

Berrar D. et al. - Practical Approach to Microarray Data Analysis
.pdf64 |
Chapter 2 |
Kerr M.K., Churchill G.A. (2001b). Experimental design for gene expression microarrays. Biostatistics 2:183-201.
Kim J.H., Kim H.Y., Lee Y.S. (2001). A novel method using edge detection for signal extraction from cDNA microarray image analysis Exp Mol Med 33:83-8.
Lee M.L., Kuo F.C., Whitmore G.A., Sklar J. (2000). Importance of replication in microarray gene expression studies: statistical methods and evidence from repetitive cDNA hybridizations. Proc Natl Acad Sci 97:9834-9.
Liao B., Hale W., Epstein C.B., Butow R.A., Garner H.R. (2000). MAD: a suite of tools for microarray data management and processing. Bioinformatics 16:946-7.
Nadon R. (2002). Shoemaker J. Statistical issues with microarrays: processing and analysis. Trends Genet 18:265-71.
Rockett J.C., Christopher Luft J., Brian Garges J., Krawetz S.A., Hughes M.R., Hee Kirn K., Oudes A.J., Dix D.J.(2001). Development of a 950-gene DNA array for examining gene expression patterns in mouse testis. Genome Biol 2:Research0014.1-0014.9.
Sherlock G., Hernandez-Boussard T., Kasarskis A., Binkley G., Matese J.C., Dwight S.S., Kaloper M., Weng S., Jin H., Ball C.A., Eisen M.B., Spellman P.T., Brown P.O., Botstein D., Cherry J.M. (2001). The Stanford Microarray Database. Nucleic Acids Res 29:152-5.
Snedecor G.W., Cochran W.G. (1989). Statistical Methods. 8th edition. Iowa State University Press, Ames. 503 pp.
Tran P.H., Peiffer D.A., Shin Y., Meek L.M., Brody J.P., Cho K.W. (2002). Microarray optimizations: increasing spot accuracy and automated identification of true microarray signals. Nucleic Acids Res 30:e54.
Tseng G.C., Oh M.K., Rohlin L., Liao J.C., Wong W.H. (2001). Issues in cDNA microarray analysis: quality filtering, channel normalization, models of variations and assessment of gene effects. Nucleic Acids Res 29:2549-57.
Wolfinger R.D., Gibson G., Wolfinger E.D., Bennett L., Hamadeh H., Bushel P., Afshari C., Paules R.S. (2001). Assessing gene significance from cDNA microarray expression data via mixed models. J Comput Biol 8:625-37.
Yang Y.H., Dudoit S., Luu P., Lin D.M., Peng V., Ngai J., Speed T.P. (2002). Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30:e15.
Chapter 3
MISSING VALUE ESTIMATION1
Olga G. Troyanskaya, David Botstein, Russ B. Altman
Department of Genetics. Stanford University School of Medicine, Stanford, CA, 94305 USA, e-mail: olgat@smi.stanford.edu, botstein@genome.stanford.edu, russ.altman@stanford.edu
1.INTRODUCTION
Gene expression microarray data are usually in the form of large matrices of expression levels of genes (rows) under different experimental conditions (columns), frequently with some values missing. Missing values have a variety of causes, including image corruption, insufficient resolution, or simply dust or scratches on the slide. Missing data may also occur systematically as a result of the robotic methods used to create the arrays. Our informal analysis of the distribution of missing data in real data sets shows a combination of all of these reasons, with none dominating. Experimentalists usually manually flag such suspicious data points and exclude them from subsequent analysis. However, many analysis methods, such as principal components analysis or singular value decomposition, require complete matrices to function (Alter et al., 2000, Raychaudhuri et al., 2000).
Of course, one solution to missing data problem is to repeat the experiment, but this strategy can be expensive. Such approach has been used in the validation of microarray analysis algorithms (Butte et al., 2001). Alternatively, as most often done in day-to-day practice, missing log2 transformed data are replaced by zeros or, less often, by an average expression over the row, or “row average”. This approach is not optimal, since these methods do not take into consideration the correlation structure of the data. Many analysis techniques that require complete data matrices, as well as other analysis methods such as hierarchical clustering, k-means
1 Parts of the work presented in this chapter were originally published in Bioinformatics (Troyanskaya et al., 2001).
66 Chapter 3
clustering, and self-organizing maps, may benefit from using more accurately estimated missing values.
The missing value problem is well addressed in statistics in the contexts of non-response issues in sample surveys and missing data in experiments (Little and Rubin 1987). Common methods include iterative analysis of variance methods, filling in least squares estimates (Yates 1933), randomized inference methods, and likelihood-based approaches (Wilkinson 1958). An algorithm similar to nearest neighbors was used to deal with missing values in CART-like algorithms (Loh and Vanichsetakul 1988). The most commonly applied statistical techniques for dealing with missing data are model-based approaches, and there is not a large published literature concerning missing value estimation for microarray data.
To address the problem of missing value estimation in the context of microarray data, we introduce KNNimpute, a missing value estimation method we developed to minimize data modeling assumptions and take advantage of the correlation structure of the gene expression data. The work that introduced KNNimpute as well as another method SVDimpute (Troyanskaya et al., 2001) was the first published comprehensive evaluation of the issue of missing value estimation specifically as applied to microarray data. Since then, a few other works have at least in part addressed this issue (e.g., Bar-Joseph et al., 2002). Many statistical packages for microarray analysis also have routines for missing value estimation. An important issue in choosing the method to use for estimation is evaluation of the method's performance on microarray data. In this context, issues such as data sets used in evaluation (data type, data size, diversity of data sets, etc.) and quality of the evaluation metric are important.
This chapter will focus on KNNimpute. We suggest specific guidelines for the use of KNNimpute software and recommend specific parameter choices. This chapter also presents results of comparative evaluation of KNNimpute with a singular value decomposition (SVD) based method (SVDimpute), the method of replacing missing values with zeros, and a row average method.
2.ALGORITHMS
2.1KNNimpute Algorithm
The KNN-based method takes advantage of the correlation structure in microarray data by using genes with expression profiles similar to the gene of interest to impute missing values. Let us consider gene i that has one missing value in experiment j. Then this method would estimate the value (i, j) with a weighted average of values in experiment j of K other genes that

3. Missing Value Estimation |
67 |
have a value present in experiment i and expression most similar to j in all experiments other than i. In the weighted average, the contribution of each gene is weighted by similarity of its expression to that of gene A. The exact neighborhood of K neighbors used for estimation in KNNimpute is recalculated for each missing value for each gene to minimize error and memory usage.
We examined a number of metrics for gene similarity (Pearson correlation, Euclidean distance, variance minimization) and Euclidean distance appeared to be a sufficiently accurate norm (data not shown). This finding is somewhat surprising, given that the Euclidean distance measure is often sensitive to outliers, which could be present in microarray data. However, we found that log-transforming the data seems to sufficiently reduce the effect of outliers on gene similarity determination.
2.2SVDimpute Algorithm2
In this algorithm, we use singular value decomposition (3.1) to obtain a set of mutually orthogonal expression patterns that can be linearly combined to approximate the expression of all genes in the data set. Following previous work (Alter et al., 2000), we refer to these patterns, which in this case are identical to the principal components of the gene expression matrix, as eigengenes (Alter et al., 2000, Anderson 1984, Golub and Van Loan 1996).
The matrix  contains eigengenes, and their contribution to the expression in the eigenspace is quantified by the corresponding eigenvalues on the diagonal of matrix
contains eigengenes, and their contribution to the expression in the eigenspace is quantified by the corresponding eigenvalues on the diagonal of matrix  We can then identify the most significant eigengenes by sorting the eigengenes based on their corresponding eigenvalue. It has been shown by (Alter et al., 2000) that several significant eigengenes are sufficient to describe most of the expression data, but the exact number (k) of most significant eigengenes best for estimation needs to be determined empirically by evaluating performance of SVDimpute algorithm while varying
We can then identify the most significant eigengenes by sorting the eigengenes based on their corresponding eigenvalue. It has been shown by (Alter et al., 2000) that several significant eigengenes are sufficient to describe most of the expression data, but the exact number (k) of most significant eigengenes best for estimation needs to be determined empirically by evaluating performance of SVDimpute algorithm while varying  We can then estimate a missing value j in gene i by first regressing this gene against the k most significant eigengenes and then use the coefficients of the regression to reconstruct j from a linear combination of the k eigengenes. The
We can then estimate a missing value j in gene i by first regressing this gene against the k most significant eigengenes and then use the coefficients of the regression to reconstruct j from a linear combination of the k eigengenes. The value of gene i and the
value of gene i and the  values of the k eigengenes are not used in determining these regression coefficients.
values of the k eigengenes are not used in determining these regression coefficients.
SVD can only be performed on complete matrices; thus we originally substitute row average for all missing values in matrix A, obtaining A' .
2 For more on singular value decomposition, see Chapter 5.
3 Details of these experiments are presented in (Troyanskaya et al., 2001)
68 |
Chapter 3 |
We then utilize an expectation maximization (EM) method to arrive at the final estimate. In the EM process, each missing value in A' is estimated using the above-described algorithm, and then the procedure is repeated on the newly obtained matrix, until the total change (sum of differences between individual values of A and A' ) in the matrix falls below the empirically determined threshold of 0.01.
3.EVALUATION
3.1Evaluation Method
Evaluation was performed on three microarray data sets (DeRisi et al., 1997; Gasch et al., 2000; Spellman et al., 1998). Two of the data sets were timeseries data (DeRisi et al., 1997, Spellman et al., 1998), and one contained a non-time series subset of experiments from Gasch et al. (2000). One of the time-series data sets contained less apparent noise (Botstein, personal communication) than the other. We further refer to those data sets by their characteristics: time series, noisy time series, and non-time series.
We removed any rows and columns containing missing expression values from each of the data sets, yielding “ complete” matrices. Then, between 1% and 20% of the data were deleted at random to create test data sets, and each method was used to recover the introduced missing values for each data set. To assess the accuracy of estimation, the estimated values were compared to those in the original data set using the root mean squared (RMS) difference metric. This metric is consistent across data sets because the average data value in data sets is the same after centering. This experimental design allowed us to assess the accuracy of each method under different conditions (type of data, fraction of data missing) and determine the optimal parameters (number of nearest neighbors or eigengenes used for estimation) for KNNimpute and SVDimpute.
3.2Evaluation Results
3.2.1KNNimpute
KNNimpute is very accurate, with the estimated values showing only 6-26% average deviation from the true values, depending on the type of data and fraction of values missing (Figure 3.1). In terms of errors for individual values, approximately 88% of the data points are estimated with RMS error under 0.25 with KNN-based estimation for a noisy time series data set with 10% entries missing (Figure 3.2). Under low apparent noise levels in time series data, as many as 94% of the values are estimated within 0.25 of the
3. Missing Value Estimation |
69 |
original value. Notably, this method is successful in accurate estimation of missing values for genes that are expressed in small clusters (groups of coexpressed genes), while other methods, such as row average and SVD, are likely to be more inaccurate on such clusters because the clusters themselves do not contribute significantly to the global parameters (such as top eigenvalues) upon which these methods rely.
The algorithm is robust to increasing the percentage of values missing, with a maximum of 10% decrease in accuracy with 20% of the data missing (Figure 3.1). However, a smaller percentage of missing data does make imputation more precise (Figure 3.1). We assessed the variance in RMS error over repeated estimations for the same file with the same percent of missing values removed. We performed 60 additional runs of missing value removal and subsequent estimation using one of the time series data sets. At 5% values missing and K = 123, the average RMS error was 0.203, with variance of 0.001. Thus, our results appear reproducible.
KNNimpute is relatively insensitive to the exact value of K within the range of 10 to 20 neighbors (Figure 3.1). When a lower number of neighbors is used for estimation, performance declines primarily due to overemphasis of a few dominant expression patterns. When the same gene is present more than once on the arrays, the method appropriately gives a very strong weight to other clones for that gene in the estimation (assuming that the multiple clones exhibit similar expression patterns). The deterioration in performance at larger values of K (above 20) may be due to noise present in microarray data outweighing the signal in a “neighborhood” that has become too large and not sufficiently relevant to the estimation problem.
Thus, the optimal value for K likely depends on the average cluster size for the given data set.

70 |
Chapter 3 |
Microarray data sets typically involve a large number of experiments, but sometimes researchers need to analyzedata sets with small numbers of arrays. KNNimpute is robust to the number of experiments in the data set

3. Missing Value Estimation |
71 |
and can accurately estimate data for matrices with as low as six columns (Figure 3.3). However, we do not recommend using this method on matrices with less than four columns.
3.2.2SVDimpute and Row Average
SVD-based estimation provides considerably higher accuracy than row average on all data sets (Figure 3.4), but SVDimpute yields best results on time-series data with a low noise level, most likely reflecting the signalprocessing nature of the SVD-based method1. In addition, the performance of SVDimpute is very sensitive to the exact choice of parameters (number of top eigengenes) used for estimation and deteriorates sharply as the parameter is varied from the optimal value (which is unknown for a non-synthetic data set)4.
Estimation by row (gene) average performs better than replacing missing values with zeros, but yields drastically lower accuracy than either KNNor SVDbased estimation (Figure 3.4). As expected, the method performs most poorly on non-time series data (RMS error of .40 and more), but error on other data sets is also drastically higher than both of the other methods. This is not surprising because row averaging assumes that the expression of a gene in one of the experiments is similar to its expression in a different experiment, which is often not true in microarray experiments. In contrast to SVD and KNN, row average does not take advantage of the rich information provided by the expression patterns of other genes (or even duplicate runs of the same gene) in the data set. An in-depth study was not performed on column average, but some experiments were performed with this method, and it does not yield satisfactory performance (results not shown).
4 Detailed results presented in (Troyanskaya et al., 2001)

72 |
Chapter 3 |
4.PRACTICAL GUIDELINES FOR USE OF KNNimpute SOFTWARE
4.1Software Description
KNNimpute5 software is a C++ program that can be used on any UNIXor LINUX-based platform. The program requires a C++ compiler (preferably gcc), and can be used on a Windows platform with a UNIX emulator, for example Cygwin6. Detailed instructions for installation and use of the KNNimpute software are supplied with the download.
4.2Recommendations for Software Use
KNNimpute software requires data input in a tab-delimited text file formatted in preclustering data format (pcl), described at http://genomewww5.stanford.edu/MicroArray/help/formats,shtml#pcl. It is essential to follow the format specifications exactly. To minimize the effect of outliers, it is important to log-transform the data set prior to imputation (a different transformation or other data processing steps may be equally appropriate).
5 KNNimpute can be downloaded at http://smi-web.stanford.edu/projects/helix/pubs/impute/
6 http://www.cygwin.com/
3. Missing Value Estimation |
73 |
The only parameter the user can vary is K, the number of nearest neighbors used in estimation. We recommend setting K in the range of 10-20, with lower K preferable for very large data sets to minimize the amount of time required for estimation. For smaller data size, any K between 10 and 20 is appropriate. We do not recommend the use of KNNimpute with very small (as compared to the number of genes in the genome) data sets, where the method may not be able to identify groups of similarly expressed genes for accurate estimation. However, small data sets that consist of tight clusters of functionally similar genes may be appropriate for estimation. The method should not be used on data sets containing fewer than four experiments.
While KNNimpute provides robust and accurate estimation of missing expression values, the method requires a sufficient percent of data to be present for each gene to identify similarly expressed genes for imputation. Therefore, we recommend exclusion from the data set any genes or arrays with a very large fraction of missing values (for example, genes or arrays with more than 25% of values missing).
4.3Performance of KNNimpute
For a data set with m genes and n experiments and number of nearest neighbors set to k, the computational complexity of the KNNimpute method is approximately  assuming
assuming 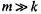 and fewer than 20% of the values missing. Thus, if the size of the dataset doubles in the number of genes, the algorithm will take approximately four times as long to run, while the same increase in the number of experiments would lead to doubling of the original running time. The KNNimpute software (implemented in C++) takes 3.23 minutes on a Pentium III 500 MHz computer to estimate missing values for a data set with 6,153 genes and 14 experiments, with 10% of the entries missing. However, for very large data sets (more than 20,000 genes) the program may need to run overnight or even longer, depending on the performance characteristics of the specific computer system used.
and fewer than 20% of the values missing. Thus, if the size of the dataset doubles in the number of genes, the algorithm will take approximately four times as long to run, while the same increase in the number of experiments would lead to doubling of the original running time. The KNNimpute software (implemented in C++) takes 3.23 minutes on a Pentium III 500 MHz computer to estimate missing values for a data set with 6,153 genes and 14 experiments, with 10% of the entries missing. However, for very large data sets (more than 20,000 genes) the program may need to run overnight or even longer, depending on the performance characteristics of the specific computer system used.
5.CONCLUSIONS
KNNimpute is a fast, robust, and accurate method of estimating missing values for microarray data. Both KNNimpute and SVDimpute methods far surpass the currently accepted solutions (filling missing values with zeros or row average) by taking advantage of the structure of microarray data to estimate missing expression values.
We recommend KNNimpute over SVDimpute method for several reasons. First, the KNNimpute method is more robust than SVD to the type of data for which estimation is performed, performing better on non-time