

Genomics: The Science and Technology Behind the Human Genome Project. |
Charles R. Cantor, Cassandra L. Smith |
|
Copyright © 1999 John Wiley & Sons, Inc. |
|
ISBNs: 0-471-59908-5 (Hardback); 0-471-22056-6 (Electronic) |
9 Enhanced Methods for Physical Mapping
WHY BETTER MAPPING METHODS ARE NEEDED
In Chapter 8 we described the original top-down and bottom-up approaches that have led to the construction of a fair number of macrorestriction maps and ordered libraries. These methods are quite laborious, and it would be difficult to replicate them on very large numbers of mammalian genomes. New methods will be needed that are much more powerful
if we are ever to be able to explore the full range |
of evolutionary diversity and the full |
range of human diversity by genome analysis. It seems |
fairly clear that in the future we |
will want the ability to go into any individual genome |
and obtain samples suitable for se- |
quencing of large contiguous blocks of DNA. At least from the present perspective, this could require prior mapping studies to prepare the samples needed for subsequent sequencing. The key will be to develop approaches that allow a rapid focus on a particular region of interest and then a rapid collection and ordering of samples suitable for direct sequencing. It would be especially desirable if methods could eventually be developed that focus directly on map differences between individuals or species. Many future studies will doubtlessly be interested only in differences between two otherwise fairly homologous samples. Today techniques for effective differential mapping are unknown, and we
will largely focus instead on methods for making direct mapping approaches much more efficient.
LARGER YEAST ARTIFICIAL CHROMOSOMES (YACs)
YACs are a major tool currently used for making ordered libraries of large insert clones. The basic design and generation of YACs was described in Chapter 8. A major issue with YACs has been the size of the DNA insert. The first YAC libraries made had average insert sizes of 200 to 300 kb. This is a vast improvement over cosmid clones when used in schemes for rapid walking. However, since the first libraries, the sizes of YACs have con-
tinued to grow steadily. At |
least two improvements in YAC design have assisted this. |
Early on, in YAC development |
it was noted that mammalian DNA was not always rich in |
sequences that could serve serendipitously as yeast replication origins (autonomously replicating sequences). The original YAC vectors had only a single origin in one arm of the YAC vector. Requiring that this origin replicate the entire chromosome places potentially severe kinetic constraints on the viability of the chromosome. This problem can be alleviated considerably by building authentic YAC origins into both vector arms.
A second technique for increasing |
the |
size of YAC inserts has been to |
size-fractionate |
the DNA to be cloned both before and |
after ligation of YAC vector arms. The ligation is |
||
normally carried out in a melted agarose |
sample. Under these conditions |
Mb DNA is |
|
285
286 |
ENHANCED METHODS FOR PHYSICAL MAPPING |
|
|
quite |
susceptible to shear breakage, which increases as |
the square |
of the length of the |
DNA. Any DNA that is fragmented by shear breakage or |
nuclease contamination during |
||
the ligation procedure, and also any contaminating vector arms, will be eliminated by the |
|||
second |
size-fractionation. This is important because, otherwise, large |
numbers of vector |
|
arms will contaminate the true YACs. Since these carry the selectable markers, and can recombine with yeast chromosomal DNA, they lead to a high background of useless clones. Several groups have reported the construction of YAC libraries with average insert sizes of 500 to 700 kb and even larger. However, the greatest success has been seen with a continuing effort at Genethon to make larger and larger YACs. This has resulted in a se-
ries of libraries with average insert sizes of 700 kb, 1.1 Mb, 1.3 Mb, and 1.4 Mb. The largest insert libraries resulted from an extensive effort at Genethon. These had average
insert sizes in excess of 1 Mb. The protocols for producing these megaYACs do not seem to be reproducible at this stage. Instead, by having the same team concentrate on the repeated construction of YAC libraries, the quality of these libraries appeared to improve on
average, for unknown reasons |
as the team gained more experience. All of these libraries |
||||||||
are made from PFG-fractionated |
|
|
Eco |
R I partial digests of genomic DNA. Usually DNA is |
|||||
transformed into yeast by electroporation. |
|
|
|
|
|
|
|
||
The major problem with megaYACs (and most YAC libraries for that matter) are re- |
|||||||||
arranged clones. These include chimeric clones, clones with deletions, and clones with in- |
|
||||||||
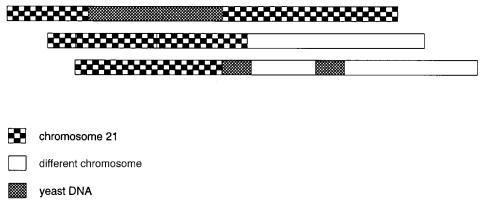
sertions of yeast DNA. These are illustrated in Figure 9.1. Deletions and yeast insertions |
|||||||||
make it difficult to use the YACs directly |
as DNA sources for finer mapping or sequenc- |
||||||||
ing. However, such clones are still useful for the kinds of mapping strategies we will de- |
|||||||||
scribe later in this chapter. Chimeric clones |
are more of a problem because they can lead |
||||||||
to serious errors in mapping if they are not detected. The chimeric clones appear to con- |
|||||||||
tain two or more disconnected genomic regions. In some YAC libraries more than 50% of |
|||||||||
the clones are chimeras. |
|
|
|
|
|
|
|
|
|
There are two potential origins for |
the |
occurrence |
of chimeras. Some |
may |
arise |
||||
during ligation, especially |
if the |
insert |
DNA |
is |
at |
too |
high a concentration relative to |
||
the amount of YAC arms present. Co-ligation can be reduced substantially by more com- |
|
||||||||
plex cloning strategies than |
used in |
early YAC |
library |
construction (Wada et |
al., |
1994). |
|||
Figure 9.1 |
Some common artifacts in YAC cloning: chimeras, deletions, and insertion of yeast se- |
quences. |
|
|
|
|
LARGER YEAST ARTIFICIAL CHROMOSOMES (YACs) |
|
287 |
For example, suppose that genomic DNA is |
partially digested with |
Mbo |
I in agarose, |
||
which produces fragments ending in |
|
|
|
|
|
|
|
|
5 ——— |
|
|
|
|
|
3 ———CTAG |
|
|
The single-stranded ends of the resulting sample are then partially filled in treatment with |
|
|
|||
Klenow fragment DNA polymerase and dpppA and dpppG only. The resulting genomic |
|
|
|||
DNA fragments will then have ends like |
|
|
|
||
|
|
|
5 ————GA |
|
|
|
|
|
3 ————CTAG |
|
|
and thus they cannot be ligated to each other. In parallel, the YAC cloning vector is di- |
|
|
|||
gested to completion with |
Bam |
H I to generate telomeres (see Box 8.1) and then digested |
|
|
|
with |
Sal I to yield fragments that end in |
|
|
|
|
|
|
|
5 ————G |
|
|
|
|
|
3 ————CAGCT |
|
|
The |
ends of these are then partially |
filled in |
by treatment with Klenow DNA polymerase |
|
|
in the presence of only dpppT and dpppC. This yields fragments ending in
5 ———-GTC
3 ———-CAGCT
Now the vector arms produced cannot ligate to each other, but they are still capable of
ligating to the genomic DNA fragments prepared as described above. |
|
|
|
||||
A second source |
of chimeras will arise from recombination. |
In preparing |
yeast for |
||||
DNA transformation, |
usually a |
small fraction of |
the cells |
is rendered competent |
to |
pick |
|
up DNA, and it is not at all |
uncommon for these |
cells to |
pick up |
several YACs. |
Usually |
||
the YACs will separate in subsequent cell divisions. Occasionally a cell will stably maintain two different YACs. However, since mitotic recombination is very prevalent in yeast, two different YACs can recombine at shared DNA sequences, and as a result two chimeric
daughters are produced. If each of these retains a centromere, |
they will usually segregate |
to separate daughter cells, each of which will now maintain |
a different single chimeric |
YAC (Fig. 9.2). Evidence for such recombination between two YACs has been obtained in at least one case where the two original inserts corresponded to DNA of known sequence, and thus the site of the recombination event could be identified. Alternatively, a dicentric YAC and an acentric YAC could be produced by a recombination event. In this case the latter clone will be lost, and the former may break unless one of the centromeres becomes inactivated.
Human DNA is likely to be a very favorable recombination target in yeast because of the large amount of interspersed repeated DNA sequences. This can also lead to instabilities within a single YAC which may lose part of its insert by an intramolecular recombination event. Just how prevalent these rearrangements are in particular libraries is not
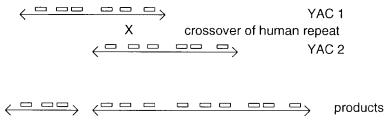
288 ENHANCED METHODS FOR PHYSICAL MAPPING
Figure 9.2 Recombination between human repeated sequences (shown as boxes) as a mechanism for the production of chimeric YACs.
known, but there are reasons to think that these are serious problems. The yeast strains used for almost all current YAC library construction have not had their recombination functions disabled. When a recombination deficient yeast host was used, a dramatic de-
crease in the fraction of chimeras was observed (Ling et al., 1993). Additional indirect evidence that recombination and not co-cloning is the major cause of chimeric YACs comes
from |
observations on libraries made from hybrid cell lines. In most |
of these cases, the |
amount of chimerism is very low. This presumably arises because the |
human DNA is |
|
much |
more dilute in these samples, and human-rodent recombination is |
much less effi- |
cient because most repeated sequences are not well conserved between the two species. |
||
|
When YACs are prepared directly from DNA obtained from flow-sorted chromosomes, |
|
the frequency of chimeras is also quite low. This is partly a result of the low DNA concentrations, which will diminish coligation and recombination events. However, the decreased chimera frequency also is likely to reflect the fact that in these samples all of the DNA preparation and manipulation was carried out in agarose. Agarose will reduce the number of DNA fragments with broken, unligatable ends, which are highly recombino-
genic in yeast.
HOW FAR CAN YACs GO? |
|
|
|
Larger insert clones facilitate mapping projects |
in |
several ways. The number of clones |
|
one needs to order to fill the |
minimum tiling path |
is |
reduced. This greatly simplifies the |
process of clone ordering. If one has a fingerprinting method that requires a certain absolute amount DNA for demonstrating an overlap, the larger the clone the smaller a fraction of the clone this amount represents. Finally larger clones can easily be used to order clones one-half to one-third their size. Thus, as we will describe later, by having a tiered set of samples, the whole process of ordering them is greatly facilitated. The limit of the
tier is determined by the largest stable and reliable clones that are |
available. |
|
|
|
|||
The |
chromosomes of |
S. cerevisiae |
range |
in size |
from |
250 |
kb to 2.4 Mb. Thus the |
largest |
current YACs |
are in the midsize range |
of yeast chromosomes. |
It is |
not |
clear |
|
whether there is a size limit to yeast chromosomes. One issue already mentioned is the frequency of replication origins in the insert DNA. Early studies with artificial chromosomes in yeast indicated that stability, measured as retention over many generations of growth, actually increased sharply as a function of size, but these studies were not carried out up to the size range of current megaYACs. At some point the amount of foreign DNA that any organism can tolerate becomes limited by its competition for binding of key cellular enzymes or regulatory proteins. Where this limit occurs in yeast is unknown.
|
|
|
|
|
|
|
|
HOW |
FAR |
CAN |
YACs GO? |
289 |
Some hints |
that |
S. |
cerevisiae |
can tolerate really large amounts of foreign DNA are |
|
|||||||
available from studies with amplifiable YACs. The basic scheme behind such cloning vec- |
|
|
|
|
|
|||||||
tors is shown in Figure 9.3 |
a. Yeast centromeres can be inactivated if transcription occurs |
|
||||||||||
across them. To take advantage of this effect, a YAC vector arm has been designed with a |
|
|
|
|
||||||||
strong, regulatable promoter extending toward a centromere. This vector is used to trans- |
|
|
|
|
||||||||
form DNA into yeast in the ordinary way. The yeast is allowed to grow in the presence of |
|
|
|
|
|
|||||||
selectable markers on the vector arms, first in the absence of transcription from the regu- |
|
|
|
|
||||||||
latable promoter. Then the promoter is activated, and growth is continued in the presence |
|
|
|
|
|
|||||||
of selection. What happens is that with centromeres inactivated, the YACs segregate un- |
|
|
|
|
||||||||
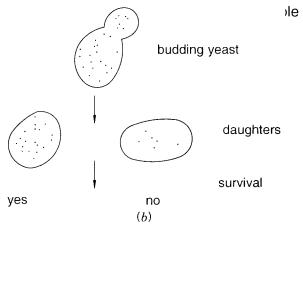
evenly into daughter cells (Fig. 9.3 |
|
b ). Those daughters that receive many copies of the |
|
|||||||||
YACs have a selective advantage; those that receive very few copies are killed by the se- |
|
|
|
|
||||||||
lection. The result is to increase, progressively, the average number of YACs per viable |
|
|
|
|
||||||||
cell. This process continues up to the point |
where there are 10 to 20 copies of each YAC |
|
|
|
||||||||
per cell. At this point the YAC DNA is 20 to 40% of the entire yeast DNA. This technique |
|
|
|
|||||||||
appears to be very promising, for it produces cells that are much easier to screen by hy- |
|
|
|
|
||||||||
bridization than ordinary single-copy YACs. However, it has not yet been applied to li- |
|
|
|
|
||||||||
braries of megaYACs. |
|
|
|
|
|
|
|
|
|
|
||
One potential improvement over standard YAC cloning methods might come from the use |
|
|
|
|
|
|||||||
of S. pombe |
rather |
than |
S. cerevisiae. |
The former yeast |
has about the |
same |
genome |
size |
as |
|
||
the latter. However, |
S. pombe |
has only three chromosomes that range in size from 3.6 to 5.8 |
|
|||||||||
Mb. Based solely on this observation, it seems reasonable to speculate that |
|
|
|
|
S. pombe |
ought to |
||||||
be able to accommodate large YACs if there were a way to get them into the cell. One poten- |
|
|
|
|
|
|||||||
tial complication is that the centromeres of |
|
S. pombe |
and |
S. cerevisiae |
are very different |
in |
||||||
size. |
S. cerevisiae |
has a |
functional |
centromere |
that covers only a |
few hundred |
base pairs. |
|
|
|
||
Figure 9.3 |
Amplifiable YACs. ( |
a ) Vector |
used that allows regulation |
of |
centromere function. ( |
b ) |
Random segregation in mitosis leads to selective |
survival of |
cells with large numbers |
of |
YACs |
|
|
(dots). |
|
|
|
|
|
|
290 |
ENHANCED METHODS FOR PHYSICAL MAPPING |
|
|
|
|
|||||
In contrast, while the irreducible minimum centromere of |
|
|
|
|
S. pombe |
is unknown, past experi- |
||||
ence suggests that it could well be in excess of 100 kb. This would make the construction of |
|
|||||||||
cloning vectors for use in |
|
S. pombe |
very cumbersome. |
|
|
|
|
|||
An alternative cloning system for large DNA where considerable recent progress has |
|
|||||||||
been made uses bacterial artificial chromosomes or BACs (see Box 8.2). This system em- |
|
|||||||||
ploys single-copy |
E. coli |
F-plasmids as vectors for DNA inserts. Natural F-factors can be |
||||||||
a megabase in size. Thus BACs ought to have capacities in this size range if the resulting |
|
|||||||||
DNAs can be transfected into |
|
E. coli |
efficiently. The first BACs were rather small with in- |
|||||||
serts mostly in the 100to 200-kb size range. However, recently larger BACs, with sizes |
|
|||||||||
from greater than 300 kb, have been reported. It remains to be seen how much this range |
|
|||||||||
can be enhanced by further modification and protocol optimization. BACs have the intrin- |
|
|||||||||
sic advantage that the background |
|
|
E. coli |
DNA |
is only a |
third that of background yeast |
||||
DNA. It is also relatively easy |
to purify the BAC DNA away from the host genomic |
|
||||||||
DNA. |
Powerful bacterial |
genetic |
procedures can be used to manipulate the BAC se- |
|
||||||
quences in vivo, and it is fair to say that comparable |
procedures can be used to manipu- |
|
||||||||
late YACs within their host cells. A key feature for both systems is that we now know the |
|
|||||||||
entire DNA sequence of both |
|
E. coli |
and |
S. cerevisiae. |
|
Procedures are likely to be devel- |
||||
oped and in place soon for direct DNA sequencing of large insert clones in one or both of |
|
|||||||||
these organisms. By knowing all of the host DNA sequence ahead of time, it will be pos- |
|
|||||||||
sible to design sequencing or PCR primers in an intelligent, directed way. Any accidental |
|
|||||||||
host sequence that results from primer errors or homology will be immediately apparent |
|
|||||||||
once the putative clone sequence is compared with that of its host genome. Another bac- |
|
|||||||||
terial large DNA cloning system that is in widespread use is the P1 artificial chromosome |
|
|||||||||
(PACs), described in Box 2.3. |
|
|
|
|
|
|
|
|
||
VECTOR OBSOLESCENCE |
|
|
|
|
|
|
|
|
||
Based on past experience with ordinary recombinant |
DNA |
procedures |
over |
the |
past |
|
||||
decade, the highly desirable vector |
of today is an inefficient, undesirable vector tomor- |
|
||||||||
row. There is no way we can predict what the optimal vectors will be like five years from |
|
|||||||||
now; what bells and whistles they must have to facilitate the rapid mapping and sequenc- |
|
|||||||||
ing procedures that will then be in use. To demonstrate how cloudy our crystal ball is in |
|
|||||||||
this respect, within five years the development of rapid methods to screen clones for pos- |
|
|||||||||
sible functions is very likely. Just what form these |
screens will take, and what require- |
|
||||||||
ments they will impose on cloning vectors, are entirely unknown. |
|
|
|
|
|
|||||
Imagine that tomorrow a vast improvement in some cloning vector has been achieved. |
|
|||||||||
All of the current map data and samples do not use this vector. How will we transfer the |
|
|||||||||
enormous number of samples used in |
genomic mapping from yesterday’s obsolete vec- |
|
||||||||
tors to the new ones? Certainly it will not be efficient to do this clone by clone. New |
|
|||||||||
strategies are badly needed that allow flexibility in the handling of samples to allow mass |
|
|||||||||
recloning or rescreening of entire ordered or partially ordered libraries to retain useful or- |
|
|||||||||
der information but equip the clones with the newly desired features. It is fair to say that |
|
|||||||||
today, while the problem is recognized, creative solutions to it are still lacking. We will |
|
|||||||||
either need clever selection, very effective automation for large numbers of separate sam- |
|
|||||||||
ples, |
or very effective multiplexing |
that will |
allow many samples to be |
handled |
together |
|
||||
and then sorted out in some very simple way afterward.
|
HYBRID MAPPING STRATEGIES: CROSS-CONNECTIONS BETWEEN LIBRARIES |
291 |
||||
One |
consequence of |
the |
virtual certainty of |
vector obsolescence is that it |
is desirable |
|
to minimize the numbers of samples archived for storage and subsequent redistribution. |
|
|||||
Instead, it seems more efficient to develop procedures that will allow desired clones to be |
|
|||||
pulled easily from whatever new libraries are made. The advantage of PCR-based ap- |
|
|||||
proaches is obvious in this regard. These approaches require storing only DNA sequence |
|
|||||
information that allows primers to be made whenever they are needed to assay for a given |
|
|||||
sequence in a sensitive |
way. Whatever library a |
desired DNA sequence is in, |
it should |
|
||
then be possible to find |
it in a relatively quick and inexpensive way, by PCR assays on |
|||||
pools of clones or hybridization assays on arrays of clones from that library, as we will |
|
|||||
describe later in this chapter. In this way no large numbers of samples need be stored for |
|
|||||
long time |
periods nor |
for |
mass distribution. |
Only pools of samples will |
have to |
be |
archived. |
|
|
|
|
|
|
HYBRID MAPPING STRATEGIES: CROSS-CONNECTIONS
BETWEEN LIBRARIES
In any physical mapping project, sooner or later there is the need to handle a number of
different types of samples. These include cell lines, radiation hybrids, |
and large restric- |
||
tion fragments for regional assignments and gaining an overview, as well as various clone |
|||
libraries such as megaYACs, YACs, P1, and cosmid clones that actually form the eventual |
|||
basis for DNA sequencing. Past projects have tended to concentrate on ordering at most a |
|||
few of these samples across |
the chromosome, and then they resorted to using |
other types |
|
of samples in selected regions where these were needed to address particular problems. |
|||
Based on these |
experiences, |
it now seems evident that much of the labor in handling all |
|
of these types of samples is preparing a dense set of labeled DNA probes or PCR primers |
|||
(e.g., STSs |
vide infra |
) that are needed for most fingerprinting or mapping activities. |
|
Once one has such probes or primers, an attractive scheme for ordering them is shown |
|||
in Figure 9.4. The labeled probes or primers are used to interrogate, simultaneously, all |
|||
samples that are of potential interest for the chromosome of question. If |
a dense enough |
||
set of probes |
or primers exists, and if the clone libraries are highly |
redundant, we will |
|
show that the result of the interrogation should be to order all the samples of interest in |
|||
parallel. Ordering by any of the methods currently at our disposal involves finding over- |
|||
lap information. The larger the number of different samples used in the |
overlap proce- |
||
dure, the more likely one or more will cover the key region needed to form an informative |
|||
overlap. This approach is neither top down or bottom up; in most respects it combines the |
|||
best features of the two extreme views of map construction. They key issue is how to im- |
|||
plement such a |
strategy in |
an efficient way. There are three basic issues: how to handle |
|
the probes, how to handle the samples, and how to do the interrogation. |
|
||
Tens of thousands of DNA samples are involved in most large-scale mapping efforts. |
|||
These cannot be handled routinely as individual liquid DNA preparations. One viable ap- |
|||
proach is to make dense arrays of DNA spots on filters. As an example consider the filter
shown schematically in Figure 9.5. It consists of 2 |
104 individual cosmids. This must |
|
be prepared by an automated arraying device (Box 9.1). The key fact is that once the sam- |
||
ples are prepared, the device can make as many copies of the filter as needed with rela- |
||
tively little additional effort. If each cosmid has an average of 4 |
104 base pairs of DNA, |
|
the array represents a total of 8 |
108 base pairs. This is |
a fivefold redundant coverage of |
292 ENHANCED METHODS FOR PHYSICAL MAPPING
cross connections between DNA samples
radiation hybrids
restriction fragments
large insert clones
small insert clones
probe hybridization, PCR, or fingerprinting
Figure 9.4 Mapping by making cross-connections between a set of different DNA samples. The key variables are the density of available probes and the density or coverage of available samples.
an average, 150 Mb, human chromosome. It is sufficient for most mapping exercises that one can contemplate. For example, hybridization of a labeled probe to the filter will identify any cosmids that contain corresponding DNA sequences. All the cosmids can be examined in a single experiment, and if the signal to noise in the hybridization is good, the resulting data should be fairly unequivocal. We assume that any repeated DNA sequences
in the probe will be competed out by methods described earlier in the book.
The alternative approach to making arrays is to make pools of samples. This has the potential advantage that the DNA is handled in homogeneous solution, which facilitates
some screening procedures like PCR. It also |
has the advantage |
that a |
relatively small |
number of pools can replace a very large number of |
individual samples |
or spots |
on an ar- |
ray. Procedures for constructing these pools intelligently will be a major theme of this chapter. However, regardless of how they are made, a disadvantage of pools is that a sin-
gle interrogation will not usually identify unique clone targets identified by a probe. Instead, one usually has to perform several successive probings or PCRs in order to deter-
mine which elements of particular pools |
were responsible |
for |
positive signals generated |
by the probe. |
|
|
|
Hundreds to thousands of probes or |
primers are used |
in |
a large-scale mapping effort. |
The complexity of these samples is almost as great as that of the clones themselves. In the past most probes or primer pairs have been handled individually. Probes have consisted of small DNA clones, cosmids, YACs, large DNA fragments, or radiation hybrids. It is necessary to compete out any repeated DNA in these probes to prevent a background level of hybridization that would be totally unacceptable. PCR tricks abound that can be used to

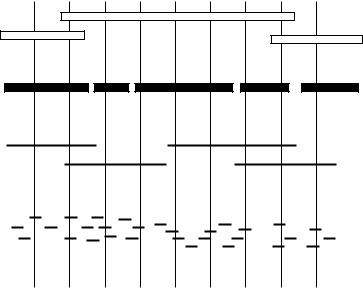
Figure 9.5 |
|
An example of |
a dense sample array. Cosmid clones from |
a chromosome |
19-specific |
|
|
library were ( |
a ) arrayed by 36-fold compression from 384-well microtitre plates and ( |
b ) probed by |
|||||
hybridization |
with a randomly |
primed pool of five cosmids. (Unpublished work |
of A. Copeland, J. |
|
|||
Pesavento, |
R. Mariella, |
and |
D. Masquelier of LLNL. Photographs kindly |
provided by |
Elbert |
||
Branscomb.) |
|
|
|
|
|
|
|
293

294 ENHANCED METHODS FOR PHYSICAL MAPPING
BOX 9.1
AUTOMATED MANIPULATION OF SAMPLE ARRAYS
A considerable background of experience exists in automated handling of liquid sam- |
|
||||
ples |
contained |
in microtitre plates. While it is by no means clear that this is |
the opti- |
|
|
mal format for mapping and sequencing automation, the availability of laboratory ro- |
|
||||
bots |
already |
capable of |
manipulating these plates has resulted in most |
workers |
|
adopting this format. A typical microtitre plate contains 96 wells in an 8 by 12 format, |
|
||||
which is about 3 |
5 cm in size (Fig. 9.6 bottom). Each well holds about 100 |
l (liq- |
|||
uid) of sample. Higher-density plates have recently become available: An 18 by 24 |
|
||||
sample plate, the same size as the standard plate with proportionally smaller sample |
|
||||
wells, seems easily adapted to current instruments and protocols (Fig. 9.6 top). A four- |
|
||||
fold |
higher-density 36 |
by 48 sample plate has been made, but it is not yet |
clear that |
|
|
many existing robots have sufficient mechanical accuracy and existing detection systems sufficient sensitivity to allow this plate to be adopted immediately.
Figure 9.6 Typical microtitre plates: 384-well plate (top), 96-well plate (bottom).
(continued)