

med_genetika
.pdfЦитогенетический метод является основным современным генетическим методом исследования, который позволяет поставить точный диагноз наследственной болезни. Он состоит в визуальном исследовании хромосомного набора пациента с определением структурного дефекта. Чаще всего исследуют кариотип лимфоцитов периферической крови, клеток костного мозга, фибробластов кожи и клеток эмбриона. Обычно при помощи светового микроскопа изучают метафазные хромосомы, окрашенные по методу Гимза (дифференцированная окраска). Пробоподготовка заключается в заборе материала, выделении клеток, их культивировании, обработке ФГА и колхицином.
Для анализа хромосом используют рутинные, а чаще дифференцированные методы окрашивания (рис.5).
Рис. 5. Схематическое изображение хромосом человека (гаплоидный набор) при дифференцированном окрашивании
Алгоритм проведения цитогенетического исследования лимфоцитов состоит из следующих этапов:
1.Взятие материала для исследования (чаще всего 1-2 мл периферической крови из локтевой вены)
2.Центрифугирование цельной крови, получение фракции лейкоцитов
21
3.Культивирование лейкоцитов в питательной среде, стимуляция митоза лимфоцитов фитагемагглютинином (ФГА), остановка деления на стадии метафазы колхицином
4.Приготовление мазка, фиксация, окрашивание
5.Микроскопирование и анализ хромосом.
Классификация и номенклатура равномерно окрашенных хромосом человека (рутинный способ) была выработана на международных совещаниях, созывавшихся в Денвере (1960), Лондоне (1963) и Чикаго (1966). Согласно рекомендациям этих конференций, хромосомы располагаются в порядке уменьшения их длины (крупные, средние, мелкие). В зависимости от местоположения центромеры их делят на метацентрические (перетяжка находится в центре хромосомы и плечи ее почти одинаковые), субметацентрические (перетяжка смещена от центра, имеются короткое и длинное плечи), акроцентрические (центромера расположена у самого края хромосомы и короткое плечо практически не выражено). По этим двум критериям (длина и положение центромеры) все хромосомы разделены на семь групп, которые были обозначены буквами английского алфавита от А до G. Все пары хромосом было предложено нумеровать арабскими цифрами. Такая
система записи называется идиограмма. |
|
|
|
Группа А (1-3) |
– самые крупные хромосомы. 1 и 3 |
– метацентрические, |
2 – |
субметацентрическая. |
|
|
|
Группа B (4-5) – |
длинные субметацентрические хромосомы. |
|
|
Группа C (6-12,Х) – средние субметацентрические. |
|
|
|
Группа D – (13-15) – средние акроцентрические. |
|
|
|
Группа Е – (16-18) – короткие субметацентрики. |
|
|
|
Группа F – (19-20) – короткие метацентики. |
|
|
|
Группа G – (21-22) – короткие акроцентрики. |
|
|
|
Половые хромосомы – Х-хромосома – средняя |
субметацентрическая, |
Y- |
|
хромосома |
выделяется как самостоятельная. |
|
|
В каждой хромосоме согласно Парижской международной номенклатуре
различают область центромеры, длинное плечо – q, |
короткое плечо – p. В |
каждом плече выделяют области (1,2,3,…), внутри |
областей – сегменты |
(1,2,3,4,5,…) что позволяет верифицировать морфологическую картину полос, получаемых при дифференцированном окрашивании, и локализовать имеющийся структурный дефект.
Молекулярно-биохимические методы направлены на выявление в организме химических веществ, которые образуются при определенных наследственных нарушениях и являются маркерами болезней. Биохимические показатели (первичный белковый продукт гена, накопление патологического метаболита внутри клетки и во внеклеточных жидкостях больного) отражают сущность болезни более адекватно, чем клинические симптомы, не только в диагностическом, но и в генетическом аспекте. Именно поэтому, несмотря на сложность и высокую стоимость биохимических методов, им принадлежит ведущая роль в диагностике моногенных наследственных болезней.
22
Современные сверхточные технологии (жидкостная хроматография, массспектрометрия, магнитная резонансная спектроскопия, ДНК-идентификация и др.) позволяют выявить вариации в структуре гена, полипептидной цепи, промежуточных метаболитов, конечных продуктов реакции.
Популяционно-статистические методы исследования основаны на законе Харди-Вайнберга (закон генетической стабильности популяций). Смысл этого закона заключается в том, что при определенных условиях соотношение частот доминантных и рецессивных генов, сложившееся в генофонде популяции, сохраняется неизменным в ряду поколений. Популяций, полностью отвечающих требованиям закона Харди-Вайнберга, в природе не существует. В естественных популяциях, в том числе и человеческих, происходит мутационный процесс, естественный отбор и миграция. Однако изменение частот аллелей под действием эволюционных факторов осуществляется очень медленно, и поэтому популяционно-статистический метод успешно применяется при исследовании распространенности признаков, генов и наследственных заболеваний в человеческих популяциях. Метод позволяет выявить роль наследственных и средовых факторов в возникновении наследственной патологии и фенотипического полиморфизма.
Близнецовый метод позволяет определить роль генетического вклада в развитии патологического признака (болезни). Исследуется наличие и выраженность интересующего признака у однояйцовых (монозиготных) и двуяйцовых (дизиготных) близнецов. Монозиготные близнецы генетически идентичны и имеют высокую парную частоту (конкордантность) для наследственных заболеваний, а дизиготные близнецы имеют в среднем около 50% общих генов и низкую конкордантность для наследственных болезней. Если этиология заболевания не связана с генотипом, то конкордантность для однояйцовых и двуяйцовых близнецов будет примерно одинаковой.
Метод моделирования наследственных болезней человека на лабораторных животных имеет все преимущества экспериментального исследования: возможность работы на линейных животных, получение любых гибридов, изучение клеток и тканей в динамике заболевания, разработка и применение новых методов терапии и др. Именно на модельных животных успешно проводят исследования по клеточной инженерии, генотерапии, разрабатываются новые молекулярно-генетические методы диагностики наследственных болезней.
Дерматоглифика исследует рисунок кожных складок на ладонях и стопах, которые коррелируют с наследственной патологией. В настоящее время установлена наследственная обусловленность кожных узоров, хотя характер наследования окончательно не выяснен. Изучение людей с хромосомными болезнями выявило у них специфические изменения кожного рисунка пальцев и ладоней, что позволяет использовать методы дерматоглифики и пальмоскопии в диагностике этих заболеваний. Менее изучены дерматоглифические отклонения при генных болезнях. Однако описаны
23
специфические отклонения этих показателей при шизофрении, миастении, лимфоидной лейкемии.
2. Хромосомные болезни Хромосомные болезни (синдромы) – это группа наследственных
болезней, характеризующихся множественными пороками развития.
Хромосомные болезни, как правило, представляют собой спорадические случаи в семье, возникшие в результате спонтанных мутаций в половых клетках одного из родителей. Лишь 3-5% являются формами, передающимися из поколения в поколение.
Хромосомные нарушения являются следствием хромосомного дисбаланса во всех или в большинстве клеток организма. Хромосомный дисбаланс нарушает нормальное физическое и психическое развитие организма. Нарушения развития имеют широкий спектр: от гибели зиготы на первых стадиях дробления до вполне совместимых с постнатальным существованием сравнительно небольших отклонений в физическом, психическом или половом статусе ребенка. Степень аномалий развития коррелирует со степенью хромосомных нарушений: чем больше хромосомного материала утрачено или приобретено, тем сильнее отклонение в развитии, тем раньше в онтогенезе оно проявляется. Нехватку генетического материала переносят тяжелее, чем его избыток.
Хромосомный дисбаланс может быть представлен нарушением числа хромосом (геномные мутации) или нарушением строения какой-либо хромосомы (хромосомные мутации). В зависимости от того, где имеется мутация (в системе аутосом или в системе половых хромосом), все хромосомные синдромы подразделяются на две группы: синдромы, связанные
с аутосомными аномалиями и синдромы, связанные с аномалиями половых хромосом. В каждой группе различают синдромы, обусловленные числовыми или структурными нарушениями в кариотипе.
К числовым нарушениям кариотипа относятся полиплоидии и анеуплоидии. Полиплоидия – это увеличение числа хромосом, кратное гаплоидному набору (n), т.е. вместо нормального диплоидного набора в клетках имеется триплоидия (3n) или тетраплоидия (4n). Такие нарушения чаще всего несовместимы с нормальным развитием зародыша, и эмбрионы с этой аномалией элиминируются пренатально, поэтому полиплоидию можно обнаружить в основном у человека в материале спонтанных абортов. Единичные случаи триплоидии описаны у живорожденных детей, которые
погибали в первые дни после рождения.
Анеуплоидия – это увеличение или уменьшение числа хромосом в связи с утратой одной хромосомы (2n-1) или с наличием добавочной хромосомы (2n+1). Хромосомный набор (2n-1)=45 называется моносомией, а (2n+1)=47 – трисомией. У человека возможно наличие двух или трех добавочных хромосом в кариотипе – это явление называется полисомией и наблюдается только при аномалиях половых хромосом. К анеуплоидии относится и хромосомный
24
мозаицизм – явление, при котором у одного пациента имеются клетки с разным набором хромосом (45 и 46, 45 и 47 и др.). Полные моносомии и трисомии у больных образуются в результате нерасхождения хромосом в гаметогенезе их родителей и являются, главным образом, следствием новой мутации. Это связано с первичным нерасхождением хромосом, когда клетка, вступающая в мейоз, имеет нормальный кариотип (46 хромосом). В редких случаях числовые нарушения являются унаследованными в результате вторичного нерасхождения, когда клетка, вступающая в мейоз, имеет трисомию (47 хромосом).
Трисомии являются следствием простого нерасхождения: оно происходит либо в первом, либо во втором делении мейоза.
Полисомии являются следствием двойного или последовательного нерасхождения. Двойное нерасхождение – это одновременное нерасхождение в оогенезе и сперматогенезе, последовательное – нерасхождение сначала в первом, а затем и во втором делениях мейоза.
Хромосомный мозаицизм возникает вследствие нерасхождения или отставания хроматид в анафазе при митотическом делении клеток зародыша в раннем эмбриогенезе, в результате чего у одного индивидуума образуются клетки с разным набором хромосом.
Структурные перестройки (хромосомные аберрации) связаны с нарушением целостности (разрывом) одной или нескольких хромосом. При структурных аномалиях в кариотипе имеет место либо утрата, либо избыток хромосомного материала какой-либо хромосомы. При этом величина и морфология этой хромосомы изменяются по сравнению с ее нормальным гомологом.
Утрата части хромосомы называется делецией (del). Делеции бывают интерстициальными и концевыми. Концевые делеции обоих плеч ведут к образованию кольцевой хромосомы (r).
Удвоение части хромосомы (избыток хромосомного материала)
называется дупликацией (dup).
Одной из причин возникновения делеции или дупликации является неравный кроссинговер.
Сочетание в одной хромосоме делеции одного плеча и дупликации другого приводит к образованию изохромосомы (i), которая содержит в своем составе два одинаковых плеча: оба p-плеча или оба q-плеча. Изохромосома образуется в мейозе в результате поперечного деления центромеры.
Синдромы, обусловленные структурными перестройками, могут быть следствием новой мутации во время гаметогенеза у одного из родителей пробанда, либо быть унаследованными в результате родительской сбалансированной хромосомной перестройки.
Сбалансированная хромосомная перестройка – это такая аномалия кариотипа, при которой хромосомного дисбаланса в клетках нет, но порядок расположения некоторых участков хромосом отличается от исходного, т.е. в пределах одной или нескольких хромосом часть хромосомного материала
25
перенесена в другое место. Фенотип обладателей сбалансированных перестроек нормальный.
К сбалансированным хромосомным перестройкам относятся инверсии и транслокации.
Инверсия (inv) – это поворот участка хромосомы на 180 градусов в результате двух разрывов в ней. Различают перицентрическую и парацентрическую инверсии.
Перицентрическая инверсия – это такая перестройка, при которой разрывы происходят в разных плечах хромосомы и инвертированный участок включает в себя центромеру.
Парацентрическая инверсия происходит в пределах одного плеча. При перицентрической инверсии морфология измененной хромосомы отличается от соответствующего нормального гомолога, а при парацентрической инверсии изменяется лишь рисунок сегментации соответствующего плеча по сравнению с нормальным гомологом.
К транслокациям (t) относятся реципрокные и робертсоновские. Реципрокная транслокация – это взаимный обмен участками между
двумя негомологичными хромосомами в результате разрывов в этих хромосомах. У носителей реципрокных транслокации в кариотипе имеются две структурно измененные хромосомы.
Робертсоновская транслокация (или транслокация типа центрического слияния) – это перестройка, в результате которой две акроцентрические хромосомы вследствие разрывов в центромерах соединяются в одну. В результате у носителя этой перестройки в кариотипе имеется 45 хромосом, так как одна транслокационная хромосома состоит из длинных плеч двух акроцентрических хромосом.
Фенотип пациентов со сбалансированными хромосомными перестройками не изменен, но в результате сегрегации перестроенных хромосом в мейозе у них могут образоваться гаметы с хромосомным дисбалансом, что повлечет за собой появление потомства с хромосомной аномалией. Последняя может явиться причиной спонтанного аборта, мертворождения или рождения ребенка с хромосомным заболеванием.
Частота хромосомных нарушений у человека.
Популяционная частота хромосомных нарушений человека составляет 6 – 7 на 1000 новорожденных. При этом на аномалии в системе половых хромосом приходится 2,6 на 1000, на трисомию аутосом –1,6 на 1000, на структурные перестройки (сбалансированные и несбалансированные) – 2,4 на 1000 новорожденных.
2.1 Синдромы, обусловленные аномалиями в системе аутосом.
Эти синдромы связаны как с числовыми, так и со структурными изменениями хромосом. Из числовых нарушений у живорожденных определяют только трисомии по 21, 18, 13, 8, 9, 22 хромосомам. Трисомии по остальным аутосомам, а также все аутосомные моносомии – это летали и
26
поэтому могут быть обнаружены только у спонтанных абортусов. Трисомии по указанным шести аутосомам являются сублеталями, так как они также могут быть причиной элиминации плода. Структурные перестройки аутосом (делеции и дупликации) в виде частичных моносомий или частичных трисомий известны по длинным и коротким плечам большинства аутосом и являются причиной соответствующих синдромов.
Для аутосомных синдромов характерны множественные и грубые пороки внутренних органов, резкая задержка психомоторного развития, грубый дефект интеллекта в сочетании с многочисленными микроаномалиями (дизморфиями), и, как правило, незначительная продолжительность жизни больных, а также возможна ранняя диагностика.
Синдромы Дауна, Эдвардса и Патау вызваны полной трисомией аутосом. 2.1.1 Болезнь Дауна – трисомия 21. Описан в 1866 г J.Down.
Популяционная частота – 1:700. 94% случаев представлены простой полной трисомией 21, транслокационная форма составляет 4%, мозаичная –2%. Мозаицизм – это одновременное наличие у одного и того же лица клеток с нормальным кариотипом и патологическим. И если при простой трисомии и транслокационном варианте клиническая картина болезни практически неразличима, то при мозаичном варианте выраженность клинических симптомов, в том числе и степень олигофрении, зависит от соотношения нормального и патологического клона клеток: чем меньше процент нормальных клеток с 46 хромосомами, тем более выражена клиническая картина. Это одна из причин обязательного кариотипирования больного синдромом Дауна и его родителей. От цитогенетического диагноза зависит и тактика лечения больного, и медико-генетический прогноз потомства у его родителей. У девочек и мальчиков патология встречается одинаково часто. С увеличением возраста женщины риск рождения у нее ребенка с болезнью Дауна возрастает (в возрасте матери до 20 лет – 1:2325, в возрасте 45 лет и старше – 1:45). В большинстве случаев в семье регистрируют одного больного, в очень небольшом числе семей наблюдаются повторные случаи. Монозиготные близнецы обычно конкордантны, в то время как большинство дизиготных близнецов дискордантны. Мужчины с синдромом Дауна бесплодны, однако описано 17 женщин, у которых были дети (среди детей встречались нормальные; с синдромом Дауна; умственно отсталые без синдрома Дауна; мертворожденные).
Этиология и патогенез.
Чаще всего причиной нерасхождения хромосом в 21-й паре является нарушение оогенеза. Лишь в 21-25% случаев возникновение трисомии связано с аномальным сперматогенезом. Предложена модель возникновения трисомии 21 в 1-м мейотическом делении у матери. Согласно этой гипотезе, снижение уровня гормонов ведет к более медленному протеканию мейоза и уменьшению числа хиазм. Преждевременная терминализация хиазм способствует раннему разделению бивалентов, причем мелкие хромосомы, в частности 21, имеют высокий шанс разделиться раньше срока.
27
Причина умственной отсталости до сих пор не ясна. В патогенезе умственной отсталости при трисомии 21 первостепенное значение придается онтогенетической незрелости ЦНС, в частности, недостаточной миелинизации нервных волокон. В последние годы установлено важное значение производных фолиевой кислоты, которые участвуют в цикле монокарбоновых кислот и через них – в синтезе метилаз, необходимых для миелинизации нервных волокон и правильного функционирования ацетилхолинэстеразы в качестве нейромедиатора. Согласно существующим гипотезам в патологический процесс вовлекаются не только гены 21 хромосомы, но и гены других хромосом. При этом происходят вторичные нарушения хромосомного баланса, ведущие к апоптозу и потере нейронов. Причем морфологические отклонения в формировании головного мозга фиксируются уже в пренатальном периоде. А умственная отсталость ассоциируется с процессами нейродегенерации. Возникновение процессов нейродегенерации объясняют нарушениями синаптических связей и биохимическими отклонениями от нормы. Согласно современным представлениям апоптоз обусловлен локальными изменениями в синапсах, которые приводят к нарушениям обмена бета-амилоида (предшественник протеина АРР), Cu, Zn супероксидазной дисмутазы (SODI). Раннее начало деменции связывают с аполипопротеинами, дефицитом эстрогена и высоким уровнем абета–1-42- пептидом.
Это четко очерченное состояние, несмотря на значительную изменчивость отдельных признаков. В целом, высокая изменчивость фенотипических проявлений характерна для всех хромосомных синдромов. У опытного клинициста диагноз редко вызывает сомнение (рис. 6). Но, несмотря на внешнюю узнаваемость синдрома, диагноз всегда требует цитогенетического подтверждения. При рождении у больных с синдромом Дауна отмечается некоторая тенденция к пренатальной гипотрофии. В период новорожденности надежными диагностическими признаками являются: плоский профиль лица, монголоидный разрез глаз, эпикант, колобомы, плоская переносица, глазной гипертелоризм, деформация и уменьшение ушных раковин, макроглоссия, короткая шея с избыточной кожей, поперечная складка на ладонях.
28
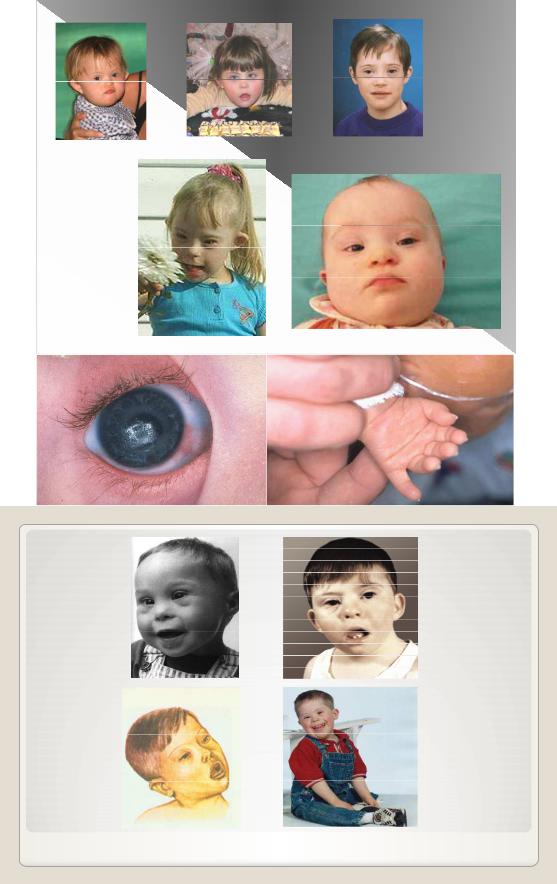
Рис. 6. Фенотипические проявления болезни Дауна
29
Как правило, имеется выраженная мышечная гипотония в сочетании с разболтанностью суставов, отсутствует рефлекс Моро. Сочетание клинических симптомов или значительной их части с глухим голосом и плохо развитой речью, а, самое главное, с врожденным слабоумием делает диагноз синдрома Дауна несомненным. Помимо поражения центральной нервной системы, ведущего к умственной недостаточности, в патологический процесс вовлекаются многие другие органы и системы. Из пороков внутренних органов часты врожденные пороки сердца (40-60%), желудочно-кишечного тракта в виде атрезии двенадцатиперстной кишки, слепой кишки, ануса (12%); эндокринной системы (патология щитовидной железы) –35%, опорнодвигательного аппарата (до 80%), тугоухость (40-70%), патология зрения (глаукома, катаракта, частые конъюнктивиты), иммунологическая недостаточность, проявляющаяся в частых инфекционных заболеваниях. Нередко синдром Дауна сочетается с лейкозом.
Однако главным симптомом, определяющим инвалидность с детства, является олигофрения разной степени тяжести (дебильность, имбецильность, идиотия). В связи с чем длительное время это заболевание называлось «монголоидная идиотия». Многие годы генез этого заболевания был неясен, его связывали с нарушениями эмбриогенеза. Делались попытки связать это заболевание с тератогенезом. Только в 1959 г. синдром Дауна был отнесен к хромосомным болезням, когда французскими цитологами (Lejeune, Turpine, Gautier) было доказано, что в его основе лежит наличие одной дополнительной хромосомы.
Ежегодно в России рождается около 2400 детей с синдромом Дауна. Продолжительность жизни больных снижена: 30% из них погибают в конце первого года жизни, 45% - в конце третьего года. В 20 раз повышен риск смерти от острого лейкоза, причины которого неизвестны. Существуют три гипотезы: высокий риск анеуплоидии, связанный с митотическими нарушениями в стволовых клетках крови, сниженная резистентность к лейкозогенным вирусам и, как показывают экспериментальные данные, низкая эффективность системы репарации. Являясь инвалидами с детства, эти дети нуждаются в пожизненном уходе, реабилитации, медицинской коррекции. Постоянный контакт с ребенком, обучение его простым элементам самообслуживания (одевание, овладение гигиеническими навыками, исполнение несложных поручений и т.п.) позволяет адаптировать ребенка к жизни в обществе. По своему характеру такие дети доброжелательны, улыбчивы, проявляют нежность к своим родителям и обслуживающему персоналу. Еще И.И. Штильбанс (1965) отмечал, что «дети с болезнью Дауна ласковы, добродушны, послушны, но временами упрямы; они пугливы, любопытны и любят подражать окружающим, благодаря чему их можно приучить помогать окружающим по хозяйству, одеваться, однако к систематическому труду они не способны. Несложные житейские навыки обычно усваиваются многими из них».
30