

|
HYBRID MAPPING STRATEGIES: CROSS-CONNECTIONS BETWEEN |
LIBRARIES |
295 |
||
BOX |
9.1 |
(Continued) |
|
|
|
|
Liquid-handling robots such as the Beckman Biomec, the Hewlett-Packard Orca |
|
|||
chemical robot, among others, can use microtitre plates singly or several |
at |
a time. |
|
||
More complex |
sets of plates can be handled by storage and retrieval |
from |
vertical |
|
|
racks called microtitre plate hotels. A number of custom-made robots have also been |
|
||||
built to handle specialized aspects of microtitre plate manipulation efficiently, such as |
|
||||
plate duplication, custom sample pooling, and array making from plates. All of these |
|
||||
instruments share a number of common design features. Plate wells can be filled or |
|
||||
sampled singly |
with individual pipetting devices, addressable in the |
|
|
x-yplane. Rows |
|
and columns can be sampled or fed by multiple-headed pipetors. Entire plates can be |
|
||||
filled in a single step by 96-head pipetors. This is done, for instance, when all the wells |
|
||||
must |
be filled with the same sample medium for cell growth or the same buffer solu- |
|
|||
tion for PCR.
|
Most standard biochemical and microbiological manipulations can be carried out in |
|
|||||||||||||||||||||
the wells of the microtitre plates. A sterile atmosphere can be provided in various ways |
|
||||||||||||||||||||||
to allow colony inoculation, growth, and monitoring by absorbance. Temperature con- |
|
|
|||||||||||||||||||||
trol allows incubation or PCR. Solid state DNA preparations such as agarose plug |
|||||||||||||||||||||||
preparations of large DNA, or immobilized magnetic microbead-based preparations of |
|
|
|||||||||||||||||||||
plasmid DNA or PCR samples are all easily adapted to a microtitre plate format. The |
|||||||||||||||||||||||
result is that hundreds to thousands of samples can be prepared at once. Standard liq- |
|
||||||||||||||||||||||
uid phase preparations of DNA are much more difficult to automate in the microtitre |
|
||||||||||||||||||||||
plate format because they usually require centrifugation. While centrifuges have been |
|
||||||||||||||||||||||
built that handle microtitre plates, loading and unloading them is tedious. |
|
|
|
|
|
||||||||||||||||||
|
For |
microbiological |
samples, |
automated |
colony |
pickers |
have |
been |
built |
that |
|||||||||||||
start |
with |
a |
conventional |
array |
of |
clones |
or |
plaques |
|
in |
an |
ordinary petri |
dish |
(or |
|||||||||
a rectangular dish for more accurate mechanical positioning), optically |
detect the |
||||||||||||||||||||||
colonies, pick them one at a time by poking with a sharp object, and transfer them to a |
|
||||||||||||||||||||||
rectilinear array in microtitre plates. The rate-limiting step in most automated handling |
|
||||||||||||||||||||||
of |
bacteria |
or |
yeast |
colonies |
appears |
to |
be |
sterilization |
of |
the |
sharp |
object |
used |
||||||||||
for |
picking, |
|
which |
must |
be |
done |
after |
each |
step |
|
to |
avoid |
cross-contamination. |
||||||||||
With liquid handling, cross-contamination can also be a problem in many applications. |
|
||||||||||||||||||||||
Here one has the choice of extensive rinsing of |
the pipet tips between each sample, |
|
|||||||||||||||||||||
which is time-consuming, or |
the |
use |
of |
disposable |
pipet |
tips, |
which |
is |
very |
costly. |
|||||||||||||
As we gain more experience with this type of automation, more clever designs are sure |
|
||||||||||||||||||||||
to emerge that improve the throughput by parallelizing some of the steps. A typical ex- |
|
||||||||||||||||||||||
ample would be to have multiple sample tips or pipetors so that some are being steril- |
|
||||||||||||||||||||||
ized or rinsed off line while others are being used. At present, most of the robots that |
|||||||||||||||||||||||
have been developed to aid mapping and sequencing are effective but often painfully |
|
||||||||||||||||||||||
slow. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dense sample arrays on filters are usually |
made by using offset printing to com- |
|||||||||||||||||||||
press microtitre plate arrays. An example is shown in Figure 9.7. Here a 96-pin tool is |
|||||||||||||||||||||||
used to sample a 96-well microtitre plate of DNA samples, and stamp its image onto a |
|||||||||||||||||||||||
filter, for subsequent hybridization. Because the pin tips are much smaller than the mi- |
|
||||||||||||||||||||||
crotitre plate wells, it is possible to intersperse the impressions of many plates to place |
|||||||||||||||||||||||
all of these within the same area originally occupied by a single plate. With presently |
|||||||||||||||||||||||
available robots, usually the maximum compression |
attained |
is |
16-fold |
(a |
4 |
|
|
4 array |
|||||||||||||||
of 96 well images). This leads to a 3 |
|
|
|
|
5 cm filter area with about 1600 samples. Thus |
||||||||||||||||||
a 10 |
10 |
cm filter can hold about |
10 |
|
|
|
|
|
4 samples, |
more than |
enough for most current |
||||||||||||
(continued)

296 ENHANCED METHODS FOR PHYSICAL MAPPING
BOX 9.1 |
(Continued) |
|
|
|
Figure 9.7 |
|
|
Making a dense array of samples by offset spotting of a more dilute array. |
|||||
applications. Most dense arrays that have been made thus far for mapping projects are |
|||||||||||
random. That is, no information is available about any of the DNA samples at the time |
|
||||||||||
the |
array |
is made. As the map develops, the array becomes much more informative, |
|
||||||||
but the |
|
x, yindexes of each sample have only historical significance. |
|
|
|
|
|||||
|
Once |
a map has been completed, it is convenient to reconfigure the array |
of |
sam- |
|||||||
ples so that they are placed in the actual order dictated by the map. While this is not |
|||||||||||
absolutely necessary, it does allow for visual inspection of a hybridization to be in- |
|||||||||||
stantly interpretable in many cases. There is no difficulty in instructing a robot to re- |
|||||||||||
configure an array. This procedure needs to be done only once, however |
slow |
the |
|
||||||||
process, and then replicas of the new configuration can be made rapidly. The great mo- |
|
|
|||||||||
tivation |
for achieving |
more |
compressed arrays is sample storage. Many |
clones |
and |
||||||
most |
DNA |
samples are stored at low temperature. Large libraries of |
samples |
can |
|||||||
rapidly saturate all available temperature-controlled laboratory storage |
space, |
espe- |
|||||||||
cially if a large number of replicas is made for subsequent distribution. |
|
|
|
|
|||||||
|
|||||||||||
select out human material from hybrid cell lines or to reduce the complexity of a probe to |
|||||||||||
desirable levels. Some of these techniques were described in Chapter 4; |
others |
will be |
|||||||||
dealt with in Chapter 14. It is worth noting that randomly chosen oligonucleotides often |
|||||||||||
make very effective probes for fingerprinting. For example, a randomly chosen 10-mer |
|||||||||||
should detect 1 out of every 25 cosmid clones. A key issue that we will |
address |
later in |
|||||||||
this chapter is whether it is possible to increase the efficiency of handling probes by pool- |
|||||||||||
ing them instead of using them individually. |
|
|
|
|
|||||||
SCREENING |
BY |
PCR |
VERSUS HYBRIDIZATION |
|
|
|
|
||||
A key variable in contemporary mapping efforts is whether the connections between probes |
|
|
|||||||||
and samples are made by hybridization or by PCR. The two techniques have compensating |
|
|
|||||||||
disadvantages |
and |
advantages, |
and |
the choice of which to use will depend on the nature of |
|
||||||
the samples available and the complexity of the mapping project. We will consider these dif- |
|
||||||||||
ferences at the level of the target, the probe, and the organization of the project. |
|
|
|
|
|||||||
The sensitivity of PCR at detecting small amounts of target is unquestionably greater |
|||||||||||
than hybridization. The advantage of being able to use smaller samples |
is |
that |
with |
||||||||
presently |
available methods, |
it |
is much easier to automate the preparation |
of relatively |
|||||||
small DNA samples. It is also cheaper to make such samples. When pools of targets are used, the greater sensitivity of PCR allows more complex pools with larger numbers of samples to be used. What matters for detection is the concentration of the particular target
|
|
|
|
|
|
|
SCREENING |
BY |
PCR |
VERSUS |
HYBRIDIZATION |
297 |
||||||
DNA that will be detected by one method or the other. With PCR this |
concentration |
can |
|
|||||||||||||||
be almost arbitrarily low, so long as there are not contaminants that will give an unaccept- |
|
|
||||||||||||||||
able PCR background. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Hybridization has the advantage when a large number of |
physically |
discrete |
samples |
|
|
|||||||||||||
(or pools) must be examined simultaneously. We have already described the use of dense |
|
|
|
|||||||||||||||
arrays of samples on filters to process in parallel large numbers of targets in hybridization |
|
|
||||||||||||||||
against a single probe. These arrays can easily contain 10 |
|
|
|
|
|
|
|
|
|
4 samples. In comparison, typi- |
|
|||||||
cal PCR reactions must be done in individually isolated liquid samples. Microtitre plates |
|
|||||||||||||||||
handle |
around 10 |
2 |
of these. Devices |
have |
been built |
that |
can |
do |
thousands of |
simultane- |
|
|||||||
ous PCRs, but these are large; with objects on such a scale one could easily handle 10 |
5 |
|||||||||||||||||
|
||||||||||||||||||
samples at once by hybridization. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
At the level of the probe, PCR is more demanding than hybridization. Hybridization |
|
|||||||||||||||||
probes can be made by PCR or by radiolabeling any DNA sample available. Typical PCR |
|
|
|
|||||||||||||||
analyses require a knowledge of the DNA sequence of the target. In contrast, hybridiza- |
|
|||||||||||||||||
tion requires only a sample |
of the DNA that corresponds to one element |
of |
the |
target. |
|
|||||||||||||
PCR primers require custom synthesis, which is still expensive and time-consuming al- |
|
|
||||||||||||||||
though recent progress in automated synthesis has lowered the unit cost of these materials |
|
|
|
|||||||||||||||
considerably. PCR with large numbers of different primers is not very convenient because |
|
|
|
|||||||||||||||
in most current protocols the PCR reactions must be done individually or at most in small |
|
|
||||||||||||||||
pools. Pooling of probes (or using very complex probes) is a powerful way to speed up |
|
|
||||||||||||||||
mapping, as we will illustrate later in this chapter. However, in PCR, primers are difficult |
|
|||||||||||||||||
to pool. The reason is that with increasing numbers of PCR primers, the possible set of |
|
|||||||||||||||||
reactions rises as the square of the number of primers: Each primer, in principle, could |
|
|||||||||||||||||
amplify with any other primer if a suitable target were present. Thus the expected back- |
|
|
||||||||||||||||
ground will increase as the square of the number of primers. The desired signal will in- |
|
|
||||||||||||||||
crease only linearly with the number of primers. Clearly this rapidly becomes a losing |
|
|
||||||||||||||||
proposition. In contrast, it is relatively easier to use many hybridization probes simultane- |
|
|||||||||||||||||
ously, since here the background will increase only linearly with the number of probes. |
|
|
|
|
||||||||||||||
Both PCR and hybridization schemes lend themselves to |
large |
scale |
organized |
|
||||||||||||||
projects, but the implications and mechanics are very different. With hybridization, filter |
|
|||||||||||||||||
replicas of an array can be |
sent out to a set of distant users. However, the power |
of |
the |
|
||||||||||||||
array increases, the more about it one knows. Therefore, for an array to have optimal im- |
|
|
||||||||||||||||
pact, it is highly desirable that all results of probe hybridizations against it be compiled in |
|
|||||||||||||||||
one centrally accessible location. In practice, this lends itself |
|
to schemes in place in |
|
|||||||||||||||
Europe where the array hybridizations are actually done in central locations, and data are |
|
|
||||||||||||||||
compiled there. Someone who wishes to interrogate an array with a probe, mails in that |
|
|
||||||||||||||||
probe to a central site. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
With |
PCR screening, |
the key |
information |
is the |
sequences |
of |
the |
DNA |
primers. |
|
||||||||
This information can easily be compiled and stored on a centrally |
accessible |
database. |
|
|
||||||||||||||
Users simply have to access this database, and either make the primers needed or obtain |
|
|
||||||||||||||||
them from others who have already made them. This allows a large number of individual |
|
|
|
|
||||||||||||||
laboratories to use the map, as it develops, and to participate in the mapping without any |
|
|
||||||||||||||||
kind of elaborate distribution scheme for samples and without centralized experimental |
|
|
||||||||||||||||
facilities. The PCR-based screening approach has thus far been |
|
more |
popular |
in |
the |
|
||||||||||||
United States. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
In the long run it would |
be nice to have a hybrid approach that |
blends the |
advantages |
|
|
|||||||||||||
of both PCR and hybridization. One way to think about this |
|
would be to use small |
|
|||||||||||||||
primers |
or |
repeated |
sequence |
primers |
to reduce |
the |
complexity |
of |
complex |
probes |
by |
|
|
|
||||

298 |
|
ENHANCED METHODS FOR PHYSICAL MAPPING |
|
|
|
|
|
|
|||||
sampling |
either useful portions of pools of probes or |
large insert |
clones |
such as YACs. |
|
||||||||
This would greatly increase the efficiency of PCR for handling complex pools and also |
|
|
|||||||||||
decrease the cost of making large numbers of custom, specific PCR primers. What is still |
|
||||||||||||
needed for an optimal method is spatially resolved PCR. The idea |
is |
to |
have a method |
|
|||||||||
that would allow one to probe a filter array of samples directly by PCR without having to |
|
||||||||||||
set up a separate PCR reaction for each sample. However, in order for this to be efficient, |
|
||||||||||||
the |
PCR products from each element of the array have |
to be kept from |
mixing. While |
|
|||||||||
some progress at developing in situ PCR has been reported, it is not yet clear that this |
|
||||||||||||
methodology is generally applicable for mapping. One nice approach is to do PCR inside |
|
|
|||||||||||
permeabilized cells, and then the PCR products |
are |
retained inside |
the |
cells. However, |
|
||||||||
this procedure, thus far, cannot be carried out to high levels of amplification (Teo and |
|
||||||||||||
Shaunak, 1995). |
|
|
|
|
|
|
|
|
|
|
|||
TIERED |
SETS |
OF |
SAMPLES |
|
|
|
|
|
|
|
|
|
|
In cross-connecting different sets of DNA samples |
and |
libraries, |
it |
is |
helpful |
to |
have |
|
|||||
dense sets of targets that span a range of sizes in an orderly way. The rationale behind this |
|
||||||||||||
statement is illustrated in Figure 9.8. The figure shows an attempt to connect two sets of |
|
||||||||||||
total digest fragments or clones by cross-hybridization or some other kind of complemen- |
|
|
|||||||||||
tary fingerprinting. When large fragment |
|
|
J is used as a probe, it detects three smaller frag- |
|
|||||||||
ments |
a, b,and |
c but does not indicate their order. When the smaller fragments |
a, b,and |
c |
|||||||||
are |
used |
as probes, |
b detects only |
J,which means |
that |
|
b |
is the central small fragment; |
a |
||||
and |
b |
each detect additional flanking larger fragments, so they must be external. A gener- |
|
||||||||||
alization of this argument indicates that it is more efficient if one has access to a progres- |
|
||||||||||||
sion of samples where average sizes diminish roughly by factors of three. Less sharp size |
|
||||||||||||
decreases will mean an unnecessarily large number |
of different sets of samples. Larger |
|
|||||||||||
size decreases will mean that ordering of each smaller set will be too ambiguous. |
|
|
|
||||||||||
|
The same kinds of arguments are applicable in more complex cases where overlapping |
|
|
|
|||||||||
sets of fragments or clones exist. Consider the example shown in Figure 9.9. The objec- |
|
||||||||||||
tive is to subdivide a 500 kb YAC into cosmid clones so that these can be used as more |
|
||||||||||||
convenient sources of DNA for finding polymorphisms or for sequencing. The traditional |
|
|
|
||||||||||
approach to this problem would have been to subclone the purified YAC DNA into cos- |
|
|
|||||||||||
mids. However, this involves a tremendous amount of |
work. The more modern approach |
|
|
|
|||||||||
is to start with a YAC contig flanking the clone of interest. This contig will automatically |
|
||||||||||||
arise in |
the context of producing an ordered YAC |
library. With a fivefold redundant |
array |
|
|||||||||
of YACs, the contig will have typically five members in this region. Therefore, if these |
|
||||||||||||
members |
are used separately for hybridization or fingerprinting, they will divide the re- |
|
|||||||||||
gion into intervals that average about 100 kb in size. Each interval will serve as a bin to |
|
||||||||||||
assign cosmids |
to the |
region. Since the intervals |
are |
less than three times |
the cosmid |
in- |
|
||||||
Figure 9.8 Clone ordering when tiered sets of clones are available.
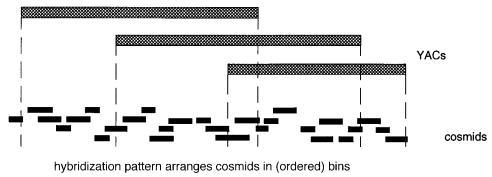
|
|
|
|
TIERED SETS OF SAMPLES |
299 |
sert size, the ordering should be reasonably effective. However, the key point in the over- |
|
||||
all strategy is that one need not make the YAC contig in advance. Cross-connecting the |
|
||||
various sets of samples in parallel will eventually provide all of the information needed to |
|
||||
order all of them. |
|
|
|
||
The example shown in Figure 9.9 is a somewhat difficult case because the size step |
|
||||
taken, from 500 kb clones to 40 kb clones, is a big one. It would be better to have one or |
|
||||
two tiers of samples in between; say 250 kb YACs and 100 kb P1 clones. This will com- |
|
||||
pensate quite a bit for the inevitable irregularities in the distribution and coverage of each |
|
||||
of the libraries in particular regions. An analogy that may not be totally farfetched is that |
|
||||
the intermediate tiers of samples can help strengthen |
the ordering process in the |
same |
|
||
manner as intermediate levels in a neural net can enhance its performance (see Chapter |
|
||||
15). |
|
|
|
|
|
YAC and cosmid libraries involve large numbers of clones, and it would be very ineffi- |
|
||||
cient to handle such samples one at a time. We have already shown that cosmids can be |
|
||||
handled |
very |
efficiently as dense arrays of DNAs on filters. YACs are less easily handled |
|
||
in this manner. The amount of specific sample DNA is much less, since typical YACs are |
|
||||
single-copy clones in a 13-Mb genome background, |
while cosmids are multicopy |
in a |
|
||
4.8-Mb background. Thus hybridization screening of arrays of YAC clones has not always |
|
||||
been very successful. It would greatly help if there were an effective way to purify the |
|
||||
YAC DNA away from the yeast genomic DNA. Automated procedures already have been |
|
|
|||
developed to purify total DNA from many YAC-bearing strains at once (Box 9.1). Now |
|
||||
what is needed is a simple automatable method for plucking the YACs out of this mixture. |
|
||||
An alternative to purification would be YAC amplification. As described earlier in this |
|
||||
chapter, this is possible but not yet widely used, and |
the amount of amplification is |
still |
|
||
only modest. Perhaps the most effective method currently available for increasing our |
|
||||
sensitivity of working with YAC DNA is PCR. |
In Chapter 14 we will illustrate how |
|
|||
human-specific PCR, based on repeating sequence primers like |
Alu ’s, can be used |
to see |
|||
just the human DNA in a complex sample. Almost every YAC is expected to contain mul- |
|
||||
tiple |
Alu |
repeats. It is possible to do hundreds of PCR reactions simultaneously with com- |
|
||
monly available thermal cyclers, and ten to perhaps a hundred times larger number |
of |
|
|||
samples is manageable with equipment that has been designed and built specially for this |
|
||||
purpose. |
|
|
|
|
|
Figure 9.9 |
Division of a YAC into a contig of cosmids by taking advantage of other YACs known |
to be contiguous. |
|

300 |
ENHANCED METHODS FOR PHYSICAL MAPPING |
|
|
|
||||||
SIMPLE |
POOLING |
STRATEGIES |
FOR |
FINDING A |
CLONE |
|
|
|
||
OF INTEREST |
|
|
|
|
|
|
|
|
|
|
A major problem in genome analysis is to find in a library the clones that correspond to a |
|
|||||||||
probe of interest. This is typically the situation one faces in trying to find clones in a re- |
|
|||||||||
gion marked by a specific probe that is suspected to be near a gene of interest. With cos- |
|
|||||||||
mid clones, one screens a filter array, as illustrated earlier. With YAC clones, filter arrays |
|
|||||||||
have not worked well in many hands, and instead, PCR is used. But with today’s methods |
|
|||||||||
it is inconvenient and expensive to analyze the YACs in a large library, individually, by |
|
|||||||||
PCR. For example, a single coverage 300-kb human genomic YAC library is 10 |
4 clones. |
|||||||||
Fivefold coverage would require 5 |
|
|
104 clones. This number of PCR reactions is still |
|||||||
daunting. However, if the objective is to find a small number of clones in the library that |
|
|||||||||
overlap a single probe, there are more efficient schemes for searching the library. These |
|
|||||||||
involve pooling clones and doing PCR reactions |
on |
the |
pools instead of |
on individual |
|
|||||
clones. |
|
|
|
|
|
|
|
|
|
|
One of the simplest and most straightforward |
YAC |
pooling schemes |
involves |
three |
|
|||||
tiers of samples (Fig. 9.10). The YAC library is normally distributed in 96-well microtitre |
|
|||||||||
plates (Fig. 9.6 a ). Thus 100 plates would be required to hold 10 |
|
4 clones. Pools are made |
||||||||
from the clones on each plate. This can be done by sampling them individually or by us- |
|
|||||||||
ing multiple headed pipetors or other tools as described in Box 9.1. Each plate pool con- |
|
|||||||||
tains 96 samples. The plate pools are combined ten at a time to make super pools. First, |
|
|||||||||
ten super pools are screened by PCR. This takes 10 PCR reactions (or 50 if a fivefold re- |
|
|||||||||
dundant library is used). Each positive superpool is then screened by subsequent, separate |
|
|||||||||
PCR reactions of each of the ten plate pools |
it contains. In turn, each plate pool that |
|
||||||||
shows a positive PCR is divided into 12 column pools (of 9 YACs each) and, separately, 9 |
|
|||||||||
row pools (of 12 YACs each), and a separate PCR analysis is done on each of these sam- |
|
|||||||||
ples. In |
most cases |
this should |
result |
in a unique |
row-column positive combination |
that |
|
|||
serves to identify the single positive YAC that has been responsible for the entire tier of PCR amplifications. Each positive clone found in this manner will require around 41 PCR
Figure 9.10 |
Three-tier pooling strategy for finding a clone of interest in a YAC library. |

|
|
|
SEQUENCE-SPECIFIC |
TAGS |
301 |
|
reactions. This is a vast improvement over the 10 |
4 reactions |
required if YACs are exam- |
|
|||
ined one at a time. For a fivefold redundant library, if the super pools are kept the same |
|
|
||||
size, then |
the total number of PCRs needed will be 81 per |
positive clone. This is |
still |
|
|
|
quite reasonable. |
|
|
|
|
|
|
SEQUENCE-SPECIFIC TAGS |
|
|
|
|
|
|
The power of the simple pooling approach just described had a strong effect on early |
|
|
|
|||
strategies developed for genome mapping. In reality the power is greatest when individual |
|
|
|
|||
clones are sought, and it diminishes considerably when the goal |
is to order a whole li- |
|
|
|
||
brary. However, the attractiveness of PCR-based screening has led to much consideration |
|
|
|
|||
about the kinds of PCR primers that would be suitable for genome analysis. Since PCR |
|
|
||||
ordinarily requires known sequences, the issue really becomes what kinds of DNA se- |
|
|
|
|||
quences should be used for finding genes or for genome mapping. A number of different |
|
|
|
|||
types of approaches are currently being used; these are described below. |
|
|
|
|
||
STS. |
This is short for a sequence tagged site. The original notion was that any |
arbi- |
|
|
||
trary bit of known DNA sequence could be used as a probe if it successfully gener- |
|
|
||||
ated useful PCR primers. One early notion was to take all of the existing polymor- |
|
|
||||
phic genetic probes and retro-fit these as STSs by |
determining |
a partial |
DNA |
|
|
|
sequence, and then developing useful PCR primers based on this sequence. This ap- |
|
|
|
|||
proach was never executed on a large scale, which is probably good in retrospect |
|
|
||||
because recently developed genetic markers are far more useful for mapping than |
|
|
|
|||
earlier ones, and the process for finding these automatically includes |
the develop- |
|
|
|||
ment of unique sequence tags suitable for PCR. |
|
|
|
|
|
|
STAR. |
This stands for sequence tagged rare restriction site. The great utility |
of such |
|
|
||
probes has already been described in considering strategies for efficient restriction |
|
|
||||
mapping or ordering total digest libraries. The appeal of STARs is that they allow |
|
|
||||
precise placement of probes on physical maps, even at rather early stages in map |
|
|
||||
construction. The disadvantage of STARs, mentioned earlier, is that many cleavage |
|
|
||||
sites for rare restriction nucleases turn out to be very |
G |
|
C rich, |
and thus PCR |
in |
|
these regions is more difficult to perform. For any kind of probe, clone ordering is most efficient if the probes come from the very ends of DNA fragments or clone in-
serts. As shown in Figure 9.11, this allows PCR to be done by using primers in the vector arms in addition to primers in the insert. Thus the number of unique primers
that must be made for each probe is halved. STARs in total digest libraries naturally come from the ends of DNA fragments, and thus they promote efficient mapping strategies.
Figure 9.11 |
Use of vector primers in PCR to amplify the ends of clones like YACs. |
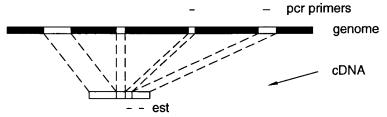
302 |
ENHANCED METHODS FOR PHYSICAL MAPPING |
|
|
|
|
|
|
|||||||
STP |
or STRP. |
|
|
|
These abbreviations refer to sequence tagged polymorphism or poly- |
|
||||||||
|
morphic sequence tag. A very simple notion is involved here. If the PCR tag is a |
|
||||||||||||
|
polymorphic |
sequence, then |
the genetic and physical maps can be |
directly |
aligned |
|
|
|||||||
|
at the position of this tag. This allows the genetic and physical maps to be built in |
|
||||||||||||
|
parallel. A possible limitation |
here is that some of the most |
useful |
genetic probes |
|
|||||||||
|
are tandemly repeating sequences, and a certain subset of these, usually very simple |
|
|
|||||||||||
|
repeats |
like |
(AC) |
|
n , tend to give extra unwanted amplification products in typical |
|||||||||
|
PCR protocols. However, it seems possible to find slightly more complex repeats, |
|
||||||||||||
|
like |
(AAAC) |
n , that are |
equally useful as genetic probes but |
show fewer |
PCR |
arti- |
|||||||
|
facts. |
|
|
|
|
|
|
|
|
|
|
|
|
|
EST. |
This |
stands for |
expressed sequence tag. It could really refer to any piece of cod- |
|
||||||||||
|
ing DNA sequence for which PCR primers have been established. However, in |
|
||||||||||||
|
practice, EST almost always refers to a segment of the DNA sequence of a cDNA. |
|
||||||||||||
|
These |
samples |
are |
usually obtained by starting with an existing cDNA library, |
|
|||||||||
|
choosing clones at random, sequencing as much of them as can be done in a single |
|
|
|||||||||||
|
pass, and then using this sequence information to place the clone on a physical map |
|
|
|||||||||||
|
(through somatic cell genetics or FISH). There are many advantages to such se- |
|
||||||||||||
|
quences as probes. One knows that a gene is involved. Therefore the region of the |
|
||||||||||||
|
chromosome is of potential interest. The bit of DNA sequence obtained may be in- |
|
|
|||||||||||
|
teresting: |
It |
may |
match something already in the database or |
be |
interpretable |
in |
|
||||||
|
some way (see Chapter 15). In general, the kinds of PCR artifacts observed with |
|
||||||||||||
|
STPs, STRPs, and STARs are much less likely to occur with ESTs. |
|
|
|
||||||||||
Despite |
their |
considerable |
appeal |
there |
are a number of potential problems |
in |
dealing |
|
|
|||||
with ESTs as mapping reagents. As shown in Figure 9.12, cDNAs are discontinuous sam- |
|
|
||||||||||||
ples of genomic DNA. They will typically span many exons. This can be very confusing. |
|
|
||||||||||||
If the EST crosses a large |
intron, the probe will show PCR amplification, but genomic |
|
|
|||||||||||
DNA or a YAC clone will not. A common strategy for EST production uses largely un- |
|
|
||||||||||||
translated DNA sequence at the 3 |
|
-ends of the cDNA clones. It is relatively easy to clone |
||||||||||||
these regions, |
and |
they |
are more polymorphic than the internal coding region. |
|
||||||||||
Furthermore cDNAs from gene families will tend to have rather different |
3 |
|
|
|
|
-untranslated |
||||||||
regions, and thus one will avoid some of the problems otherwise encountered with multi- |
|
|
||||||||||||
ple positive PCR reactions |
from members of a gene family. These 3 |
|
|
|
|
|
-end sequences will |
|||||||
also tend to contain only a single exon in front of the untranslated region. However, all of |
|
|
||||||||||||
these advantages carry a price: The 3 |
-end sequence |
is |
less |
interesting and |
interpretable |
|||||||||
than the remainder of the cDNA. |
|
|
|
|
|
|
|
|
||||||
Figure 9.12 A potential problem with ESTs as mapping reagents is that an EST can cross one or more introns.

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
POOLING IN MAPPING STRATEGIES |
303 |
||||||||||
Eugene Sverdlov and his colleagues in Moscow have developed an efficient procedure |
|
|
|
|||||||||||||||||||||||
for preparing chromosome-specific cDNA libraries. Their three-step procedure is an ex- |
|
|
|
|||||||||||||||||||||||
tension of simpler procedures that were tried earlier by others. This procedure uses an ini- |
|
|
|
|||||||||||||||||||||||
tial |
Alu |
-primed PCR reaction to make a cDNA copy of the hnRNA produced in a hybrid |
|
|
|
|||||||||||||||||||||
cell containing just the chromosome of interest. The resulting DNA is equipped with an |
|
|
|
|||||||||||||||||||||||
oligo-G tail, and then a first round PCR is carried out using an oligo-C containing primer |
|
|
|
|||||||||||||||||||||||
and an |
Alu |
primer. Then a second |
round of PCR is done |
with a nested |
|
|
|
|
Alu |
primer. The |
||||||||||||||||
PCR primers are also designed so that |
the first round introduces one restriction site and |
|
|
|
||||||||||||||||||||||
the second round another. The resulting products are then directionally cloned into a vec- |
|
|
|
|||||||||||||||||||||||
tor requiring both sites. In studies to |
date, Sverdlov and his coworkers have found that |
|
|
|
||||||||||||||||||||||
this scheme produces a diverse |
|
set |
of |
highly enriched human cDNAs. Because these |
|
|
|
|||||||||||||||||||
come from |
|
|
Alu s in hnRNA, they will contain introns, and this gives them potential advan- |
|
|
|
||||||||||||||||||||
tages |
as |
mapping |
probes |
when |
compared |
with conventional cDNAs. As with the 3 |
|
|
- |
|||||||||||||||||
cDNAs discussed above, cDNA from hnRNAs will be |
more |
effective |
than |
ordinary |
|
|
|
|||||||||||||||||||
cDNAs in dealing with gene families and in avoiding cross-hybridization with conserved |
|
|
|
|||||||||||||||||||||||
exonic sequences in rodent-human cell hybrids. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
POOLING |
IN MAPPING STRATEGIES |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
All of the above methods for screening |
individual samples in a complex library are fine |
|
|
|
||||||||||||||||||||||
for finding genes. However, they are very inefficient in ordering a whole library. The |
|
|
|
|||||||||||||||||||||||
schematic result of a successful screen for a gene-containing clone in an arrayed library is |
|
|
|
|||||||||||||||||||||||
shown |
in Figure |
9.13 |
|
|
a. |
A single positive clone is detected, presumably containing the |
|
|
|
|||||||||||||||||
sample |
of |
interest. However, from the viewpoint of information |
retrieval, |
this |
result is |
|
|
|
||||||||||||||||||
very weak. A sample array is potentially an extremely informative source of information |
|
|
|
|||||||||||||||||||||||
about |
the |
order of the samples it contains. A single positive hybridization extracts the |
|
|
|
|||||||||||||||||||||
minimum possible amount of |
information |
from the array and |
requires a time-consuming |
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(a) |
|
(b) |
Figure 9.13 |
Hybridization to arrays. ( |
a ) Typical pattern seen when screening a |
library for a par- |
ticular gene. ( |
b ) Ideal pattern of hybridization in interrogating a dense sample array. |
|
|
304 |
|
ENHANCED METHODS FOR PHYSICAL MAPPING |
|
|
|
|
|||||
experiment. From the viewpoint of information theory, a much better designed interroga- |
|
||||||||||
tion of the array would produce the result shown in Figure 9.13 |
|
|
|
b. In this ideal black-and- |
|||||||
white case, roughly half the clones would be scored positive and half negative if we are |
|
||||||||||
considering only positive and negative hybridization results. When the amount |
of |
signal |
|
||||||||
can |
be |
quantitated, much more |
information is potentially available from a single |
test of |
|||||||
the array through all the gray levels seen for each individual clone. |
|
|
|
|
|||||||
|
The |
practical issue is how |
to take advantage of the power of arrays, or pools |
of |
samples, |
||||||
in a maximally efficient way so that all of the clones are ordered accurately with a minimum |
|
||||||||||
number of experiments. The answer will depend critically on the nature |
of the |
errors in- |
|
||||||||
volved in interrogating arrays or pools. We start by considering fairly ideal cases. A detailed |
|||||||||||
analysis |
of the effects of errors in real applications is |
still not available, but |
the power of |
||||||||
these approaches is so great that it appears that a reasonable error rate can be tolerated. |
|
||||||||||
|
The goal is to design probe or sample pools and arrays that allow roughly half of the tar- |
|
|||||||||
gets to be scored positive in each hybridization or PCR. There are a number of different ways |
|
||||||||||
to try to accomplish this. One general approach is to increase the amount of different DNA |
|
||||||||||
sequences in the probes or the targets. The other general approach is to increase the fraction |
|
||||||||||
of potential target DNA that will be complementary to probe sequences. In principle, both |
|
||||||||||
approaches can be combined. A very simple strategy is to use complex probes. For example, |
|
||||||||||
purified large DNA fragments can be used as hybridization probes. DNA from hybrid cell |
|
||||||||||
lines or radiation hybrids can be used as probes. In some or all of these cases, it is helpful to |
|||||||||||
employ human-specific PCR amplification so that the probe contains sufficient concentra- |
|
||||||||||
tions of the sequences that are actually serving to hybridize with specific samples in the tar- |
|
||||||||||
get. |
|
|
|
|
|
|
|
|
|
|
|
|
The logic behind the use of large DNA probes is that they automatically contain conti- |
||||||||||
nuity information, and sets of target clones detected in such a hybridization should lie in |
|||||||||||
the same region of the genome. It is far more efficient, for example, to assign an array of |
|||||||||||
clones |
to |
chromosome regions |
by hybridizing with |
DNA |
purified |
from |
those |
regions, |
|
||
than it |
is to assign the regional location of clones one at a time by hybridizing |
to a panel |
|||||||||
of |
cell |
lines. The key technical advances that makes these new strategies |
possible |
are |
|||||||
PCR amplification of desired sequences and suppression hybridization of undesired re- |
|
||||||||||
peated sequences. |
|
|
|
|
|
|
|
|
|||
|
An alternative to large DNA probes is simple sequence probes. Oligonucleotides of |
||||||||||
lengths 10 to 12 will hybridize with a significant fraction of arrayed cosmids or YACs. |
|||||||||||
Alternatively, one could use simple sequence PCR primers, as we discussed in Chapter 4. |
|||||||||||
There is no reason why probe |
molecules must be used individually. Instead, one |
could |
|
||||||||
make pools of probes and use |
this pool directly in hybridization. By constructing differ- |
||||||||||
ent sets of pools, it is possible, after the fact, to sort out which members of |
which pools |
||||||||||
were responsible for positive hybridization. This turns out to be more efficient in princi- |
|||||||||||
ple. Any sequences can be used in pools. One approach is to select arbitrary, nonoverlap- |
|||||||||||
ping sets of single-copy sequences. Another approach |
is to build |
up pools from |
mixtures |
|
|||||||
of short oligonucleotides. |
|
|
|
|
|
|
|
|
|||
|
Another basic strategy for increasing the efficiency of mapping is to use pools of sam- |
||||||||||
ples. This |
is a necessary part of PCR screening |
methods, but |
there |
is no |
reason why |
it |
|||||
also could not be used for hybridization analyses. We will describe some of the principles
that |
are involved in pooling strategies in the next few sections. These principles apply |
|||
equally whether pools |
of probes or pools of samples are |
involved. |
Basically it would |
|
seem |
that one should be |
able to combine simultaneously sample |
pooling |
and probe pool- |