30мм (в любом соотношении).
Общая масса загружаемых шаров составляет
25% от объёма барабана, при этом расстояние
от нижней кромки загрузочного люка до
поверхности шаровой загрузки барабана
должно составлять (1000 30)мм.
При вибрационном движении барабана
мельницы стальные шары внутри мельницы
смешивают и измельчают шихту [3].
30)мм.
При вибрационном движении барабана
мельницы стальные шары внутри мельницы
смешивают и измельчают шихту [3].
Для лучшего перемешивания компоненты шихты засыпают в загрузочный люк вибромельницы слоями: оксид железа, оксид марганца, цинковые белила и т.д.
Для улучшения поверхностно-активных свойств шихты при помоле в конце загрузки в мельницу добавляют 200мл триэтаноламина, разбавленного с дистиллированной водой в соотношении 1:1. помол шихты производят в течении 1-2 часов.
Как загрузка, так и помол шихты осуществляют при включённой вентиляции.
По окончании помола через верхний люк мельницы отбирают пробу шихты для анализа её химического состава и удельной поверхности. В случае несоответствия химического состава заданному осуществляют корректировку состава шихты введением необходимых компонентов. Корректировка шихты производится по компоненту, которого по данным анализа в составе больше[4].
Как в случае корректировки шихты, так и в случае несоответствия регламенту величины удельной поверхности шихты( 0,7-0,9м2/г), производят дополнительный помол в течении одного часа. Затем анализ шихты повторяют.
Шихту, соответствующую требованиям регламента по составу и удельной поверхности, через нижний люк вибромельницы выгружают в барабаны и подают на туннельную печь для ферритизации [5].
3.2.3 Ферритизация шихты
Для ферритизации шихты используется электрическая туннельная печь «Диффузор». Нагрев продукта в рабочем пространстве электропечи осуществляется силитовыми нагревательными элементами, расположенными на боковых стенках нагревательной камеры по зонам. Туннельная нагревательная камера имеет семь температурных зон.
Таблица 3.1 Температурные зоны туннельной нагревательной камеры
|
Температура по зонам печи,°С |
Скорость нагрева по зонам печи,град/час |
|
1зона-370 |
143,9 |
|
2зона-510 |
213,8 |
|
3зона-720 |
158,9 |
|
4зона-980 |
218,4 |
|
5зона-980 |
0 |
|
6зона-980 |
0 |
|
7зона-980 |
0 |
Марганец-цинковую шихту подают в туннельную печь в керамических кюбелях по семь кг, которые продвигаются по зонам печи при помощи толкателя с интервалом в 20 минут.
Таким образом, шихта при прохождении через туннельную печь медленно нагревается до температуры ферритизации 9800С, а в конце печи медленно
остывает. Ферритизованную шихту из кюбелей ссыпают в барабаны и подают на стадию вторичного помола на вибромельницу М-400.
3.2.4 Помол ферритизованной шихты.
Перед вторичным помолом шихты, её взвешивают в барабанах на платформенных весах для определения веса нетто, который должен быть равен запланированной на стадии приготовления шихты величине массы марганец-цинкового ферритового порошка, с учётом потерь при ферритизации и потерь в виде россыпей при транспортировке[7].
Таблица 3.2. Характеристика исходного сырья для производства ферритовых изделий
|
Сырье |
Стандарт,ТУ,регламент |
Показатели для проверки |
Регламентир. показатели |
|
1. Fе2О3 Для магнитомягких ферритов»чистый»МТВ, МР-1 |
ТУ 14-15-228-90 |
Массовая доля,% Fe 2 O3 Si Са |
менее 99,2 не бол. 3х10-2 не бол. 2х10-2 |
|
2.Железо (III) оксид для ферритов «чистый» ММ-
|
ТУ-6-09-4783-83
|
Массовая доля, % : Fе2О3 Si Са |
не менее 99,2 8х10-3-2х10-2 8х10-3-2х10-2
|
|
3. Марганца (II, III) оксид, х.ч. |
ГОСТ 202-84 ТУ 6-02-916-79 |
Массовая доля, % : Мn |
90 не более 2х10не менее 99,6 |
Процесс помола ферритизованной шихты аналогичен процессу помола шихты на стадии её приготовления.Измельчённый марганец-цинковый порошок анализируют на соответсвие её требованиям ТУ на удельную поверхность и процентного содержания его основных компонентов.В случае удовлетворительного
3.2.5 Методы синтеза ферритовых порошков
Трудности получения ферритов с воспроизводимыми свойствами в основном связаны с самой природой этих материалов, являющихся фазами переменного состава. Многие свойства ферритов определяются не только соотношением основных компонентов, но и термодинамическими параметрами синтеза, важнейшими из которых являются температура и давление кислорода в газовой фазе. Поэтому необходимо установить и использовать на практике соотношения между внешними параметрами и состоянием системы. Большое значение имеет изучение равновесных диаграмм состояния, природы атомных дефектов и механизма их возникновения, исследование термодинамических и термохимических свойств ферритов, установление кинетических закономерностей и механизмов формирования фаз переменного состава[7].
Многие физические свойства ферритов являются структурно-чувствительными и зависят от условий формирования структуры при спекании и изменения ее в присутствии микропримесей или в ходе дополнительной термообработки.
Ферритовые порошки чаще всего получают керамическим методом, в котором при повышенной температуре осуществляется твердофазная реакция с образованием однофазного продукта:
xMeOk+yMeOl+…+z/2 Fе2О3+mO2=MeXMeY…Fe2Om
Степень ферритизации порошковой смеси существенно зависит от свойств исходных оксидов и условий механической гомогенизации. По данным рентгенофазового анализа, для одной и той же ферритовой композиции изменение указанных условий при постоянных температуре и продолжительности нагрева может приводить к колебаниям степени ферритизации от 20 до 80%. Присутствие в продуктах реакции исходных оксидов отрицательно сказывается на последующих процессах спекания. Вместе с тем повышение температуры, способствующее увеличению степени ферритизации, крайне нежелательно из-за существенного снижения активности порошков.Повторное измельчение и дополнительные обжиги,гомогенизирующие смесь и облегчающие диффузию,часто
К числу таких изменений относится загрязнение смеси материалом мельницы при его истирании, гидратация оксидов, частичное их восстановление или окисление. Таким образом, используемые в керамической технологии приемы гомогенизации ферритовых порошков неизбежно приводят к дополнительному появлению неоднородностей. Так, если при помоле шихта загрязняется оксидов, образующими легкоплавкую эвтектику с основными компонентами системы, то качество ферритовой шихты резко ухудшается из-за анизотропного роста зерен при спекании и сопутствующего ему изменения магнитных характеристик [6].
Необходимо также отметить еще один существенный недостаток, присущий керамическому методу, — наличие остаточной химической неоднородности однофазных продуктов твердофазного синтеза, которые приводят к флуктуациям физических свойств, чувствительных к изменениям химического состава феррита[7].
Известное развитие получили варианты керамического метода, в котором в качестве исходных сырьевых материалов используют смеси сульфатов, нитратов, карбонатов и гидроксидов, подвергаемые термическому разложению после тщательного смешения и измельчения.
При правильном выборе режима разложения (скорости и продолжительности нагрева) процессы образования оксидов и ферритизацию удается совместить в сравнительно узком температурном интервале. Оксиды, получаемые разложением солей, в момент образования имеют высокую степень дефектности и большую подвижность элементов структуры, повышенную реакционную способность[7].
Так, термическое разложение смеси:
0,4 моля NiSO4· 7H2O + 0,6 моля ZnSO4 · 7H2O + 2 моля FeSO4 · 7H2O
приводит
к получению никельцинкового феррита
 .При
этом температурный интервал разложения
практически совпадает
с образованием однофазного ферритового
продукта. Метод,
основанный на использовании солевых
смесей, не всегда решает
до конца задачу получения ферритов с
гомогенным распределением компонентов
[6].
.При
этом температурный интервал разложения
практически совпадает
с образованием однофазного ферритового
продукта. Метод,
основанный на использовании солевых
смесей, не всегда решает
до конца задачу получения ферритов с
гомогенным распределением компонентов
[6].
Интенсивно развиваются методы получения ферритовых порошков из твердых растворов солей и гидроксидов. В таких растворах, а также в продуктах их термического разложения ферритообразующие компоненты находятся в более высокой степени смешения, чем в системе, образованной из смеси солей.
Существующие методы получения твердых растворов солей (гидроксидов) основаны на равновесной или неравновесной кристаллизации.
Равновесную кристаллизацию осуществляют: 1) методом изотермического и изоконцентрационного снятия микропересыщений и 2) методом изотермического и изоконцентрационного испарения растворителя.
Методы неравновесной кристаллизации используют один из перечисленных ниже приемов: 1) соосаждение в форме малорастворимых соединений, 2) замену растворителя, 3) распылительную сушку, 4) криохимическую кристаллизацию.
3.2.5.1 Метод изотермического и изоконцентрационного снятия микропересыщений
К раствору, насыщенному при температуре T1, прибавляют при интенсивном перемешивании небольшие порции другого раствора, имеющего температуру Т2 (Т2>T1) и более высокую концентрацию. Если концентрации компонентов подобраны соответствующим образом, то при быстром снятии микропересыщений в микрообъеме из раствора выпадают кристаллы, в которых соотношение солевых компонентов отвечает получаемому фериту[6].
3.2.5.2 Метод изотермического и изоконцентрационного испарения
Непрерывное испарение растворителя из раствора, равновесного с кристаллами заданного состава. По мере удаления растворителя и отбора выпадающих кристаллов концентрация маточного раствора постоянно корректируется добавлением насыщенного раствора, соотношение солевых компонентов в котором отвечает кристаллизуемой соли [7].
3.2.5.3 Методы соосаждения
Основаны на осуществлении химических реакций, приводящих к соосаждению железа с другими феррито-образующими компонентами в форме нерастворимых или мало растворимых солей или гидроксидов
x(Me 2+)+y(Me 2+)+(1-x-y)Fе2+ +RO2-=(MeXMeYFe 1-x-y)RO
Очевидно, что однородность солевых твердых растворов, кристаллизующихся в сильно пересыщенной (неравновесной) системе, образующейся при смешении раствора легкорастворимых солей ферритообразующих компонентов с осадителем, зависит от растворимости и скорости кристаллизации отдельных солевых компонентов. При значительной разности этих величин ожидать получения совершенно однородных кристаллов практически невозможно[7].
3.2.5.4 Метод замены растворителя
Основан на эффекте «высаливания» — резком уменьшении растворимости солей в водно-органических смесях по сравнению с чистой водой. Метод замены растворителя удобней всего применять к солям, которые обладают высокой растворимостью в воде и практически нулевой растворимостью в водно-органических смесях с малым содержанием органического компонента. Органические вещества, используемые в качестве высаливающего агента, должны обладать неограниченной взаимной растворимостью с водой и относительно высоким давлением паров при комнатной температуре, способствующим быстрому испарению молекул органического вещества с поверхности кристаллизованной солевой массы. Этим условиям в полной мере отвечает ацетон, который чаще всего используют для гомогенизации ферритообразующих компонентов методом замены растворителя (можно использовать также этиловый, изопропиловый спирт и др.)[6].
3.2.5.5 Распылительная сушка
Диспергирование исходного раствора в потоке теплоносителя. В большинстве известных вариантов распылительной сушки теплоносителем является нагретый воздух. Естественно, что малая скорость испарения растворителя крайне нежелательна из-за возможности дробной кристаллизации солевых компонентов из раствора. Вместе с тем дробную кристаллизацию можно устранить лишь при очень больших скоростях испарения растворителя. Метод распылительной сушки был успешно применен для получения марганец-цинковых ферритов из сульфатных растворов при относительно невысоких температурах теплоносителя в зоне распыления (280—300° С)[6].
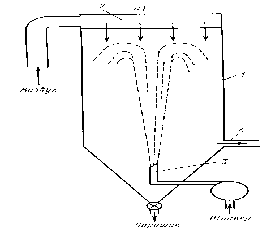
Рисунок 3.2.Схема распылительной сушилки с фонтанирующими форсунками.
1 - камера сушилки,2 - устройство для подачи горячего воздуха,3 - распылительное устройство,4 - устройство для улавливания пылеобразующих продуктов.
3.2.5.6 Криохимический метод
Распыление растворов солей ферритообразующих компонентов в жидкий, не смешивающийся с растворителем хладоагент. Быстрое замораживание отдельных капель раствора позволяет получить собственно криохимический продукт, представляющий собой мелкие криогранулы, имеющие, как правило, сферическую форму с достаточно равномерным распределением исходных солевых компонентов по их объему. Удаление растворителя из продукта криохимического синтеза производят путём сублимации льда при низких давлениях и температурах, не превышающих температур плавления криогранул. Схема метода представлена на рис. 3.3.
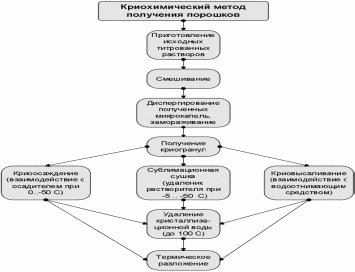
Рисунок 3.3.Схема криохимического метода получения ферритообразующих компонентов.
Равновесные методы кристаллизации имеют довольно узкую область применения, ограниченную возможностью образования солевых твердых растворов. Среди солевых твердых растворов, в состав которых входят ферритообразующие компоненты, необходимо отметить твердые растворы солей типа шенитов, оксалоаммониевые комплексные соли и т. д.
Равновесные методы кристаллизации неприменимы, если солевые компоненты не образуют твердые растворы[7].
Так, большинство сульфатов или нитратов ферритообразующих компонентов не образуют между собой солевые твердые растворы. Поэтому применение равновесных методов к таким системам не обеспечит их гомогенизации — продукт кристаллизации будет представлять собой механическую смесь индивидуальных сульфатов или нитратов.
Для ферритообразования соосажденных гидроксидов предложена следующая процедура: сразу же после осаждения гидроксид железа превращается в липидокрокит пропусканием воздуха через водную суспензию гидроксидов
2Fe(OH)2 + ½ O2 = 2FeOOH = H2O
Образующаяся рентгеноаморфная масса при старении дегидратируется и переходит в упорядоченное состояние, свойственное структуре феррита. Скорость ферритизации зависит как от степени неоднородности растворов, так и от способности отдельных гидроксидов к дегидратации. Если хотя бы один из компонентов системы легко дегидратируется, это свойство в известной мере присуще и твердому раствору. А устойчивость к дегидратации обоих компонентов системы затрудняет процесс старения и образования ферритов[6].
3.2.5.7 Электрохимический метод
Имеет несомненное преимущество перед обычным методом соосаждения гидроксидов. Он позволяет периодический процесс осаждения сделать по существу непрерывным, так как при электролизе происходит саморегенерация осадителя.
При получении ферритов анодным растворением металлов отпадает необходимость в их предварительной очистке, поскольку переход отдельных компонентов в осадок происходит лишь при определенных значениях электродного потенциала. Так как рН раствора в процессе осаждения практически не изменяется, как это имеет место при химическом осаждении, можно ожидать, что полученные осадки гидроксидов или оксидов отличаются высокой степенью гомогенности [3].
3.2.5.8 Оксалатный метод
Оксалаты двухвалентных металлов изоморфны друг другу и образуют непрерывные или ограниченные ряды твердых растворов. Из числа распространенных ферритообразующих элементов лишь медь и литий не входят в эти растворы. Термическое разложение смешанных оксалатов на воздухе при сравнительно невысоких температурах (Т < 500° С) приводит к образованию феррита по схеме:
3(Me 1/3Fe 2/3)C2O4x 2H2O+2O2=MeFe2O4+6CO2+6H2O
Серьезный недостаток оксалатного метода получения ферритовых порошков — значительное различие скорости осаждения индивидуальных оксалатов. Даже при больших пресыщениях, создаваемых смешением достаточно концентрированных растворов, содержащих ионы Me2+ и C2O42-, отдельные оксалаты переходят в осадок дифференцированно и весь цикл кристаллизации завершается лишь через несколько часов[6].
В соосажденных оксалатах первые порции кристаллов обогащены легко кристаллизующимися компонентами, а последние — трудно кристаллизующимися. Вместе с тем оптимизация условий осаждения оксалатов дала возможность в ряде случаев получить достаточно однородные ферритовые порошки.
Помимо метода соосаждения, оксалаты можно использовать в методах распылительной сушки, замены растворителя и криохимической кристаллизации.
Получение смешанных оксалатов взаимодействием суспензии оксидов, гидроксидов или карбонатов двухвалентных металлов с щавелевой кислотой. Такое взаимодействие уже при комнатной температуре протекает практически до конца образования недиссоциированных Н2О или летучих СО2 продуктов. Достоинством метода является отсутствие каких-либо загрязнений (неизбежных при осаждении оксалатов из растворимых солей) и полное соответствие исходного состава материала составу продукта. К сожалению, оксид железа не может быть соосажден в форме оксалата и вводится дополнительно.
Соосаждение ферритообразующих катионов бикарбонатом аммония лежит в основе внедренного в промышленность способа получения марганец-цинковых ферритовых порошков [3].
Сульфаты и нитраты большинства ферритообразующих элементов хорошо растворимы в воде и практически не образуют солевых твердых растворов. Сульфаты и нитраты не нашли применения в методах замены растворителя и распылительной сушки, так как ферритовые порошки, полученные этими методами, имеют значительные флуктуации состава. Вместе с тем применение метода криохимической кристаллизации позволяет в случае необходимости получать оптически прозрачную керамику из порошков, синтезированных в свою очередь из высокооднородных механических смесей сульфатов[7].
Возможность использования сульфатов и нитратов для гомогенизации ферритообразующих компонентов в полной мере относится и к другим растворимым солям, включая хлориды, бромиды, формиаты и ацетаты.
Соли типа шенитов с общей формулой Me2+ N2+ (RO42-)2 · 6H2O использовали для гомогенизации ферритообразующих компонентов методами как равновесной, так и неравновесной кристаллизации.
Способность к образованию непрерывных рядов твердых растворов при любых соотношениях компонентов, по-видимому, связана с большим размером элементарной ячейки шенитов, превосходящим этот параметр для других солей. Другое преимущество шенитов — хорошая растворимость в воде, резко изменяющаяся с изменением температуры. Оба свойства шенитов легли в основу равновесных методов получения изоморфных смесей. Одновременно с этим твердые растворы солей типа шенитов с успехом используют в методах замены растворителя и криохимической кристаллизации. Эффективность шенитных твердых растворов как исходных веществ для получения высокогомогенных ферритовых порошков была показана на примерах Мg—Мn, Ni—Со, Ni—Znи других ферритов.
Таким образом, одна и та же ферритовая композиция может быть получена различными методами. При выборе конкретной формы исходных соединений и конкретного метода гомогенизации ферритообразующих компонентов следует учитывать следующие факторы:
1)простоту и доступность метода;
2)экономичность;
3)воспроизводимость;
4)возможность получения ферритового порошка с высокойхимической однородностью;
5) возможность получения ферритового порошка с высокой активностью к спеканию [3].